

Abstract
Friedreich's ataxia (FRDA) is the most common hereditary ataxia, affecting ∼3 in 100 000 individuals in Caucasian populations. It is caused by intronic GAA repeat expansions that hinder the expression of the FXN gene, resulting in defective levels of the mitochondrial protein frataxin. Sensory neurons in dorsal root ganglia (DRG) are particularly damaged by frataxin deficiency. There is no specific therapy for FRDA. Here, we show that frataxin levels can be upregulated by interferon gamma (IFNγ) in a variety of cell types, including primary cells derived from FRDA patients. IFNγ appears to act largely through a transcriptional mechanism on the FXN gene. Importantly, in vivo treatment with IFNγ increases frataxin expression in DRG neurons, prevents their pathological changes and ameliorates the sensorimotor performance in FRDA mice. These results disclose new roles for IFNγ in cellular metabolism and have direct implications for the treatment of FRDA.
INTRODUCTION
Friedreich's ataxia (FRDA) is a devastating orphan disease. Symptoms usually appear late in the first decade or early in the second decade of life, and include features of both peripheral and cerebellar ataxia. Cardiac involvement is very frequent and premature death is often caused by cardiac insufficiency due to dilated cardiomyopathy. Approximately 10% of patients also develop diabetes mellitus (1).
FRDA is caused by defective frataxin expression. Frataxin is a mitochondrial protein, synthesized as a 210-amino acid precursor that is proteolytically processed into a 130-amino acid mature polypeptide (2,3). Frataxin binds iron and it is involved in the assembly of iron-sulfur clusters (ISC) (4,5), prosthetic groups incorporated into several key metabolic enzymes (6). Frataxin-defective cells in fact have reduced activity of ISC-containing enzymes, such as aconitase and succinate dehydrogenase, a general imbalance in intracellular iron distribution and increased sensitivity to oxidative stress. The cells mostly affected by frataxin reduction are the large sensory neurons of dorsal root ganglia (DRG) (7).
There is currently no specific therapy to prevent the progression of the disease (8). Here, we show that frataxin can be upregulated by interferon gamma (IFNγ), a cytokine involved in multiple aspects of iron metabolism and the immune response (9). Most importantly, in vivo treatment with IFNγ increases frataxin levels in DRG neurons and substantially prevents DRG neuronal degeneration and neurological dysfunction in FRDA mice.
RESULTS
During the course of a proteomic screening for proteins differentially expressed in cells derived from FRDA patients, a serendipitous observation suggested that IFNγ might upregulate frataxin. Different IFNγ-responsive cell lines were then exposed to recombinant IFNγ and frataxin accumulation was quantitated after 24h by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and immunoblot analysis. As shown in Figure 1, IFNγ induces the accumulation of frataxin in human cervical carcinoma HeLa cells (Fig. 1A) and in the monocytic leukemia cell line U937 (Fig. 1B) in a dose-dependent manner. Similarly, IFNγ can promote frataxin expression in the human glioblastoma cell line U118 (Fig. 1C). To verify that IFNγ could induce frataxin accumulation in non-transformed cells, resting peripheral blood mononuclear cells (PBMC) from normal individuals were exposed to IFNγ and frataxin accumulation was quantitated by SDS–PAGE and immunoblot analysis. Figure 1D shows that IFNγ can induce frataxin accumulation in resting PBMC in a dose-dependent manner. Together, these data indicate that IFNγ is able to upregulate frataxin levels in a variety of cell types.
Figure 1.

IFNγ induces frataxin accumulation in multiple cell types. HeLa cells (A), U937 cells (B), U118 cells (C) and PBMC isolated from healthy donors (D) were cultured for 24h in the presence of the indicated concentrations of IFNγ, and then whole cell lysates were analyzed by SDS–PAGE and blotted with anti-frataxin and anti-actin mAbs. Representative blots are shown, three to six independent experiments for each cell type were performed.
