

SUMMARY
Neuromodulatory input, acting on G-protein coupled receptors, is essential for the induction of experience-dependent cortical plasticity. Here we report that G-coupled receptors in layer II/III of visual cortex control the polarity of synaptic plasticity through a pull-push regulation of LTP and LTD. In slices, receptors coupled to Gs promote LTP while suppressing LTD; conversely, receptors coupled to Gq11 promote LTD and suppress LTP. In vivo, the selective stimulation of Gs- or Gq11-coupled receptors brings the cortex into LTP-only or LTD-only states, which allows the potentiation or depression of targeted synapses with visual stimulation. The pull-push regulation of LTP/LTD occurs via direct control of the synaptic plasticity machinery and it is independent of changes in NMDAR activation or neuronal excitability. We propose these simple rules governing the pull-push control of LTP/LTD form a general metaplasticity mechanism that may contribute to neuromodulation of plasticity in other cortical circuits.
INTRODUCTION
Mechanisms for bidirectional synaptic plasticity such as NMDAR-dependent forms of long-term potentiation (LTP) and depression (LTD) are essential for experience-dependent modification of cortical function (Buonomano and Merzenich, 1998). A widespread consensus model states that the patterns of NMDAR activation and the ensuing increase in intracellular Ca are sufficient to encode the polarity of synaptic changes: changes in Ca above or below a modification threshold resulting in LTP or LTD respectively (Malenka and Bear, 2004). Indeed, in support of this idea, alterations in LTP and LTD induction are often accounted for by changes in NMDAR function. Recent studies indicate, however, that neuromodulators also play a role in determining the polarity of NMDAR-dependent synaptic plasticity through mechanisms that are not fully understood (see (Pawlak et al., 2010).
Experience-induced plasticity depends not only on the patterns of sensory input, but also on neuromodulatory signals related to the behavioral and emotional state of the animal (Bear and Singer, 1986; Conner et al., 2003; Gu, 2002; Hu et al., 2007; Kilgard and Merzenich, 1998). Indeed, visual cortical plasticity depends crucially on the integrity of the cholinergic, adrenergic and serotonergic systems (Bear and Singer, 1986; Gu and Singer, 1995). This permissive function was originally attributed to increased neural excitability and sensory responsiveness (Bear and Singer, 1986; Thomas et al., 1996b). However, neuromodulatory systems have only modest effects on the tuning and signal to noise ratio of visual responses (Ego-Stengel et al., 2002; Zinke et al., 2006), and most plausibly they gate experience-induced plasticity by directly controlling synaptic plasticity mechanisms such as LTP and LTD. Hence understanding the neuromodulation of LTP and LTD is of great significance.
Previous research on the neuromodulation of plasticity uncovered the simple principle that receptors coupled to the Gs-protein selectively gate and promote LTP, whereas the receptors coupled to Gq11 promote LTD (Choi et al., 2005; Kirkwood et al., 1999; Scheiderer et al., 2004; Seol et al., 2007). Importantly, although individually Gs- and Gq11-coupled receptors respectively enable LTP or LTD only, when co-applied they enable spike-timing dependent bidirectional changes (Seol 2007). This suggests that the interaction between the signaling of these two types of receptors is not simply additive. Here we show that receptors coupled to different G-proteins can also selectively suppress LTP or LTD. As a consequence of these opposite actions, G-protein-coupled receptors (GPCRs) regulate LTP and LTD in a pull-push manner: receptors coupled to the adenylyl cyclase signaling pathway via Gs promote LTP and suppress LTD, whereas receptors coupled to phospholipase C via Gq11 promote LTD and suppress LTP. We propose that this neuromodulator-based metaplasticity allows rapid dynamic control of the polarity and gain of NMDAR-dependent synaptic plasticity independent of changes in NMDAR function. We also show that this mechanism can be recruited in vivo and can be used to selectively potentiate or depress targeted synapses.
RESULTS
Selective suppression of LTP and LTD by α1 and β-adrenergic-receptor agonists
Previously we found that neuromodulator receptors coupled to Gs and Gq11 respectively gate the induction of associative LTP and LTD in layer II/III pyramidal cells of visual cortex (Seol et al., 2007). Since the outcome of associative paradigms can be influenced by changes in cellular and network excitability (Pawlak et al., 2010), we decided to study neuromodulation of plasticity with the more efficacious pairing paradigm, and used β and α1 adrenergic receptors as models of Gs and Gq11 coupled receptors, respectively. We studied pairing-induced synaptic plasticity (depolarization to 0 mV to induce LTP, or to - 40 mV, to induce LTD) in two independent pathways converging onto a cell (see methods, supplementary Figure 1). One pathway was not conditioned (Figure 1 open circles) and served as a control to monitor the acute postsynaptic effects of the neuromodulators (Seol et al, 2007). In control conditions (Fig1A), the pairing paradigms induced robust homosynaptic LTP (paired pathway: 163.3 ± 22.8%, non-paired pathway: 95.1 ± 4.4%; paired t-test: p = 0.0017, n=15 slices) and LTD (paired: 77.5 ± 2.8%, non-paired: 100.5 ± 3.9%; paired t-test: p < 0.0001). Pairing did not affect paired-pulse depression, indicating that LTP and LTD are unlikely to be mediated by changes in release probability (Figure 1A). When the pairings were delivered during the end of a bath application of isoproterenol (ISO: 10 μM, 10 min) to activate β-adrenergic receptors LTP induction was robust (paired t-test: p=0.0039) but LTD was impaired (paired t-test: p=0.3507, Figure 1B). On the other hand, bath application of the α1 receptor agonist methoxamine (MTX: 5 μM, 10 min, Figure 1C) produced the opposite effects of isoproterenol: the induction of LTP was impaired (paired t-test: p = 0.5211), but the induction of LTD was robust (paired t-test, p = 0018). Co-activation of both receptors by simultaneous application of both agonists (Figure 1D) led to the induction of both LTP (paired t-test: p = 0.0022) and LTD (paired t-test: p=0.0359). An ANOVA test confirmed the significance of the differences in LTP (F(3,42) = 4.42, p = 0.0085) and LTD (F(3,38) = 14.46, p < 0.00001), and a Newman-Keuls post hoc analysis confirmed that methoxamine blocks LTP, and that isoproterenol blocks LTD.
Figure 1. Selective suppression of LTP and LTD by α1 and β-adrenergic-receptor agonists.

(A) In normal ASCF, pairing at 0 mV (arrow) induces homosynaptic LTP (top graph), while pairing at -40 mV induces homosynaptic LTD (bottom graph). (B). Bath application of the β-adrenergic agonist Isoproterenol (Iso: 10 μM, gray box) barely affects LTP (top), but suppresses LTD (bottom). (C) Bath application of the α-adrenergic agonist Methoxamine (Mtx: 5 μM, gray box) suppresses LTP (top), but not LTD (bottom). (D) Co-application of Iso+Mtx restores bidirectional plasticity. Filled circles in A-D: paired input; open circles: unpaired input. Note that neuromodulators without paired stimulation induce only transient changes in the responses (open circles in B-D). Traces are averages of ten EPSPs recorded before (thin line) and 40 min after (thick line) induction of plasticity. Scale-bars: 2.5 mV, 5 msec. Note the absence of changes in the normalized paired-pulse ratio (PPR) and membrane time constant (τ=RC), which are displayed below each group plot. Here and in subsequent figures data is presented as average ± SEM. The number of experiments is indicated in parentheses.
