

Abstract
Human apurinic/apyrimidinic (AP) endonuclease 1 (APE1) is a central participant in the base excision repair pathway, exhibiting AP endonuclease activity that incises the DNA backbone 5′ to an abasic site. Besides its prominent role as a DNA repair enzyme, APE1 was separately identified as a protein called redox effector factor 1, which is able to enhance the DNA binding activity of several transcription factors through a thiol-exchange-based reduction–oxidation mechanism. In the present study, we found that human APE1 is S-glutathionylated under conditions of oxidative stress both in the presence of glutathione in vitro and in cells. S-glutathionylated APE1 displayed significantly reduced AP endonuclease activity on abasic-site-containing oligonucleotide substrates, a result stemming from impaired DNA binding capacity. The combination of site-directed mutagenesis, biochemical assays, and mass spectrometric analysis identified Cys99 in human APE1 as the critical residue for the S-glutathionylation that leads to reduced AP endonuclease activity. This modification is reversible by reducing agents, which restore APE1 incision function. Our studies describe a novel posttranslational modification of APE1 that regulates the DNA repair function of the protein.
Keywords: APEX1, base excision DNA repair, posttranslational modification, cysteine glutathionylation, redox regulation
Introduction
Human apurinic/apyrimidinic (AP) endonuclease 1 (APE1) is a central participant in the base excision repair pathway that deals primarily with non-bulky base modifications, abasic sites, and different chemical forms of single-strand breaks.1–3 APE1 possesses AP endonuclease and 3′-phosphodiesterase activities that contribute to the repair of DNA modifications arising from spontaneous decomposition, reactions with reactive oxygen/nitrogen species, or exposure to exogenous DNA-damaging agents.1–3 Besides its predominant role as a DNA repair enzyme, APE1 was separately identified as “redox effector factor 1” (Ref-1). In this capacity, APE1/Ref-1 can enhance the DNA binding activity of several transcription factors [including AP-1 (Fos/Jun), NF-κB, HIF-1α, and p53] by modulating their reduction-oxidation (redox) status.4–7 Specifically, APE1 reduces either a disulfide bond or a sulfenic acid formed upon oxidation of cysteine (Cys) residues within the target protein to promote DNA binding and transcriptional activities. Expression of the downstream target genes is important in directing processes such as cell cycle regulation, cell proliferation, angiogenesis, apoptosis, and survival.4–7 Given the multifunctional role of APE1 in dictating the cellular response/phenotype, it is not surprising that the protein is essential for both normal and cancer cell viabilities.8–17 Delineating the regulation of APE1 function is, therefore, important in determining the biological outcome to various intracellular and extracellular stimuli.
A complex set of posttranslational modifications can regulate APE1 functions. These modifications include phosphorylation, acetylation, and ubiquitination. For instance, APE1 phosphorylation by casein kinase II completely abolishes AP endonuclease activity.18 Fritz and Kaina also reported APE1 phosphorylation by casein kinase II;19 however, they found that its phosphorylation did not affect AP endonuclease activity but instead enhanced its Ref-1 function in promoting the DNA binding activity of the AP-1 transcription factor. Additionally, APE1 is a substrate for protein kinase C, whereby phosphorylation increases APE1 in vitro redox activity.20 More recent work found that APE1 serves as a nuclear substrate for the cyclin-dependent kinase 5/p35 complexes.21 Phosphorylation of APE1 by cyclin-dependent kinase 5/p35 complexes inhibits AP endonuclease activity, leading to neurotoxin-induced neuronal cell death.
Acetylation of APE1 plays an important role in APE1-mediated transcriptional regulation.22 APE1 is acetylated at Lys6 and Lys7 by histone acetyl-transferase p300, and this acetylation markedly enhances the affinity of APE1 for the negative calcium-responsive element, leading to down-regulation of parathyroid hormone gene expression. In this case, acetylated APE1 acts as a repressor of the parathyroid hormone promoter.