We then tested whether IFNγ can upregulate frataxin in cells derived from FRDA patients. FRDA-derived GM03816 fibroblasts were exposed for 24h to different doses of IFNγ, and then frataxin was quantitated by SDS–PAGE and immunoblot analysis. Figure 2A shows that IFNγ can induce the upregulation of frataxin in frataxin-defective fibroblasts, in a dose-dependent manner. To verify that IFNγ could be effective on primary FRDA cells, freshly isolated PBMC from several FRDA patients were exposed to different doses of IFNγ for 24h. Frataxin was then quantitated by SDS–PAGE and immunoblot analysis. As shown in Figure 2B, PBMC isolated from a FRDA patient, and treated for 24h with IFNγ, exhibit significantly increased levels of frataxin expression, in a dose-dependent manner. Comparison with the levels of frataxin present in a healthy control (a brother of the patient) indicates that IFNγ induces a substantial recovery of frataxin levels. PBMC isolated from 9 out of 10 FRDA patients tested gave similar results.
Figure 2.

IFNγ induces frataxin accumulation in FRDA cells. FRDA fibroblasts (GM03816 cells) (A) and PBMC freshly isolated from an FRDA patient (B) were cultured for 24h in the presence of the indicated concentrations of IFNγ, and then whole cell lysates were analyzed by SDS–PAGE and blotted with anti-frataxin and anti-actin mAbs. The amount of frataxin present in the PBMC of a healthy sibling of the patient is also shown for comparison (HC). Nine out of 10 different FRDA patients (6 males and 4 females, GAA triplets range 350–915, age range 14–56) tested gave similar results. (C) IFNγ induces accumulation of frataxin mRNA in FRDA cells. FRDA fibroblasts were cultured for the indicated times in the presence of 500 ng/ml of IFNγ, and then frataxin mRNA was quantitated by RT–PCR. The means ± 1 SD from three independent experiments is shown. The increase in frataxin mRNA in IFNγ-treated cells, versus control-treated cells, was significant at 1h (**P < 0.001) and at 2h (*P < 0.05). (D). Actinomycin D blocks IFNγ-induced frataxin mRNA accumulation in FRDA cells. FRDA fibroblasts were pre-treated for 30 min with 5 μg/ml actinomycin D, cultured for 2h in the presence of 500 ng/ml of IFNγ and then frataxin mRNA (black columns) and PA28alpha mRNA (grey columns) were quantitated by RT–PCR. The means ± 1SD from two independent experiments is shown.
To gain insight into the mechanism of frataxin upregulation, we investigated whether IFNγ treatment modulated frataxin mRNA levels. Quantitative RT–PCR analysis showed that a significant increase in frataxin mRNA can be detected in FRDA fibroblasts as early as 1h after exposure to IFNγ, with peak accumulation at 2h and return to baseline levels after 4h (Fig. 2C). Moreover, pre-treatment with actinomycin D completely prevented IFNγ-induced frataxin mRNA accumulation (Fig. 2D). The mRNA accumulation of a control IFNγ-inducible gene, the immunoproteasome component PA28 alpha subunit (10), was similarly prevented. These results strongly suggest that IFNγ induces frataxin accumulation in FRDA cells largely by increasing frataxin transcripts, although additional mechanisms such as mRNA stabilization or protein stabilization cannot be excluded.
To investigate whether IFNγ could be effective in vivo, we utilized the YG8R FRDA mouse model (11,12). This model contains a human genomic FXN transgene, driven by the human FXN promoter, together with expanded GAA repeats within intron 1 of the FXN gene. In addition, the FXN transgenic mice have been crossed with frataxin knockout mice to ensure that the resultant YG8R FRDA mice only express human transgenic frataxin and not any endogenous mouse frataxin. Thirteen FRDA mice were treated with subcutaneous injections of 40 μg/kg IFNγ [in phosphate buffered saline (PBS)], three times/week from 8 weeks of the age for 14 weeks, while 13 FRDA mice of the same age were given PBS, using the same schedule. Every 2 weeks, motor coordination and locomotor activity were assessed. As shown in Figure 3, FRDA mice treated with IFNγ displayed significantly enhanced locomotor activity, as measured by ambulatory distance (Fig. 3A), average velocity (Fig. 3B) and vertical counts (Fig. 3C), compared with PBS-treated FRDA mice. Perhaps more importantly, motor coordination, as measured by rotarod performance, improved dramatically in IFNγ-treated mice after 10 weeks of treatment compared with PBS-treated mice (Fig. 3D). Better performances in locomotor activity and motor coordination in IFNγ-treated mice occurred independently of body weight changes (Fig. 3E).
Figure 3.