Pull-push regulation of LTP and LTD by neuromodulators
We hypothesize that Gs-coupled receptors suppress LTD and Gq11-coupled receptors suppress LTP, yet the loss of LTD/P documented in figure 1B,C could also reflect a shift in the optimal parameters for their induction. For example, visual deprivation changes NMDAR function and alters the activity threshold, but not the capacity for LTD induction (Kirkwood et al., 1996; Philpot et al., 2003). We tested this possibility by examining how neuromodulators affect the “voltage-dependence” of pairing-induced synaptic plasticity over a wide range of pairing voltages (from -60mV to 0mV). In control conditions LTP and LTD can be selectively induced by pairing with voltage values above or below a crossover point that occurs at about -20mV (Figure 2A). Isoproterenol eliminated the induction of LTD and lowered the threshold voltage for induction of LTP. On the other hand, methoxamine eliminated LTP and extended the voltage range for LTD induction. These drugs also changed -20 mV from being a membrane potential that is neutral under control conditions to one that induces LTP when Isoproterenol is present and LTD when methoxamine is present (Figure 2B), A 2-way ANOVA test confirmed the significance of the effects of the drugs (F(6,150)=4.627, p=0.0002). These results indicate that the suppression of LTP and LTD by the adrenergic agonists does not result from a change in the induction threshold because each agonist made it not just more difficult, but impossible to induce changes in one or the other polarity in the voltage range we tested
Figure 2. Pull-push regulation of LTP/D by Gs- and Gq11/11-coupled receptors.

(A) Pairing at -20 mV induces no net change in EPSP slope in normal ACSF (open circles), but induces LTP in the presence of isoproterenol (10 μM Iso: black circles) or LTD in the presence of methoxamine (5 μM Mtx: gray circles. (B) Pairing at different voltages induces LTD and LTP (measured 30-40 min after pairing) in control ASCF (open circles), but only LTP after bath applied isoproterenol (black circles), and only LTD after applied methoxamine (gray circles). (C) Pairing at -10 mV (open triangles) and -30 mV (open circles) induce little change in normal ACSF, but induce LTP and LTD after co-application of 10 μM isoproterenol and 5 μM methoxamine (black triangles and circles). (D) Co-application of isoproterenol and methoxamine enhances bi-directional changes (Iso+Mtx: black symbols; control: open symbols). (E-F) LTP-only plasticity also results after activation of the Gs-coupled PGE2 receptor with Butaprost (10 μM: black circles), whereas LTD-only plasticity results after bath application of the Gq11-coupled M1 receptor agonist McN (3 μM: grey circles; 10μM: open circles). The number of experiments is indicated in parentheses.
Co-activation of α- and β-adrenergic receptors restored bidirectional plasticity (Figure 1D), which indicates the suppressive effects mediated by one receptor type can be reversed or counterbalanced by activation of the other type. To determine whether the co-activation also affects the facilitating aspect of neuromodulation of plasticity (Seol et al., 2007), we examined the effects of co-applying 5μM methoxamine and 10μM isoproterenol on the “voltage-dependency” of pairing-induced plasticity. We found that pairing at voltages that normally yield little synaptic changes (-30mV and -10mV) result in robust LTD or LTP in the presence of both agonists (Figure 2C 2-way ANOVA: F(6,150)=4.627, p=0.0002). This increase in the slope of the voltage dependency of pairing induced plasticity indicates that the co-activation of α– and β–adrenergic receptors increases the gain of both LTP and LTD. Thus, the opposite individual effects of the α– and β–adrenergic receptors do not cancel out in a simple linear manner. Rather, the enhancement of one polarity of plasticity by a given adrenergic receptor is not affected by activation of the other receptor.
Finally we examined whether other Gs- and Gq11-coupled receptors regulate plasticity in the same way as α- and β-adrenergic receptors. We found that activation of the prostaglandin E2 receptor (EP2: coupled to Gs) with the agonist butaprost (10μM) resulted in only LTP, whereas the M1 muscarinic receptor (coupled to Gq11) agonist McN severely reduced LTP at 5 μM, and completely abolished it while enhancing LTD at 10 μM (Figure 2E, F). Altogether, the results indicate that neuromodulators 1) do not simply shift the “voltage-dependence” of LTP/D induction, but rather control LTP and LTD in a pull-push manner: promoting one polarity while suppressing the other one, and 2) this regulation is not neurotransmitter specific: two different Gq11-coupled receptors promoted LTD and suppressed LTP, whereas two Gs-coupled receptors promoted LTP and suppressed LTD.
Adrenergic metaplasticity relies on events downstream from NMDAR activation
Adrenergic receptors might control the induction of plasticity by changing the recruitment of NMDA receptors through changes in cell excitability and inhibition (Fuenzalida et al., 2007; Liu et al., 2006; Moore et al., 2009; Tully et al., 2007). To evaluate the contribution of changes in excitability (Hardingham et al., 2008) in the suppression of LTP and LTD we recorded excitatory synaptic currents (EPSC’s) with blockers of Na+, K+ and Ih in the pipette, and using low stimulation intensity to prevent (Hardingham et al., 2008) the recruitment of GABAergic response (see Figure S2). Under these experimental conditions, methoxamine still suppressed LTP (F(2,18) = 8.30, p = 0.0026) and isoproterenol still suppressed LTD (F(2,18) = 25.72, p < 0.0001. Figure S2), indicating that these effects are independent of changes in fast IPSC’s or voltage-dependent conductances.
Next we evaluated the possibility that the suppression of LTP and LTD resulted from agonist-induced postsynaptic changes in NMDAR function (Ji et al., 2008; Liu et al., 2006). We confirmed that isoproterenol enhances and methoxamine reduces the amplitude of the NMDAR-mediated synaptic currents (Figure S3A,B) without affecting the voltage dependence (Figure S3D). Importantly, in both cases the NMDAR-mediated returned to baseline values within 15 min of washing out the drugs (100.2 ± 0.6 % after Iso, n = 15,4, p = 0.163; 100.5 ± 1.9 % after methox, n = 16,5, p = 0.334. Figure S3). We took advantage of this reversibility and applied the LTP/D inducing pairings at least 15 min after washing out the agonists, when NMDAR responses are back to normal. To prevent rundown of plasticity the drugs were applied and washed out before breaking the seal to start the whole-cell recordings (Figure 3A). As shown in figure 3B-D, LTD was suppressed when the pairing was performed 23.5 ± 2.7 min after washing out isoproterenol (F(3,20) = 124.92, p < 0.0001), and LTP was suppressed in cells pretreated (18.9 ± 3.0 min before pairing) with methoxamine (F(3,20) = 197.78, p < 0.0001). These results indicate that 1) each receptor primes synapses into a prolonged suppressive state of LTP or LTD that outlasts the changes in NMDAR function, and 2) the acute changes in NMDAR are not necessary for the suppression of LTP and LTD.
Figure 3. Adrenergic metaplasticity can be primed and is independent of NMDAR.

(A-D) Priming the adrenergic suppression of LTP and LTD . (A) Experimental temporal scheme: the seal was made and ruptured after the 10 min agonist application (10μM Iso, or 5 μM Metx). The time course of the changes in NMDAR-mediated responses are depicted below (thin line). (B-D) Pairing at 0 and -40 mV induced respectively LTP (filled circles) and LTD (open circles) in control conditions (B), but no LTD when the cells were pretreated with isoproterenol (C), and no LTP after pretreatment with methoxamine (D). (E-F) The priming of the adrenergic suppression does not require co-activation of NMDA. Bath application of APV (50μM) at the time of the agonist application (E) does not affect the induction of LTP and LTD (F), nor it affects the suppression of LTD by isoproterenol (G) or LTP by nethoxamine (H). The number of experiments is indicated in parentheses. (I-K) the duration of the priming depends on agonist exposure. (I) LTD magnitude (measured 60 min after pairing) induced at different times after a 10 min isoproterenol exposure. Open circles: individual experiments; filled circles: averages for the time intervals (in min) 21<t<40, 41<t<60, 60<t100. (J-L) One-hour agonist exposure causes a long-lasting suppression of LTD and LTD. (J) Experimental diagram. (K, L) Individual experiments showing the magnitude of LTP (open triangles) and LTD (inverted triangles) induced after prolonged wash out of isoproterenol (K) or methoxamine (L). Filled triangles: experiments in which methoxamine or Isopropterenol were respectively applied (10 min) right before induction of LTD (K) or LTD (L).