A recent study from Busso et al. demonstrated a novel APE1 regulatory mechanism that involves posttranslational ubiquitination.23 They identified that three lysine residues (Lys24, Lys25, and Lys27) within the N-terminal region of APE1 are ubiquitin acceptors. Ubiquitination of APE1 is regulated by the p53 and ubiquitin E3 ligase mouse double minute 2 signaling pathway and leads to degradation of the APE1 protein. In addition, Meisenberg et al. demonstrated that human E3 ubiquitin ligase UBR3 also ubiquitylates APE1 at lysine residues 7, 24, 25, 27, 32, and 35.24 In this study, the cellular levels of APE1 protein are increased in Ubr3 knockout cells, resulting in genome instability. Since there is a close correlation between the degree of APE1 expression and genotoxin cytotoxicity, ubiquitination of APE1 could regulate cellular sensitivity to DNA-damaging agents. Taken together, posttranslational modification of APE1 can affect its functions in DNA repair, redox regulation, and gene transcription, as well as its protein stability.25
In addition to the above modifications, APE1 was shown to be regulated through redox modification of cysteine residues. Many redox-active cysteines undergo more than one kind of oxidative modification.26,27 Qu et al. reported S-nitrosation of APE1 at Cys93 and Cys310.28 S-Nitrosation is a redox-based posttranslational modification that involves oxidative addition of nitric oxide (NO) to cysteine residues within a target protein.29,30 APE1 S-nitrosation results in the redistribution of the protein from the nucleus to the cytoplasm. At the same time, the importin-dependent nuclear import pathway is repressed by NO stimulation, suggesting that the altered subcellular localization of the protein stems from the prevention of reimportation of exported APE1 into the nucleus. Although the authors suggested that NO-mediated APE1 nuclear export is important for understanding the effect of nitrosative stress on APE1-associated physiological and pathological processes, a direct relationship between redox modification of APE1 and DNA repair activity was not addressed.
S-Glutathionylation is another physiological reversible modification of protein cysteine residues, defined by formation of mixed disulfide bonds between a protein sulfenic acid (−SOH) and reduced glutathione (GSH).31 Such a modification results in an increase in molecular mass of the protein by 306 Da and a net increase in negative charge.32 Protein S-glutathionylation might also be achieved via reaction of oxidized glutathione, also known as glutathione disulfide (GSSG), with an accessible cysteine sulfhydryl group (−SH) when the intracellular GSSG pool is elevated during oxidative stress.31 It should be noted that the concentration of cellular GSSG is very low compared to that of GSH. Nevertheless, S-glutathionylation of cysteine residues in metabolic enzymes, kinases, phosphatases, and transcription factors has emerged as a central mechanism by which changes in the intra-cellular redox state may be transduced into functional cellular responses.26 Like phosphorylation, these types of cysteine modification can modulate enzyme activities, energy metabolism, signal transduction, redox homeostasis, calcium homeostasis, ion channel activity, cytoskeletal assembly, and protein stability.33,34
Human APE1 protein has seven cysteine residues, three of which are located within the redox-responsive domain.35 However, the involvement of cysteines in the redox regulatory and DNA repair functions of APE1 has not been fully characterized. We hypothesized that APE1 can be modified by S-glutathionylation and that this specific redox modification would control its repair and/or redox functions. We demonstrate herein that (i) human APE1 is a substrate for S-glutathionylation in vitro and in cells, (ii) site-specific S-glutathionylation of Cys99 inactivates APE1 AP endonuclease activity, and (iii) this modification can be reversed by reducing molecules, which restore APE1 incision function.
Results
APE1 is S-glutathionylated in vitro
As a first step in examining potential APE1 S-glutathionylation, purified recombinant native (untagged) APE1 protein was treated with diamide and GSH. Diamide is a highly specific thiol oxidant that oxidizes small thiol-containing molecules (e.g., GSH), as well as exposed cysteine residues of proteins.36 Thus, oxidation of APE1 and GSH by diamide could result in S-glutathionylation by driving the formation of a mixed disulfide linkage between APE1 and GSH (APE1-S-SG). S-Glutathionylation of purified APE1 was monitored by treating the protein with 5 mM diamide/10 mM GSH, followed by detection with an anti-SSG antibody, which specifically recognizes glutathionylated proteins, after nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot transfer (see Materials and Methods).
After treatment with diamide/GSH, a detectable amount of S-glutathionylated protein was observed following SDS-PAGE at a mass of ~37 kDa, which corresponds to the size of APE1 (Fig. 1a, top panel). In addition, a higher-molecular-mass species was detected around 113 kDa (top panel, lane 3, arrow), albeit to a lesser degree, presumably representing an S-glutathionylation-triggered multimeric APE1 form.37 Since S-glutathionylation can be reversed by deglutathionylation with reducing agents,37 we tested whether the observed modification could be reversed by dithiothreitol (DTT) treatment. As would be expected for S-glutathionylation, the observed modification of APE1 was fully reversed by DTT exposure (top panel, lane 4). We could not detect S-glutathionylated APE1 in the samples that were not treated with diamide/GSH (without or with DTT, top panel, lanes 1 and 2).