In vivo IFNγ treatment improves locomotor and motor coordination performances in FRDA mice. Two groups of 13 eight-week-old FRDA mice were used, one group injected subcutaneously with 40 μg/kg IFNγ, three times/week for 14 weeks and the other with PBS. Every 2 weeks, functional evaluation of locomotor and coordination activity was performed. Mean values for each functional parameter measured from all 13 FRDA mice of each group are shown at the indicated time points. Both locomotor parameters, such as ambulatory distance (A, P < 0.01), average velocity (B, P < 0.01), vertical counts (C, P < 0.001) and coordination parameters such as rotarod performance (D, P < 0.001) gave significantly different results between the two groups. Body weight was also measured at every time point (E). Squares: IFNγ-treated animals, triangles: PBS-treated animals.
We then analyzed the expression of human frataxin, as well as the pathological features, in DRG neurons from FRDA mice, following IFNγ treatment (40 μg/kg IFNγ three times/week for 14 weeks) or PBS treatment. Higher levels of human frataxin expression can be detected in DRG tissue samples from IFNγ-treated FRDA mice, compared with PBS-treated FRDA mice, as detected by immunohistochemistry (Fig. 4A–D). Accordingly, the accumulation of mature human frataxin in DRG from IFNγ-treated FRDA mice, compared with PBS-treated FRDA mice can also be detected by SDS–PAGE/western blot analysis, both in female and male mice groups (Fig. 4E and F). Most importantly, DRG degeneration is prevented by in vivo IFNγ treatment. HE staining of DRG tissue collected from FRDA mice that received PBS shows several neurons with typical vacuolar degeneration (Fig. 5A, arrows), as previously described (11,13). These features were virtually absent in FRDA mice treated with IFNγ (Fig. 5B). Quantitation and statistics of vacuolated DRG neurons in PBS and IFNγ-treated FRDA mice is shown in Figure 5C.
Figure 4.

In vivo IFNγ treatment upregulates human frataxin in DRG of FRDA mice. Immunohistochemistry of DRG tissue from PBS-treated FRDA mice (A and C) and IFNγ-treated FRDA mice (B and D), after 14 weeks of treatment. Immunostaining (see Materials and Methods) was performed in the presence (A and B) or absence (C and D) of anti-frataxin mAb, followed by anti-mouse IgG secondary antibody treatment and DAB staining. All four panels were counterstained with hematoxylin. (E). SDS–PAGE analysis of cell extracts from pooled DRG neurons from male (M, n = 3) and female (F, n = 2) FRDA mice, treated with either PBS or 40 μg/kg IFNγ three times/week for 14 weeks. Blots were analyzed for the mitochondrial protein VDAC (as negative control), for the IFNγ-inducible PA28alpha (as positive control), and for human frataxin. (F). Densitometric analysis of the results shown in (E). Data are presented as % fold induction of frataxin accumulation in DRG neurons of IFNγ-treated FRDA mice compared with frataxin levels in DRG neurons of PBS-treated FRDA mice.