We also asked whether NMDAR activation is required during the priming of the suppressive state and co-applied the NMDAR antagonist APV (100μM) with the agonists (Figure 3E) before the pairing. In control cells, pre-treated with APV only (t = 33.25 ± 4.33 min, n = 8,4), the induction of both LTP and LTD was robust (Figure 3F), indicating the successful removal of the drug. Cells pre-treated with APV and isoproterenol (24.7 ± 0.6 min, n=7,3) exhibited robust LTP and no LTD (Figure 3G), whereas cells pre-treated with methoxamine and APV (28.0 ± 1.1 min, n = 8,4) showed normal LTD but no LTP (Figure 3H). A two-way ANOVA test (p<0.001) confirmed the significance of these differences, indicating that suppression of LTP and LTD by α and β adrenergic receptors is initiated and expressed independently of changes in NMDAR function.
Subsequently we evaluated the longevity of the suppression of LTP and LTD. In the experimental setting described in figure 3A, a 10 minutes isoproterenol exposure induces a transient suppression of LTD that recovers within an hour of washout (LTD induced at 25.3±0.9 min: 101±2.9%, at 43.4±0.9 min: 90.3±5.0%, at 75.5±8.5 min: 73.6±4.4%. F(2,22)=14.83, p=0.001. Figure 3H). To explore whether the suppression could last longer we prolonged the agonist exposure. In slices incubated one hour in isoproterenol and tested at least one hour after wash out (97±7% min) LTP induction was robust (140.2±13.6%, paired t-test: p=0.017, n=9) and LTD induction was minimal (100.9±3.9%, p=0.99, n=11. Figure 3H). However robust LTD was induced if the slices were exposed methoxanime for 10 minutes prior the pairing (60.4±10.7%, p=0.008, n=7), indicating that the β-adrenergic suppression of LTD can be reversed (Figure 3H). Similarly, one hour incubation with methoxamine induced a lasting suppression of LTP (LTP: 98.73.1% after 89.3±8.0 min of wash, p=0.56, n=12; LTD: 81.33±2.1%, p<0.001, n=12) that was reversed by 10 min exposure to isoproterenol prior the pairing (163.5±14.5%, p=0.002, n=10). Altogether the results indicate that the suppression of LTD and LTD by β– and α-adrenergic receptors can be long lasting, yet reversible.
Finally, the pull-push regulation of LTP and LTD raised the question of whether the suppression of one form of plasticity depends on the up-regulation of the other form. To address this issue we studied the effects of methoxamine in a phosphomutant mouse line that expresses normal associative LTP but impaired associative LTD (Seol et al., 2007). In these mice serine at position 831 of the GluR1 subunit has been substituted by alanine to prevent phosphorylation, hence the mutation affects only the latest stages of plasticity pathway. We confirmed that the mutant has normal pairing-induced LTP compared to wild type mice (p=0.426. Figure 4A,C) but no LTD (p=0.008. Figure 4C). Interestingly, methoxamine suppressed paring-induced LTP (p=0.0506. Figure 4B,D) in both, wild type and mutant. Thus, the suppression of LTP does not require the expression of LTD.
Figure 4. Adrenergic metaplasticity is determined prior to GluR1-S831 phosphorylation.

Bath applied Methoxamine (Mtx: 5 μM, gray box in B and D) suppresses LTP in S831A phosphomutants and their wild-type littermates (A) Wild-type mice express LTP (filled circles) and LTD (open circles) in control conditions, and suppression of LTP by methoxamine (B). (C) S831A mice express LTP (filled circles) but no LTD (open circles), yet methoxamine still suppresses LTP (D). Note that the transient depression of glutamatergic responses by methoxamine was virtually absent in S831A phosphomutant mice. The number of experiments is indicated in parentheses.
Adrenergic receptors do not affect the reversal of plasticity
The results presented above indicate that the GPCR-suppresses LTP and LTD by affecting the cascade downstream from the initial NMDAR activation step. Next we checked whether the suppression occurs at the end of the cascade –at the level of AMPA receptor trafficking in and out of the synapse. To that end we exploited the facts that LTP and LTD can be both reversed by activity. The reversal of LTP (termed de-potentiation) and LTD (termed de-depression) share common downstream mechanism of expression with LTD and LTP, as they involve changes in AMPA receptor function; yet they differ in induction mechanisms, as they involve different kinase and phosphatase pathways (Hardingham et al., 2008; Lee and Huganir, 2008). We reasoned that if the GPCR-mediated suppression occurs at the expression level (AMPAR trafficking), de-potentiation and de-depression should also be affected.
The experiments were carried out in a two independent inputs setting, to allow internal controls, and using pairing conditioning (to 0mV or −40mV to induce LTP and LTD as well as to reverse them (Figure 5). First LTD was induced in both inputs, and 20 min later one input was de-depressed by pairing with 0mV while the other input was not stimulated. The second pairing effectively reversed LTD in either control conditions (de-depressed vs. non-stimulated; paired t-test: p = 0.0086. Figure 5A), and in the presence of methoxamine (paired t-test: p = 0.0368. Figure 5B), indicating that α1 adrenergic receptors do not suppress de-depression. A similar strategy was used to test the role of β-adrenergic receptors on de-potentiation: LTP induction in both pathways, followed by pairing with −40mV in one input (Figure 5E,F). The second pairing reversed LTP either in control conditions (p=0.0343. Figure 5E) or in the presence of isoproterenol (p = 0.0007. Figure 5F).
Figure 5. Adrenergic receptors do not affect the reversal of plasticity.

In all of the experiments, plasticity was evaluated in two independent pathways converging onto the same postsynaptic cell. (A) In normal ACSF, inputs recently depressed by pairing at -40 mV (downward arrow at -20 min) can be de-depressed by subsequent pairing at 0 mV (upward arrow at 0 min. de-depressed inputs: filled circles; control inputs: open circles). (B) Bath application of methoxamine (MTX: 5 μM grey box) does not interfere with de-depression (de-depressed inputs: filled circles; control inputs: open circles). (C) In normal ACSF pairing at 0 mV (upward arrow at 0 min) induced both de-depression in previously depressed inputs (filled circles), and LTP in naïve inputs (open circles). (D) Bath application of methoxamine suppresses LTP (open circles) but not de-depression (filled circles). (E-F) Pairing at -40 mV (downward arrow at 0 min: filled circles) de-potentiates previously potentiated inputs (filled circles) in either normal ACSF (E) or after bath applied isoprorenenol (F, Iso: 10 mM, grey box). LTP was induced by pairing at 0 mV (upward arrow at -20 min). Control LTP: open circles, Depotentiation: closed circles. (G,H) Pairing at -40 mV (downward arrow at 0 min) induced both depotentiation in previously potentiated inputs (filled circles), and LTD in naïve inputs (open circles) in normal ACSF (G), but only induces depotentiation after bath application of isoproterenol. The number of experiments is indicated in parentheses.
Next we compared the effects of methoxamine on LTD and de-potentiation simultaneously by first inducing LTD in one input and then applying the 0 mV pairing to both inputs. In control experiments (Figure 5C) the second pairing potentiated both the depressed input (p=0.0008) and the naïve (p=0.0038); in the presence of methoxamine (Figure 5D) the depressed inputs potentiated (p=0.0236), but not the naïve inputs (p=0.2054), confirming that α1-adrenergic receptors prevent LTP but they do not affect de-potentiation. The effects of β-adrenergic receptors on LTD and depotentiation were compared with a similar strategy: first LTP induction of one input, followed by simultaneous pairing with -40mV of both potentiated and naïve inputs. Under normal conditions both inputs became depressed (potentiated inputs: p=0.001; naive inputs: p=0.0006. Figure 5G). In contrast, in the presence of isoproterenol only the previously potentiated input became depressed (potentiated inputs: p=0.048; naive inputs: p=0.604. Figure 5H). These results confirmed that β-adrenergic receptors prevent LTD but do not affect de-depression. In sum, the absence of effects of the agonists on the reversal of synaptic plasticity indicates 1) like in CA1 and barrel cortex (Hardingham et al, 2008; Lee and Huganir, 2008) in visual cortex the mechanisms of triggering of cortical LTP and LTD are different from the mechanisms of their reversal, and 2) it is unlikely that the adrenergic receptors affected the latest steps in the plasticity cascade (like the AMPA receptors trafficking) because those steps are seemingly available for the reversal of LTP and LTD.