Fig. 1.

S-Glutathionylation of APE1 in vitro. (a) Purified recombinant APE1 protein (0.2 μg) was incubated with 5 mM diamide (DA) and 10 mM GSH as described in Materials and Methods. For deglutathionylation, S-glutathionylated APE1 was treated with 10 mM DTT. All samples were subjected to nonreducing SDS-PAGE followed by Western blot analysis with anti-SSG antibody (top) or anti-APE1 antibody (bottom). Positions of molecular mass protein standards are designated in kilodaltons, and the arrow denotes location of multi-meric protein form. The data are representative of at least three independent experiments. (b) S-Glutathionylation was carried out by disulfide exchange with GSSG. Recombinant APE1 protein (0.2 μg) was treated with either 5 mM diamide/10 mM GSH (lane 2) or 10 mM GSSG (lane 3), followed by nonreducing SDS-PAGE and Western blot analysis with anti-SSG antibody or anti-APE1 antibody. The data are representative of at least three independent experiments.
Upon the use of an antibody directed against APE1, the altered state of the protein was confirmed for the same samples that were probed with anti-SSG antibody, again following nonreducing SDS-PAGE (Fig. 1a, bottom panel). Specifically, S-glutathionylation of APE1 resulted in a slight shift of its molecular mass when compared to non-modified APE1 (bottom panel, compare lanes 1 and 3), as well as multiple higher-molecular-mass forms of APE1 (lane 3). It is likely that the two most prominent bands seen in these experiments (i.e., corresponding to proteins of ~37 and 113 kDa), which employed highly purified APE1 protein, are the same ones detected with anti-SSG (Fig. 1a, top panel, lane 3). As above, all of the retarded APE1 species, including the slightly shifted species, the intermediate product seen around 45 kDa (observed only after long film exposure), and the ~113-kDa presumed oligomeric form of the protein (Fig. 1a, bottom panel, lane 3), disappeared when DTT was added (lane 4), suggesting that APE1 undergoes reversible S-glutathionylation and/or oxidation in vitro. It is worth emphasizing that, under the treatment conditions employed here (i.e., 5 mM diamide/10 mM GSH), ~100% of APE1 was seen to exhibit at least a slight molecular mass shift [bottom panel, compare lane 1 (untreated APE1) and lane 3 (diamide/GSH-treated APE1)], indicative of modification of all of the protein within the sample.
The increased GSSG pool that accompanies cellular oxidative stress can promote protein S-glutathionylation.33 We, therefore, examined whether S-glutathionylation of APE1 could occur via GSSG incorporation into the protein. As shown in Fig. 1b, exposure to GSSG resulted in S-glutathionylation of APE1, with the slight molecular mass shift in ~100% of the protein being even more evident (lower panel, compare lanes 1 and 2). We were unable to detect the larger oligomeric forms of glutathionylated APE1 after GSSG exposure, presumably indicative of a less potent reactivity for GSSG as compared to diamide/GSH. These results suggest that GSSG treatment can induce APE1 S-glutathionylation, which is likely to be mediated by a direct thiol-exchange mechanism.
S-Glutathionylation of APE1 reduces its AP endonuclease capacity
After determining that APE1 can be S-glutathionylated in vitro, we explored whether this modification affected the AP endonuclease activity of the protein. Specifically, we treated APE1 with diamide/GSH and then assayed the protein for incision capacity by using a standard approach that employs a radiolabeled AP-site-containing oligonucleotide substrate (see Materials and Methods). As shown in Fig. 2a, S-glutathionylation of APE1 significantly reduced total AP endonuclease activity of the sample (lane 5, >90% inhibition). Since diamide can act as a strong oxidizing agent, we found—consistent with a previous report indicating that APE1 oxidation inhibits its AP site incision potential38—that treatment with diamide alone inhibited the AP endonuclease function of the protein by ~50% (lane 3), presumably by inducing the formation of disulfide bonds within or between APE1 molecules. Reduction in S-glutathionylated APE1 with DTT restored AP site incision capacity (Fig. 2a, compare lanes 5 and 6).
Fig. 2.