Figure 5.
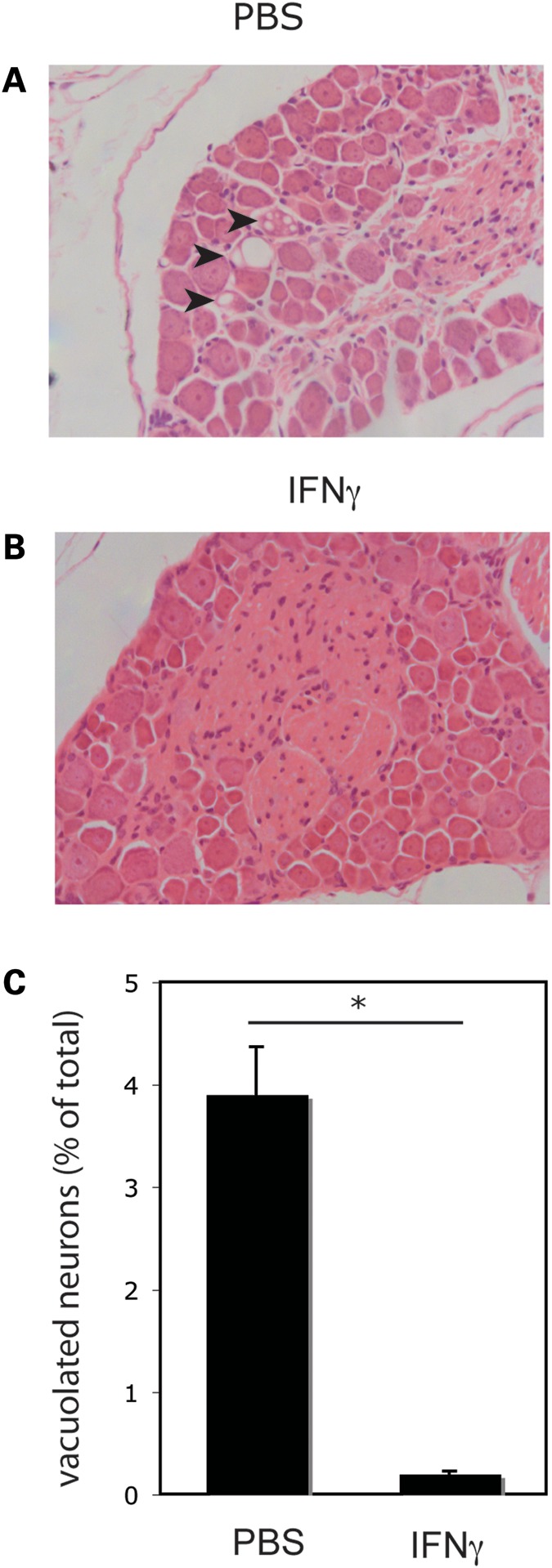
In vivo IFNγ treatment prevents degeneration of DGR neurons of FRDA mice. HE staining of DRG sections from FRDA mice treated for 14 weeks with PBS (A) or IFNγ (B). Arrows show vacuolar degeneration in the cytoplasm of three DRG neurons from PBS-treated FRDA mice. (C). Quantitation and statistical analysis of DRG neurons shown in (A) and (B). Six DRG HE sections from each of the three mice for each group (IFNγ-treated and PBS-treated, total of 18 sections per group) were analyzed and vacuolated neurons were quantitated as a percentage of total neurons counted. A significant (*P < 0.01) reduction was detected in the IFNγ-treated group compared with the PBS-treated group.
DISCUSSION
Our data indicate that exposure to IFNγ induces the upregulation of cellular frataxin. Innate immunity actively controls iron metabolism during microbial infections (14). Within hours from the detection of bacterial proliferation, a massive redistribution of bioavailable iron between the extracellular fluids and the cells of the reticulo-endothelial system, is orchestrated by inflammatory cytokines, primarily by IFNγ (15). This process, highly conserved in evolution, is mainly aimed at restricting iron access to invading pathogens, while preserving iron availability to the benefit of the host defense (16). In particular, IFNγ appears to extensively control iron distribution and availability by directly modulating the expression of a number of players in iron metabolism, including ferritin (17), the transferrin receptor (17,18), the iron exporter ferroportin (19), its peptide ligand hepcidin (20), IRP1 (21) and the iron symporter NRAMP1 (22). In general, extracellular iron is reduced, and iron is sequestered within cells. This scenario might justify a requirement for additional frataxin as a regulator of iron redistribution (23), particularly controlling the access of extra iron to the mitochondrial ISC machinery, in keeping with a recently proposed hypothesis (5).
IFNγ also triggers a variety of biological effects aimed at regulating the immune response and counteract infections (24). By directly inducing components of the reduced nicotinamide adenine dinucleotide phosphate oxidase complex and iNOS, IFNγ controls the generation of reactive oxygen species and nitric oxides (NO, including intermediates and derivatives), both required for optimal antimicrobial responses. However, NO are known to cause nitrosylation and consequent disruption of prosthetic ISC (25), suppressing the activity of ISC-containing enzymes, such as aconitase. The upregulation of frataxin might therefore be required to compensate and/or prevent possible ISC disruption, and consequent loss of function of ISC-containing proteins, in cells exposed to IFNγ. Relevant to this, we observed that the IFNγ-induced upregulation of frataxin in FRDA cells results also in a strong upregulation of aconitase activity (unpublished data).