Adrenergic suppression of LTP and LTD in Layer IV and in Hippocampal CA1 synapses
The pull-push regulation of LTP/LTD could be the primary mechanism of metaplasticity mediated by neuromodulators. Therefore, to evaluate how general the principles described above are, we tested the adrenergic suppression of LTP and LTD in two additional synapse models: the Schaffer collateral input to CA1 in the hippocampus, which is the most comprehensive synaptic model for NMDAR-dependent plasticity, and the ascending inputs from the white matter to layer IV cells (WM->IV).
The WM-IV inputs express pairing-induced NMDAR-dependent LTP/LTD (Figure 6A) for a brief postnatal critical period (Crair and Malenka, 1995; Dudek and Friedlander, 1996; Jiang et al., 2007). In slices from young individuals (P14-P15) isoproterenol selectively blocked LTD (F(3,14) = 14.79, p = 0.0003. Figure 6B), whereas methoxamine selectively blocked LTP (F(3,14) = 17.05, p = 0.0001. Figure 6C. In slices from older rats (P31-32), when plasticity is normally absent (Jiang et al., 2007), the neuromodulators did not promote either LTP (F(3,12) = 2.70, p = 0.1018 not shown) or LTD (F(3,12) = 2.63, p = 0.1066 not shown).
Figure 6. Adrenergic metaplasticity in other models of synaptic plasticity.

(A-C) White matter to layer IV pathway expresses LTP and LTD (A), as well as suppression of LTD by bath-applied isoproterenol (B), and suppression of LTP by methoxamine (C). LTP (filled circles) and LTD (open circles) were simultaneously induced in two independent pathways in the same cells using a coordinated pairing (Suppl. Figure 1). (D, E) In the Schaffer collateral input to CA1, the induction LTD of the fEPSP with LFS (1Hz, 15 min, down arrow) is reduced by pre-exposure to 10 μM isoproterenol (D), whereas the induction of LTP with TBS (arrow) is reduced by pre-exposure to 5 μM methoxamine (E). Filled circle: exposed slices; open circles interleaved controls. The grey bar in A-E depicts the agonist application duration. (F, G) The extent of the suppression depends on the agonist incubation. Experimental scheme is indicated on the top of the graphs. Left: Agonist was applied for 15 - 60 min and washed for 1 hour before testing. Right: Agonist was applied for 1 hour and washed out for 2 hours. (F) Average LTD (60 min.post-LFS) induced after isoproterenol incubation. (G) Average LTP (60 min. post-TBS) induced after methoxamine incubation. Filled circles: exposed slices; open circles interleaved controls. The number of experiments is indicated in parentheses.
Previous studies on Schaffer collateral input to CA1 have shown that activation α1- and β-adrenoreceptors respectively promote LTD and LTP (Choi et al., 2005; Thomas et al., 1996a). To evaluate the suppressive aspect of adrenergic activation we used extracellular methods to induce LTP (theta burst stimulation) and LTD (LFS: 1 Hz. 900 pulses) of the fEPSP (see methods). A brief application of isoproterenol (10uM, 10 min) transiently enhanced the EPSPs and substantially reduced the subsequent induction of LTD 20 minutes later (CTR: 60.1 ± 3/.1%, n = 10; ISO: 84.8 ± 2.9 %, n=8; p<0.001. Figure 6D). Similarly, methoxamine (5 μM, 10 min) transiently reduced the EPSP’s and reduced the magnitude of LTP (CTR:=155.4 ± 5.7%, n = 10; ISO: 119.0 ± 11.6%, n=9; p=0.016. Figure 6E). To evaluate the duration of the suppressive effects CA1 we exposed the slices to the agonists for 15, 30 or 60 min and induced plasticity one or two hour later. One hour after wash out, LTD induction was robust if the exposure to isoproterenol lasted 15 min, it was reduced if the exposure lasted 30 min, and it was minimal if the exposure lasted 60 min (2-way ANOVA: F(1, 34)=12.182, p=0.0014. Figure 6F). However, following a 60 min exposure, the level of LTD induction recovered to normal within 2 hours of wash (CTR: 79.9 ± 2.3%, n = 6; ISO: 87.9 ± 2.3 %, n=6; p<0.134. Figure 6F), indicating the reversibility of the suppression of LTD. In a similar fashion, we found that the magnitude and duration of the suppression of LTP by methoxamine (5 μM) depends on the duration of the agonist exposure (2-way ANOVA: F(1,24)=25.2, p<0.0001. Figure 6G) and it was reversed within two hours (CTR: 150.1 ± 4.1%, n = 4; MTX: 142.4 ± 4.2 %, n=11; p=0.20. Figure 6G). Altogether, these results indicate that the selective adrenergic suppression of LTP and LTD is not restricted to synapses in visual cortical layer II/III.
Neuromodulators gate experience-dependent synaptic scaling of mEPSCs
The neuromodulation of LTP and LTD is an attractive mechanism to subordinate the magnitude and polarity of plasticity to behavioral demands. To examine whether neuromodulation of plasticity is operational in vivo we exploited the fact that α1 adrenergic agonists bring synapses into an “LTD-only” state, whereas β agonists produce an “LTP-only” state (see Figure 2). We reasoned that systemic application of α1 or β agonists in conjunction with visual stimulation to drive activity in V1 should respectively depress or potentiate active synapses in the visual cortex. Thus, anesthetized rats were first injected with α1 or β agonists or vehicle (i.p., 15 mg/kg) and subjected to 1 hour of strong monocular visual stimulation to drive activity in V1 (see methods)(Girman et al., 1999). Then, the changes in synaptic strength were evaluated ex vivo by quantifying miniature EPSCs (mEPSCs) recorded from layer 2/3 pyramidal neurons located in the monocular segment of V1, either contralateral or ipsilateral to the stimulated eye (see Figure 7A,B).
Figure 7. Neuromodulators gate experience-dependent synaptic scaling of mEPSCs from visually stimulated cortex.

(A) Experimental diagram. Prior to recording, rats were injected with isoprorenol or methoxamine (15 mg/kg. i.p.) and subjected to 1 hour of monocular stimulation with drifting gratings (see methods). (B) Example mEPSCs from pyramidal neurons located in contralateral (contra) and ipsilateral (ipsi) hemispheres of a Mtx-treated animal. (C-E) Results obtained in animal treated with methoxamine (C), isoproterenol (D) or control vehicle solution (E). In each case the left panels show the average mEPSCs recorded from contralateral (stimulated, thick colored line) and ipsilateral (non-stimulated, thin black line) hemispheres superimposed with a scaled mEPSC (dashed line); the center panels show the average mEPSC amplitudes from contralateral (black filled dots) and ipsilateral (colored filled dots) hemispheres obtained from each rat (data connected by a straight line); the right panels show the cumulative distribution of mEPSC amplitude for contralateral (thick colored line) and ipsilateral (thin line) hemispheres and scaled distribution (dashed line). The inset shows mEPSC distributions. (F) CPP injection blocks the changes in average mEPSC amplitude in the stimulated cortex induced by methoxamibne (left) or Isoproterenol (right). (G-H) summary plots for average mEPSC amplitude (G) and frequency (H).
The effects of the pre-treatment with α1 agonist methoxamine and monocular stimulation are shown in Figure 7C. On average, mEPSCs recorded in the contralateral (stimulated) V1 were smaller in amplitude than the mEPSCs recorded in the ipsilateral (non-stimulated) cortex (Contra: 9.13 ± 0.07 pA, n = 22 cells; Ipsi: 11.37 ± 0.06 pA, n = 25 cells, 7 rats; p < 0.0001. Figure 7C). 4. The distribution of mEPSC amplitude distributions were significantly different (Wilcoxon test: p < 0.0001) in a multiplicative manner, that is, the distribution of all contralateral mEPSCs is similar to the distribution of all ipsilateral mEPSCs scaled down by a factor of 0.8032 (Wilcoxon test: p = 0.9151). These results are consistent with a scenario in which the stimulation activated most of the synapses and that methoxamine promoted the induction of LTD in these active synapses.
Changes in the opposite direction were observed after pre-treatment with the β agonist isoproterenol. The mEPSCs were larger in the contralateral, stimulated, V1 (Contra = 12.49 ± 0.10 pA, n = 15 cells; Ipsi = 10.55 ± 0.09 pA, n = 16 cells, 6 rats.p < 0.0001. Figure 7D), and these differences were consistently observed across individuals (paired test: p=0.007. Figure 7D). On the other hand, the differences in the mEPSC amplitude distributions were significant (Wilcoxon test p = 0.0016) and multiplicative, with the distribution of the contralateral mEPSCs similar to the ipsilateral mEPSCs scaled up by a factor of 1.183 (Wilcoxon test p = 0.782). In control rats injected with vehicle the average mEPSC amplitude was similar in the contra and ipsilateral cortices (Contra: 11.10 ± 0.10 pA, n = 11 cells; Ipsi: 10.94 ± 0.08 pA, n = 16 cells, 5 rats; Wilcoxon test: p = 0.2375. Figure 7E) in all animals tested (p=0.73, Figure 7E) indicating that visual stimulation per se, does not produce plastic changes in mEPSC amplitude. The distribution of mEPSCs in the ipsilateral (non-stimulated) cortex was similar to the distribution of the contralateral mEPSCs (Wilcoxon test p = 0.4298. Figure 7E), and also similar to the distribution of ipsilateral mEPSCs from rats treated with methoxamine or isoproterenol, supporting the idea that neuromodulators promote changes in activated synapses only. Finally, we examined the role of NMDA receptors and tested the effects of systemic injection of the competitive antagonist CPP (15 mg/kg i.p 20 min prior monocular stimulation), a dose that blocks experience-dependent plasticity without affect visual responses (Frenkel et al., 2006; Sato and Stryker, 2008). The CPP injections consistently abolished the differences in mEPSC amplitude between the contra- and ipsilateral cortices in rats treated with methoxamine (n=5; Wilcoxon test: p = 0.8489) or isoproterenol (n=5; Wilcoxon test: p = 0.9686, Figure 7F), which is consistent with a role of NMDAR in the visually induced plasticity promoted by neuromodulators. A two-way ANOVA test confirmed the significance of the differences in mEPSC amplitude across treatments (F(9,196) = 10.4139, p < 0.001 Figure 7G). The frequency of the mEPSCs, on the other hand, was not affected (Two-way ANOVA F(9,196) = 0.9163, p = 0.512 Figure 7H). Altogether the results indicate that activation of α and β adrenoreceptors can be used to globally potentiate and depress synapses in a controlled manner.
Neuromodulators in conjunction with monocular stimulation occlude the induction of LTP and LTD
The results described above (Fig7) suggest that monocular stimulation induced LTD throughout the contralateral cortex when delivered in conjunction with methoxanine, and induced LTP when delivered with isoproterenol. To further examine this idea we tested whether the treatment with neuromodulators and monocular stimulation, as it induced plasticity in vivo, occludes subsequent pairing-induced LTD or LTP in vitro. In control rats (stimulated but injected with vehicle, n=5 rats, Figure 8B) both hemispheres expressed comparable magnitude of LTP (p = 0.23) and LTD (p = 0.56). In stimulated rats injected with methoxamine (n=7 rats, Figure 8C) LTD was robust in the ipsilateral hemisphere (non-stimulated cortex) but absent in the contralateral one (p < 0.0001), consistent with the idea that LTD was already induced in these synapses. Interestingly, pairing at 0 mV potentiated synapses in the contralateral, but not in the ipsilateral, hemisphere (p<0.0001). One plausible interpretation is that the suppression of LTP by methoxamine is long lasting (see Figure 3) and persists after slice preparation, hence the lack of effects on the ipsilateral side potentiation. In that case, the potentiation induced in the contralateral side would correspond to de-depression of LTD previously induced in vivo with visual simulation. A complementary set of results was obtained when isoproterenol was injected (n= 4 rats, Figure 8D). In this case pairing with 0 mV potentiated synapses only in the ipsilapteral hemisphere (p < 0.0001), whereas pairing with -40mV depressed synapses only in the contralateral hemisphere (p < 0.0001). Altogether, the results support the idea that in vivo each agonist facilitate synaptic changes in one polarity, suppresses changes in the opposite polarity, and do not affect the reversal of plasticity.
Figure 8. Neuromodulators in conjunction with monocular stimulation occlude the induction of LTP and LTD.

(A) Experimental design. Prior to recording LTP and LTD, rats were injected with isoprorenol or methoxamine (15 mg/kg i.p.) and subjected to 1 hour of monocular stimulation with drifting gratings (see methods). (B) Normal pairing induced LTP (filled circles) and LTD (open circles) in both hemispheres (ipsi: left; contra: right) in control rats, stimulated but injected with vehicle solution. (C) In methoxamine-injected rats no LTP was induced in the ipsilateral cortex (non-stimulated), and no LTD was induced in the contralateral (stimulated) cortex. (D) In isoproterenol-injected rats, no LTD was induced in the ipsilateral cortex, and no LTP was induced in the contralateral cortex.
Blockade of β-adrenergic receptors promotes LTD in vitro and experience-dependent downscaling in vivo
We have shown that agonists for specific Gq11- and Gs-coupled receptors can bring synapses to LTD-only or LTP-only states. To complement these findings we asked whether the polarity of plasticity can also be controlled using antagonists to alter the Gs/Gq11 balance set by endogenous neurotransmitters. We focused on blocking the basal activity of β-adrenergic receptors (coupled to Gs) because we previously showed that blocking LTD induction requires antagonists against multiple Gq11-coupled receptors (adrenergic, serotonergic, cholinergic and metabotropic glutamate receptors. Choi et al, 2005). We first examined the effects of the β-antagonist propranolol (5 μM at least 30 min before baseline and throughout the experiments) on plasticity induced in vitro. At this concentration propranolol did not affect baseline responses (93±4% at 20 min, p=0.5, n=5. Data not shown) yet it severely impaired the induction of LTP with 0 mV pairing (CTR: 141.1±5.1, p<0.001, n=20; Prop: 97.5±7.1, p=0.662, n=12. Figure 9A) and promoted the induction of LTD with -20 mV pairing (82.5±8.1, p=0.068, n=12; Prop: 76.8±7.7, p=0.0039. n=11.Figure 9B). Next we examined whether systemic administration of propranolol promotes the induction of LTD in vivo using the experimental design described in Figure 7, which consist of 1 hour of monocular stimulation followed by ex vivo quantification of mEPSCs in the monocular segments of the cortices contra- and ipsilateral to the stimulated eye. In these experiments, we co-injected propranolol (10 mg/kg) with the norepinephrine re-uptake inhibitor maprotiline (10 mg/kg) to boost the endogenous level of norepinephrine. The results, shown in figure 9C, indicate that the average amplitude of all EPSCs recorded in the contralateral (stimulated) cortices was smaller than the average amplitude of the mEPSCs recorded in the ipsilateral (non-stimulated) cortices (Contra: 13.3 ± 0.39 pA, n = 23 cells; Ipsi: 15.28 ± 0.40 pA, n = 25 cells, 5 rats; p < 0.0001. Figure 9C). The distributions of mEPSC amplitude were significantly different (Wilcoxon test: p < 0.0001). The intercortical differences in average mEPSC amplitude were also consistently observed in each of the 5 rats tested (paired t-test: p= 0.0001, Figure 9C). No contra-ipsi differences were detected when monocular stimulation was delivered after injection of either only propranolol (p=0.86, 5 rats. Data not shown) or maprotiline (p=0.57, 5 rats. Data not shown). Altogether, the results indicate that blockade of β-adrenergic receptors and activation α-adrenergic receptors are comparable in promoting experience-dependent synaptic potentiation.