Most importantly, our data show that FRDA mice improve both locomotor activity and motor coordination, when treated with IFNγ, and that this is paralleled by both frataxin upregulation and neuronal preservation in DRG. IFNγ is usually produced by lymphocytes, but, intriguingly, DRG neurons can both release and respond to IFNγ in an autocrine/paracrine fashion (26). It is therefore possible that the interruption of an IFNγ-dependent feed-forward trophic mechanism (27), sustaining frataxin levels, might contribute to the exquisite sensitivity of DRG to frataxin deficiency. Exogenously administered IFNγ might reach relevant neurons in disarranged FRDA DRG (7) to rescue frataxin levels and grant cell survival. While the specific target tissues for IFNγ in FRDA remain to be further characterized, these data are immediately relevant to the search for effective treatment of FRDA. No approved cure is currently available to FRDA patients (8). Current therapeutic approaches are based on the use of anti-oxidants or iron chelators, with controversial results. Other strategies are in earlier clinical phases or pre-clinical stage (13,28). Recombinant IFNγ is approved for therapeutic use (29). Our data warrant an evaluation of IFNγ as a treatment for FRDA.
MATERIALS AND METHODS
Cell cultures
HeLa (human cervical carcinoma), U937 (monocytic leukemia) and U118 (human glioblastoma) cell lines were obtained from the European cell culture collection. Human fibroblasts derived from a FRDA patient (GM03816) were obtained from the National Institute of General Medical Sciences (NIGMS), Human Genetic Cell Repository. Recombinant human and murine IFNγ were from Peprotech.
Immunoblotting
Total cell extracts were prepared in ice-cold radio immuno precipitation assay lysis buffer. Proteins were separated on 12% SDS–PAGE, electroblotted on Protran Nitrocellulose Membranes (Whatman) and analyzed by ECL detection (GE Healthcare Life Sciences) with mAb anti-frataxin (MAB-10876 Immunological Sciences), mAb anti-actin (Sigma), mAb anti-PA28alpha (ENZO Lifesciences) and pAb anti-VDAC (Abcam).
Quantitative RT–PCR
Total RNA (500 ng) isolated from FRDA fibroblasts was extracted using TRI-zol reagent (Invitrogen) and cDNA was then prepared by using SuperScript VILO cDNA synthesis kit (Invitrogen). Levels of human FXN mRNA expression were assessed by quantitative RT–PCR using an ABI Prism 7000 sequencer and SYBR Green (Applied Biosystems) with the following primers: RTFXNFWD 5′-CATACACGTTTGAGGACTATGATGTCT-3′ and RTFXNREV 5′-TTCGGCGTCTGCTTGTTGATC-3′ (Invitrogen) and QuantiTect Primer Assay (200) (QT00095431) (Qiagen) for actin primers as housekeeping gene. Quantitative real-time PCR analysis was carried out using the 2(-Delta Delta C(T)) method. Statistical analysis was performed using a Student's t-test. In some experiments, FRDA fibroblasts were pre-treated for 30′ with 5 μg/ml of actinomycin D (Sigma Aldrich), and then treated with 500 ng/ml of IFNγ for 2h. Levels of human PA28alpha mRNA expression were assessed with the following primers: RTPA28alphaFWD 5′-TCCTTCTGCAGCGCTTGAA-3′ and RTPA28alphaREV 5′-CTCAATCCGAGGTATCTGCAGC-3′.
Animal procedures and behavioral assessments
The YG8R mice were established by cross breeding YG8 human genomic YAC transgenic mice that contain the entire FXN gene and expanded GAA repeats, with heterozygous Fxn-knockout mice (12). Thirteen YG8R mice starting at 8 weeks of age were given subcutaneous injection of 40 μg/kg murine IFNγ (PeproTech) three times/week for 14 weeks. Thirteen age-matched YG8R mice were given PBS as control, using the same schedule. Tissues were collected 4 h after the last injection. Rotarod performances were assessed using an Ugo Basile 7650 accelerating rotarod treadmill (12). Ambulatory distance, average velocity and vertical counts (mouse lifts up) were measured over 1 min periods in the dark and repeated four times for each mouse using a beam-breaker activity monitor (Medical Devices, Inc.). The mean performance for each group of mice was calculated at the indicated times. Data were analyzed by two-way analysis of variance.
DRG histology and immunohistochemistry
Histological preparations of mouse DRG were carried out by dissection of paraformaldehyde-fixed intact lumbar vertebrae, followed by decalcification treatment in Hillman and Lee's ethylenediaminetetraacetic acid, daily for 5 days. Tissues were then embedded in paraffin wax, sectioned, deparaffined and stained with hematoxylin/eosin. Immunostaining was performed on deparaffined sections after blocking with MOM mouse IgG (Vector Labs). Sections were incubated overnight with anti-frataxin mAb (1G2), followed by biotinylated anti-mouse IgG secondary antibody and an avidin-biotin complex (Vectastain Elite) reagent, and finally colored with the DAB substrate kit for peroxidase.