Figure 9. Blocking β-adrenergic receptors promotes LTD in vitro and experience-dependent down-scaling in vivo.
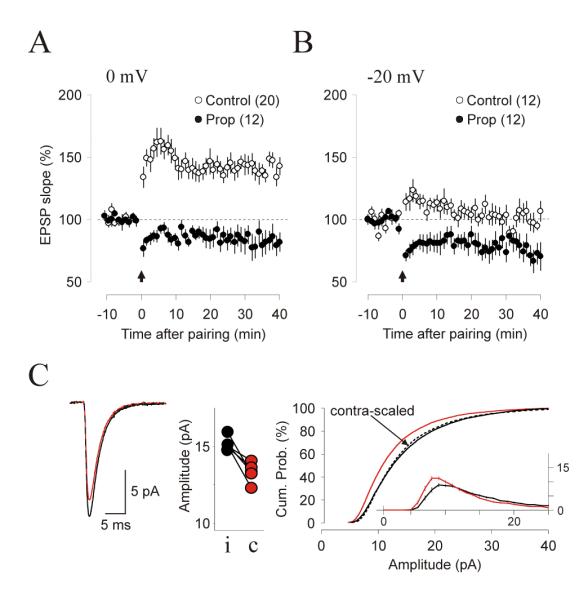
(A, B) Inclusion of 10μM propanolol in the bath blocked the induction of LTP with 0 mV pairing (A) and allowed the induction of LTD with -20 mV pairing (B). Filled circles: in propanolol; open circles: control in ACSF. (C) mEPSC recorded ex vivo from monocularly stimulated rats injected with propanalol (10 mg/Kg) and maprotiline (10mg/Kg) and stimulated monocularly as described in Figures 7 and 8. The left panel show the average mEPSCs recorded from contralateral (stimulated, thick orange line) and ipsilateral (non-stimulated, thin black line) hemispheres superimposed with a scaled mEPSC (dashed line); the center panel show the average mEPSC amplitudes from contralateral (black dots) and ipsilateral (orange dots) hemispheres obtained from each rat (data connected by a straight line); the right panels shows the cumulative distribution of mEPSC amplitude for contralateral (orange line) and ipsilateral (thin line) hemispheres and scaled distribution (dashed line). The inset shows mEPSC distributions.
DISCUSSION
Summary of findings
Neuromodulatory input is critical for the induction of experience-dependent cortical plasticity. Previous studies have shown that Gs-coupled receptors directly promote LTP induction and Gq11-coupled receptors promote LTD (Choi et al., 2005; Scheiderer et al., 2004; Seol et al., 2007). Here we report that G protein-coupled receptors also suppress the induction of LTP and LTD in a G protein-specific manner, independent of changes in neuronal excitability and NMDA receptor activation. This results in a pull-push control of LTP/D in which the polarity of the modulation (facilitation or suppression) depends on the signaling pathway activated by a G-coupled receptor. Receptors coupled to the AC signaling pathway via Gs promote LTP and suppress LTD, whereas receptors coupled to PLC via Gq11 promote LTD and suppress LTP. This pull-push control of LTP/D is operational in vivo and can be recruited to promote and control the polarity of experience dependent synaptic plasticity. We propose that rather than being simple enabling factors, neuromodulators form a metaplasticity system that allows a rapid reconfiguration of the plastic state of cortical synapses over a wide range of possibilities, from LTP-only to LTD-only states.
Mechanism of pull-push neuromodulation
The pull-push control of LTP and LTD appears to result from action at several stages of the induction cascade. We showed previously that G-coupled receptors promote the expression of LTP and LTD by changing the phosphorylation state of AMPA receptors in an NMDAR-independent manner (Seol et al., 2007). Here we show that the suppression of LTP and LTD is also independent of changes in NMDAR function. Although we cannot rule out a change in the Ca2+ signal associated NMDAR activation, the observation that receptors coupled to Gs and Gq11 suppress only one polarity (Fig, 2), argues for an action at a later stage, where the induction pathway for LTP and LTD diverge. An attractive possibility to consider is that G-coupled receptors directly suppress the activation of kinases, like CaMKII, and phosphatases, like PP1, which are essential for LTP and LTD induction (Lisman, 1989; Malenka and Bear, 2004). There are several endogenous inhibitory mechanisms that could be recruited, in principle, by neuromodulators. For example, Gs-coupled receptors, by activating PKA could suppress the activation of PP1 and block the induction of LTD (Lisman, 1989; Malenka and Bear, 2004). On the other hand, Gq11-coupled receptors, through the cascades initiated by PLC, could promote the Ca2+-dependent phosphorylation of CaMKII at the Thr305 inhibitory site (Elgersma et al., 2002; Zhang et al., 2005), and/or the phosphorylation of neurogranin, which is thought to reduce the pool of calmodulin available for CaMKII activation (Huang et al., 2004; Zhabotinsky et al., 2006). Interestingly, genetic ablation of neurogranin and constitutive inhibition of CaMKII by a Thr305D point mutation not only impairs LTP but also extends the range of stimulation frequencies for LTD induction (Huang et al., 2004; Zhang et al., 2005) in a similar fashion as activation of Gq11 receptors extend the voltage range for LTD induction with pairing paradigms (Figure 2). In sum, although the exact mechanism remain to be determined, the available data support a two-step scenario for the pull-push regulation of LTP and LTD, with facilitation occurring at the level of AMPAR phosphorylation and suppression occurring at the signaling between NMDAR activation and AMPAR regulation. A scenario of independent loci for the suppression and facilitation of LTP and LTD, with the additional assumption that the suppression caused by a given receptor can be canceled by the other receptor, could also explain why α- and β-adrenergic agonists applied individually suppress LTP and LTD respectively, but applied together enhance both LTP and LTD. For example, consider that isoproterenol enhances AMPAR insertion into the synapses following a kinase signal, while methoxamine enhances the AMPAR removal dictated by phosphatase signals. If they neutralize their negative effects on kinases and phophatases, the net effect of a co-application would be an enhanced removal or insertion of AMPARs.
Functional consequences of pull-push neuromodulation
The facilitation of LTP and LTD by Gs- and Gq11-coupled receptors, respectively, has been documented in multiple synapses (Choi et al., 2005; Katsuki et al., 1997; Kirkwood et al., 1999; Seol et al., 2007). Here we demonstrated GPCR-mediated suppression of LTP and LTD in the principal cells of layers II/III and IV in visual cortex and in the CA1 subfield of the hippocampus. A suppression of LTD by D1 dopaminergic receptors, coupled to Gs, has also been recently reported in prefrontal cortex (Zhang et al., 2009) and there are multiple reports of negative regulation of LTP by Gq11-coupled glutamate receptors (revised by (Abraham, 2008)). These findings suggest that the pull-push regulation of LTP/D that we described in layer II/III pyramidal cells is common among central synapses. Moreover, we described two properties of the neuromodulation of LTP and LTD that makes it an attractive mechanism for fast metaplasticity. The GPCR-mediated suppression of LTP/D is long lasting (see figs 4 and 7), and the suppressive effects of Gs-coupled GPCR can be reversed or neutralized by Gq11-coupled GPCR, and vice versa. Thus, by changing the Gs/Gq11 balance, neuromodulatory inputs could rapidly reset cortical synapses into states of enhanced LTP or enhanced LTD. On the other hand, a concomitant increase in Gs and Gq11 could enhance the gain for both LTP and LTD and sharpen the boundary for LTP/D. This type of metaplasticity is an attractive mechanism to gate rapid forms of cortical plasticity like perceptual learning. In addition, a basal variability in the state of the Gs/Gq11 balance might relate to the puzzling observation that comparable changes in intracellular Ca2+ might result in LTP or LTD in an unpredictable manner (Ismailov et al., 2004; Kandler et al., 1998; Nevian and Sakmann, 2006).
The pull-push metaplasticity mediated by neuromodulators differs in fundamental features from the well-documented sliding threshold model of metaplasticity. In the sliding threshold model, changes in firing rate over the course of hours or days alters the NMDAR composition at the synapse, consequently modifying the threshold activity for inducing LTP or LTD (Philpot et al., 2003). In contrast, the neuromodulation of LTP/D occurs within minutes and is independent of changes in NMDAR function. These differences likely relate to non-overlapping functions attributed to each metaplasticity mechanism: the sliding threshold would provide long-term stability to the neural circuits, whereas the neuromodulatory systems would operate in faster timescales to subordinate the rules of synaptic modification to the behavioral state of the animal.
In summary, we surmise that besides their established role in neural excitability, neuromodulators can directly control neural plasticity through the pull-push regulation of LTP/D. Thus, in behaving individuals, the polarity and gain of synaptic plasticity would not only depend on intracellular Ca2+ signals, but also on the dynamic balance of Gs- and Gq11 coupled receptors. The experiments described in figures 7, 8 and 9 indicate that this type of metaplasticity can be recruited in vivo. We showed that visual experience in conjunction with systemic application of adrenergic agonists or antagonists, predicted to bring the cortex to an LTD-only or an LTP-only state, respectively depressed and potentiated the postsynaptic strength. Whether these LTP-only and LTD-only states naturally occur in vivo is hard to evaluate, as it would require a detailed knowledge of the state of the various neuromodulatory systems. However, an LTD-only state could conceivable be achieved during REM sleep, when all neuromodulatory systems, except the cholinergic system are silent. The conjunction of an LTD-only state and high levels of activity during REM sleep could provide a cellular basis for the hypothesized sleep-mediated synaptic normalization (Vyazovskiy et al., 2008). It is also tempting to speculate that the enhancement of LTD by propranolol (as shown in figure 9) might contribute to the efficacy of the drug in blocking memory re-consolidation (Debiec and LeDoux, 2006). Finally, it is also interesting to note that the experience induced synaptic changes induced with adrenergic ligands were comparable in magnitude to homeostatic synaptic scaling induced with sensory deprivation. However the effects of the adrenergic ligands are much faster: a 15-20% increase in mini EPSC’s requires 1 hour of stimulation in isoproterenol-injected rats (Figure 7) compared to two 2 days of visual deprivation in normal rats (Desai et al., 2002; Goel et al., 2006). Whether neuromodulators play a role in natural instances of synaptic scaling, as during sleep (Vyazovskiy et al., 2008) or in response to altered sensory experience (Desai et al., 2002; Goel et al., 2006) remains to be determined.
EXPERIMENTAL PROCEDURES
Visual cortical slices (300 μm) from Long-Evans rats and C57BL/6 mice (P20-P30) were prepared as described (Seol et al., 2007). Briefly, slices were cut in ice-cold dissection buffer containing (in mM): 212.7 sucrose, 5 KCl, 1.25 NaH2PO4, 10 MgCl2, 0.5 CaCl2, 26 NaHCO3, 10 dextrose, bubbled with 95% O2/ 5% CO2 (pH 7.4) and transferred to normal artificial cerebrospinal fluid (ACSF) for at least an hour prior to recording. Normal ACSF is similar to the dissection buffer except that sucrose is replaced by 119 mM NaCl, MgCl2 is lowered to 1 mM, CaCl2 is raised to 2 mM. Visualized whole-cell recordings were made from layer II/III (>35% depth from the pia) and layer IV (~40-50% depth from the pia) regular spiking pyramidal-shaped cells with glass pipettes (4-6 MΩ) filled with intracellular solution containing (in mM): 130 (K)Gluconate, 10 KCl, 0.2 EGTA, 10 HEPES, 4 (Mg)ATP, 0.5 (Na)GTP, 10 (Na)Phosphocreatine (pH :7.25, 280-290 mOsm) to record EPSP. To record EPSCs the K-was substituted by Cs and 5 mM QX-314 (lidocaine N-ethyl bromide) was added. Only cells with membrane potentials > - 65 mV, series resistance <20 MΩ, and input resistance > 100 MΩ were studied. Cells were discarded if any of these values changed > 20% during the experiment. Data were filtered at 2 kHz and digitized at 5 kHz using Igor Pro (WaveMetrics Inc., Lake Oswego, Oregon).
Isolated glutamatergic (AMPA/NMDA) currents were evoked in the presence of picrotoxin (10 μM) and using 4 mM Ca2+ and 4 mM Mg2+ in the ACSF to reduce recruitment of polysynaptic responses. NMDAR- and AMPAR-dependent responses were discriminated based on their kinetics and voltage dependence. NMDAR-mediated currents were taken as the amplitude at Vh = +40 mV, 150 ms after the response onset, whereas the AMPAR-mediated currents were taken as the peak amplitude response recorded at Vh = -80 mV. Isolated miniature mEPSCs were recorded at -80 mV (in 1 μM TTX, 100 μM APV and 50 μM picrotoxin, Rin > 200 MΩ) and analyzed as described (Goel & Lee, 2007). See supplemental experimental procedures for more details.
Electrical stimulation and induction of plasticity
Synaptic responses were evoked in two independent pathways at 0.05 Hz with by alternated stimulation (0.2 ms; ≤ 80 μA) through two concentric bipolar electrodes (125 μm diameter; FHC, Bowdoin, ME) placed ~900 μm apart in the white matter, for layer IV recordings, and in the middle of the cortical thickness for layer II/III recordings. Stimulus intensity was adjusted to evoke simple-waveform (2-8 mV), short onset latency (< 2 ms) excitatory postsynaptic potentials (EPSPs). Input independence was confirmed by the absence of paired-pulse interactions. To induce plasticity, the recording mode was switched from current-clamp to voltage-clamp. Pairing consisted of 150 epochs (0.75 Hz) during which Vh was alternated between two target values (666 msec for each value. See supplementary figure 1). Synaptic stimulation was also alternated between pathways and delivered 100 ms after the onset of a Vh pulse. This stimulation protocol allowed us to test input specificity of plasticity or to induce plasticity independently in each pathway. Changes in synaptic strength were quantified as changes in the initial slope of the postsynaptic potential (least-squares linear regression along a 1-2 ms window) normalized by the mean baseline response obtained during the first 10 minutes of stable recordings before drug application. Unless specifically noted the pairing were performed towards the end of agonist application (minute 8-10). All drugs were purchased from sigma. To prevent oxidation, isoproterenol (Iso; 10 μM) and methoxamine (Mtx; 5 μM) were prepared freshly in ASCF containing sodium ascorbate (40 μM).
Visual Stimulation
Animals were anesthetized (pentobarbital 30-50 mg/kg) and placed unrestrained in front of a LCD screen (20 cm in front at an angle of 60° with respect to the animals’ midline) with the eye opposite to the screen covered. Visual stimulation consisted on black and white drifting bars phase-reversing at 1 Hz and rotated with step increments of multiples of 22.5°/min generated with a program written in MATLA (width, 3.72°; length 71°, contrast 100%. Mean luminance, 27 cd/m2; background luminance, 4 cd·m2; frame size 71 by 71°).). Stimulus presentations were interleaved in a randomized fashion and lasted 1 h. Rectal temperature was maintained at 37°C with a heating pad. Eye drops were administered to maintain eye moisture.
Statistical Analysis
Group plots are presented as average ± SEM. The magnitude of plasticity was taken as the average of the last 10 min of recording, beginning 20 or 30 min after conditioning stimulation. Statistical comparisons were done using ANOVA, Wilcoxon and Student’s t tests.