FUNDING
R.T. was supported in part by AtaxiaUK, National Ataxia Foundation, USA, Friedreich Ataxia Research Alliance (FARA), USA and Telethon-Italy (Grant GGP11102). M.P. was supported by AtaxiaUK, FARA, GoFAR, the Wellcome Trust (089757) and the EU FP7 (242193/EFACTS). Funding to pay the Open Access publication charges for this article was provided by Fondazione Telethon.
ACKNOWLEDGEMENTS
The authors thank Prof. Sergio Bruni, and Drs Alessandro Torgovnick, Natascia Ventura, Barbara Capuani, Roberto Di Fabio and Giulia Donadel for technical help and insightful discussion, and Lorraine Lawrence (Imperial College London) for assistance with histology.
Conflict of Interest statement. None declared.
References
- 1.Pandolfo M. Friedreich ataxia: the clinical picture. J. Neurol. 2009;256(Suppl. 1):3–8. doi: 10.1007/s00415-009-1002-3. [DOI] [PubMed] [Google Scholar]
- 2.Condò I., Ventura N., Malisan F., Rufini A., Tomassini B., Testi R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007;16:1534–1540. doi: 10.1093/hmg/ddm102. [DOI] [PubMed] [Google Scholar]
- 3.Schmucker S., Argentini M., Carelle-Calmels N., Martelli A., Puccio H. The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 2008;17:3521–3531. doi: 10.1093/hmg/ddn244. [DOI] [PubMed] [Google Scholar]
- 4.Stemmler T.L., Lesuisse E., Pain D., Dancis A. Frataxin and mitochondrial Fe-S cluster biogenesis. J. Biol. Chem. 2010;285:26737–26743. doi: 10.1074/jbc.R110.118679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adinolfi S., Iannuzzi C., Prischi F., Pastore C., Iametti S., Martin S.R., Bonomi F., Pastore A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009;16:390–396. doi: 10.1038/nsmb.1579. [DOI] [PubMed] [Google Scholar]
- 6.Yoon T., Cowan J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003;125:6078–6084. doi: 10.1021/ja027967i. [DOI] [PubMed] [Google Scholar]
- 7.Koeppen A.H. Friedreich's ataxia: pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011;303:1–12. doi: 10.1016/j.jns.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marmolino D. Friedreich's ataxia: past, present and future. Brain Res. Rev. 2011;67:311–330. doi: 10.1016/j.brainresrev.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Young H.A., Bream J.H. IFN-γ: recent advances in understanding regulation of expression, biological functions, and clinical applications. Curr. Top. Microbiol. Immunol. 2007;316:97–117. doi: 10.1007/978-3-540-71329-6_6. [DOI] [PubMed] [Google Scholar]
- 10.Strehl B., Seifert U., Kruger E., Heink S., Kuckelkorn U., Kloetzel P.M. Interferon-γ, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol. Rev. 2005;207:19–30. doi: 10.1111/j.0105-2896.2005.00308.x. [DOI] [PubMed] [Google Scholar]
- 11.Al-Mahdawi S., Pinto R.M., Ismail O., Varshney D., Lymperi S., Sandi C., Trabzuni D., Pook M. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum. Mol. Genet. 2008;17:735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- 12.Al-Mahdawi S., Pinto R.M., Varshney D., Lawrence L., Lowrie M.B., Hughes S., Webster Z., Blake J., Cooper J.M., King R., et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandi C., Pinto R.M., Al-Mahdawi S., Ezzatizadeh V., Barnes G., Jones S., Rusche J.R., Gottesfeld J.M., Pook M.A. Prolonged treatment with pimelic o-aminobenzamide HDAC inhibitors ameliorates the disease phenotype of a Friedreich ataxia mouse model. Neurobiol. Dis. 2011;42:496–505. doi: 10.1016/j.nbd.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins H.L. Withholding iron as a cellular defence mechanism—friend or foe? Eur. J. Immunol. 2008;38:1803–1806. doi: 10.1002/eji.200838505. [DOI] [PubMed] [Google Scholar]
- 15.Ganz T. Iron in innate immunity: starve the invaders. Curr. Opin. Immunol. 2009;21:63–67. doi: 10.1016/j.coi.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nairz M., Schroll A., Sonnweber T., Weiss G. The struggle for iron—a metal at the host-pathogen interface. Cell Microbiol. 2010;12:1691–1702. doi: 10.1111/j.1462-5822.2010.01529.x. [DOI] [PubMed] [Google Scholar]
- 17.Byrd T.F., Horwitz M.A. Regulation of transferrin receptor expression and ferritin content in human mononuclear phagocytes. Coordinate upregulation by iron transferrin and downregulation by interferon γ. J. Clin. Invest. 1993;91:969–976. doi: 10.1172/JCI116318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feelders R.A., Vreugdenhil G., Eggermont A.M., Kuiper-Kramer P.A., van Eijk H.G., Swaak A.J. Regulation of iron metabolism in the acute-phase response: interferon γ and tumour necrosis factor alpha induce hypoferraemia, ferritin production and a decrease in circulating transferrin receptors in cancer patients. Eur. J. Clin. Invest. 1998;28:520–527. doi: 10.1046/j.1365-2362.1998.00323.x. [DOI] [PubMed] [Google Scholar]
- 19.Van Zandt K.E., Sow F.B., Florence W.C., Zwilling B.S., Satoskar A.R., Schlesinger L.S., Lafuse W.P. The iron export protein ferroportin 1 is differentially expressed in mouse macrophage populations and is present in the mycobacterial-containing phagosome. J. Leukoc. Biol. 2008;84:689–700. doi: 10.1189/jlb.1107781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sow F.B., Florence W.C., Satoskar A.R., Schlesinger L.S., Zwilling B.S., Lafuse W.P. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J. Leukoc. Biol. 2007;82:934–945. doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]
- 21.Kim S., Ponka P. Effects of interferon-γ and lipopolysaccharide on macrophage iron metabolism are mediated by nitric oxide-induced degradation of iron regulatory protein 2. J. Biol. Chem. 2000;275:6220–6226. doi: 10.1074/jbc.275.9.6220. [DOI] [PubMed] [Google Scholar]
- 22.Alter-Koltunoff M., Goren S., Nousbeck J., Feng C.G., Sher A., Ozato K., Azriel A., Levi B.Z. Innate immunity to intraphagosomal pathogens is mediated by interferon regulatory factor 8 (IRF-8) that stimulates the expression of macrophage-specific Nramp1 through antagonizing repression by c-Myc. J. Biol. Chem. 2008;283:2724–2733. doi: 10.1074/jbc.M707704200. [DOI] [PubMed] [Google Scholar]
- 23.Richardson D.R., Huang M.L., Whitnall M., Becker E.M., Ponka P., Rahmanto Y.S. The ins and outs of mitochondrial iron-loading: the metabolic defect in Friedreich's ataxia. J. Mol. Med. 2010;88:323–329. doi: 10.1007/s00109-009-0565-x. [DOI] [PubMed] [Google Scholar]
- 24.Saha B., Jyothi Prasanna S., Chandrasekar B., Nandi D. Gene modulation and immunoregulatory roles of interferon γ. Cytokine. 2010;50:1–14. doi: 10.1016/j.cyto.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Vanin A.F. Dinitrosyl iron complexes with thiolate ligands: physico-chemistry, biochemistry and physiology. Nitric Oxide. 2009;21:1–13. doi: 10.1016/j.niox.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Neumann H., Schmidt H., Wilharm E., Behrens L., Wekerle H. Interferon γ gene expression in sensory neurons: evidence for autocrine gene regulation. J. Exp. Med. 1997;186:2023–2031. doi: 10.1084/jem.186.12.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jonakait G.M., Wei R., Sheng Z.L., Hart R.P., Ni L. Interferon-γ promotes cholinergic differentiation of embryonic septal nuclei and adjacent basal forebrain. Neuron. 1994;12:1149–1159. doi: 10.1016/0896-6273(94)90322-0. [DOI] [PubMed] [Google Scholar]
- 28.Rufini A., Fortuni S., Arcuri G., Condo I., Serio D., Incani O., Malisan F., Ventura N., Testi R. Preventing the ubiquitin-proteasome-dependent degradation of frataxin, the protein defective in Friedreich's ataxia. Hum. Mol. Genet. 2011;20:1253–1261. doi: 10.1093/hmg/ddq566. [DOI] [PubMed] [Google Scholar]
- 29.Miller C.H., Maher S.G., Young H.A. Clinical use of interferon-γ. Ann. N Y Acad. Sci. 2009;1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]