Supplementary Material
Acknowledgments
We thank Drs HK Lee and S. Hendry for valuable comments. Supported by grants from NIH
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986;320:172–176. doi: 10.1038/320172a0. [DOI] [PubMed] [Google Scholar]
- Buonomano DV, Merzenich MM. Cortical plasticity: from synapses to maps. Annu Rev Neurosci. 1998;21:149–186. doi: 10.1146/annurev.neuro.21.1.149. [DOI] [PubMed] [Google Scholar]
- Choi S, J. C, Jiang B, GH S, SS M, JS H, Shin HS, Gallagher M, Kirkwood A. Multiple receptors coupled to PLC gate LTD in visual cortex. J. Neurosci. 2005;25:11433–11443. doi: 10.1523/JNEUROSCI.4084-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Culberson A, Packowski C, Chiba AA, Tuszynski MH. Lesions of the Basal forebrain cholinergic system impair task acquisition and abolish cortical plasticity associated with motor skill learning. Neuron. 2003;38:819–829. doi: 10.1016/s0896-6273(03)00288-5. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE. Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann N Y Acad Sci. 2006;1071:521–524. doi: 10.1196/annals.1364.056. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Friedlander MJ. Developmental down-regulation of LTD in cortical layer IV and its independence of modulation by inhibition. Neuron. 1996;16:1–20. doi: 10.1016/s0896-6273(00)80136-1. [DOI] [PubMed] [Google Scholar]
- Ego-Stengel V, Bringuier V, Shulz DE. Noradrenergic modulation of functional selectivity in the cat visual cortex: an in vivo extracellular and intracellular study. Neuroscience. 2002;111:275–289. doi: 10.1016/s0306-4522(02)00011-8. [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Fedorov NB, Ikonen S, Choi ES, Elgersma M, Carvalho OM, Giese KP, Silva AJ. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron. 2002;36:493–505. doi: 10.1016/s0896-6273(02)01007-3. [DOI] [PubMed] [Google Scholar]
- Frenkel MY, Sawtell NB, Diogo AC, Yoon B, Neve RL, Bear MF. Instructive effect of visual experience in mouse visual cortex. Neuron. 2006;51:339–349. doi: 10.1016/j.neuron.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Fuenzalida M, Fernandez de Sevilla D, Buno W. Changes of the EPSP waveform regulate the temporal window for spike-timing-dependent plasticity. J Neurosci. 2007;27:11940–11948. doi: 10.1523/JNEUROSCI.0900-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girman SV, Sauve Y, Lund RD. Receptive field properties of single neurons in rat primary visual cortex. J Neurophysiol. 1999;82:301–311. doi: 10.1152/jn.1999.82.1.301. [DOI] [PubMed] [Google Scholar]
- Goel A, Jiang B, Xu LW, Song L, Kirkwood A, Lee HK. Cross-modal regulation of synaptic AMPA receptors in primary sensory cortices by visual experience. Nat Neurosci. 2006;9:1001–1003. doi: 10.1038/nn1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience. 2002;111:815–835. doi: 10.1016/s0306-4522(02)00026-x. [DOI] [PubMed] [Google Scholar]
- Gu Q, Singer W. Involvement of serotonin in developmental plasticity in kitten visual cortex. Eur. J. Neurosci. 1995;7:1146–1153. doi: 10.1111/j.1460-9568.1995.tb01104.x. [DOI] [PubMed] [Google Scholar]
- Hardingham N, Wright N, Dachtler J, Fox K. Sensory deprivation unmasks a PKA-dependent synaptic plasticity mechanism that operates in parallel with CaMKII. Neuron. 2008;60:861–874. doi: 10.1016/j.neuron.2008.10.018. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell. 2007;131:160–173. doi: 10.1016/j.cell.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Huang KP, Huang FL, Jager T, Li J, Reymann KG, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci. 2004;24:10660–10669. doi: 10.1523/JNEUROSCI.2213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismailov I, Kalikulov D, Inoue T, Friedlander MJ. The kinetic profile of intracellular calcium predicts long-term potentiation and long-term depression. J Neurosci. 2004;24:9847–9861. doi: 10.1523/JNEUROSCI.0738-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji XH, Cao XH, Zhang CL, Feng ZJ, Zhang XH, Ma L, Li BM. Pre- and postsynaptic beta-adrenergic activation enhances excitatory synaptic transmission in layer V/VI pyramidal neurons of the medial prefrontal cortex of rats. Cereb Cortex. 2008;18:1506–1520. doi: 10.1093/cercor/bhm177. [DOI] [PubMed] [Google Scholar]
- Jiang B, Trevino M, Kirkwood A. Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J Neurosci. 2007;27:9648–9652. doi: 10.1523/JNEUROSCI.2655-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandler K, Katz LC, Kauer JA. Focal photolysis of caged glutamate produces long-term depression of hippocampal glutamate receptors. Nat Neurosci. 1998;1:119–123. doi: 10.1038/368. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Izumu Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J. Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998;279:1714–1718. doi: 10.1126/science.279.5357.1714. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rioult MG, Bear MF. Experience-dependent modification of synaptic plasticity in visual cortex. Nature. 1996;381:526–528. doi: 10.1038/381526a0. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term depression in visual cortex by acetylcholine and norepinephrine. J. Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Huganir RL. AMPA receptor regulation and the reversal of synaptic plasticity -- LTP, LTD, depotentiation, and dedepression. In: Sweatt J, editor. Learning and memory: A comprehensive reference. Elsevier Press; 2008. [Google Scholar]
- Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc. Natl. Acad. Sci. USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yuen EY, Allen PB, Feng J, Greengard P, Yan Z. Adrenergic modulation of NMDA receptors in prefrontal cortex is differentially regulated by RGS proteins and spinophilin. Proc Natl Acad Sci U S A. 2006;103:18338–18343. doi: 10.1073/pnas.0604560103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Moore SJ, Cooper DC, Spruston N. Plasticity of burst firing induced by synergistic activation of metabotropic glutamate and acetylcholine receptors. Neuron. 2009;61:287–300. doi: 10.1016/j.neuron.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Wickens J, Kirkwood A, Kerr J. Timing is not everything: neuromodulation opens the STDP gate. Front. Syn. Neurosci. 2010;2:146. doi: 10.3389/fnsyn.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Espinosa JS, Bear MF. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci. 2003;23:5583–5588. doi: 10.1523/JNEUROSCI.23-13-05583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Stryker MP. Distinctive features of adult ocular dominance plasticity. J Neurosci. 2008;28:10278–10286. doi: 10.1523/JNEUROSCI.2451-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiderer CL, Dobrunz LE, McMahon LL. Novel form of long-term synaptic depression in rat hippocampus induced by activation of alpha 1 adrenergic receptors. J Neurophysiol. 2004;91:1071–1077. doi: 10.1152/jn.00420.2003. [DOI] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O’Dell TJ. Activity-dependent b-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996a;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makinson M, O’Dell TJ. Activity-dependent beta-adrenergic modulation of low frequency stimulation inducedLTP in the hippocampal Ca1 region. Neuron. 1996b;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci U S A. 2007;104:14146–14150. doi: 10.1073/pnas.0704621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci. 2008;11:200–208. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- Zhabotinsky AM, Camp RN, Epstein IR, Lisman JE. Role of the neurogranin concentrated in spines in the induction of long-term potentiation. J Neurosci. 2006;26:7337–7347. doi: 10.1523/JNEUROSCI.0729-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JC, Lau PM, Bi GQ. Gain in sensitivity and loss in temporal contrast of STDP by dopaminergic modulation at hippocampal synapses. Proc Natl Acad Sci U S A. 2009;106:13028–13033. doi: 10.1073/pnas.0900546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kirschstein T, Sommersberg B, Merkens M, Manahan-Vaughan D, Elgersma Y, Beck H. Hippocampal synaptic metaplasticity requires inhibitory autophosphorylation of Ca2+/calmodulin-dependent kinase II. J Neurosci. 2005;25:7697–7707. doi: 10.1523/JNEUROSCI.2086-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke W, Roberts MJ, Guo K, McDonald JS, Robertson R, Thiele A. Cholinergic modulation of response properties and orientation tuning of neurons in primary visual cortex of anaesthetized Marmoset monkeys. Eur J Neurosci. 2006;24:314–328. doi: 10.1111/j.1460-9568.2006.04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.