Effect of S-glutathionylation of APE1 on its AP endonuclease activity. (a) AP endonuclease activity of modified APE1 protein. Recombinant APE1 (0.2 μg) was incubated with 1 mM diamide (DA)/2 mM GSH, followed by 5 mM DTT treatment as indicated. AP-site-containing DNA substrate was incubated with unmodified or modified APE1 protein under standard reaction conditions (see Materials and Methods), and the reaction mixture was analyzed on a denaturing urea-polyacrylamide gel. S indicates uncleaved oligonucleotide substrates, and P indicates cleaved incision products. The graph represents the relative incision activity (compared to APE1 without DA, GSH, or DTT treatment) with means±SD (n=3). (b) The same samples assayed for AP endonuclease activity (a) were subjected to Western blot analysis. Equal amounts of proteins were separated by nonreducing SDS-PAGE and probed with antibodies specific to SSG or APE1. Lane designations 1–6 correspond to those in (a). Positions of molecular mass protein standards are designated in kilodaltons. The data are representative of at least three independent experiments. (c) Inhibition of AP endonuclease activity was observed by the addition of various concentrations of diamide (DA) together with 1 mM GSH (upper panel). The same samples were subjected to Western blot analysis with anti-SSG and anti-APE1 antibodies after separation by nonreducing SDS-PAGE (lower panel).
As a means to establish that inhibition was due to S-glutathionylation, we carried out nonreducing SDS-PAGE/Western blot analysis using the same samples that were assayed for AP endonuclease activity. As seen earlier, essentially all of the APE1 protein within the sample was efficiently S-glutathionylated after diamide/GSH treatment, as revealed by the slight increase in APE1 molecular mass (Fig. 2b, compare lanes 1 and 5). The modification was reversible by DTT (Fig. 2b, lane 6), and diamide alone did not induce S-glutathionylation (lane 3). These data support that the AP endonuclease activity of APE1 can be regulated by modification of a cysteine residue(s) via S-glutathionylation.
We next determined the effect of different diamide concentrations on the extent of APE1 modification and the degree of endonuclease inhibition. We fixed the GSH concentration at 1 mM and varied the diamide concentrations from 0 to 1 mM. As shown in Fig. 2c (upper panel), APE1 exhibited reduced AP endonuclease activity when the diamide concentration was 0.5 mM or higher. At the lower diamide concentrations of 0.001–0.2 mM, no significant enzyme inhibition was observed. When the same samples were applied to nonreducing SDS-PAGE/Western blot analysis to examine modification of APE1, S-glutathionylation of the protein was surprisingly not readily observed at the dose of 0.5 mM diamide (Fig. 2c, anti-SSG panel). The minimum dose of diamide required to detect APE1 S-glutathionylation was 0.7 mM (anti-SSG panel), the same concentration at which the slight mobility shift in the APE1 protein was observed (anti-APE1 panel). These data suggest that APE1 can be modified to an extent that is not easily detected by Western blot analysis and does not significantly alter protein mobility yet has the capacity to inhibit its endonuclease activity (possibly, a single S-glutathionylation event).
S-Glutathionylation of APE1 reduces AP–DNA complex stability
We next explored whether the reduced AP endonuclease activity stemmed from altered DNA binding or impaired catalysis. In particular, we determined the kinetic parameters of APE1 without or following 1 mM diamide/1 mM GSH treatment, which results in essentially 100% of APE1 being modified (see, for instance, Fig. 2c). Lineweaver–Burk plots of 1/V versus 1/[S] from incision assays conducted at 5, 10, 25, 50, and 100 nM AP–DNA revealed a kcat and KM for untreated APE1 of 43 s−1 and 65 nM, respectively, and a kcat and KM of 59 s−1 and 289 nM, respectively, for diamide/GSH-treated APE1. Consistent with decreased substrate affinity (i.e., the 4.4-fold increase in KM), electrophoretic mobility shift assays (EMSAs) found essentially no stable APE1/AP–DNA complexes after the protein was exposed to diamide and GSH (Fig. 3). Notably, the reduced protein/DNA complex formation following diamide/GSH treatment was largely reversible by DTT.
Fig. 3.

Effect of S-glutathionylation on APE1 DNA binding. After treatment with 1 mM diamide (DA), 1 mM GSH, and/or 1 mM DTT (as indicated), AP-site-containing DNA (100 fmol) was incubated with 2 ng of APE1 protein and then resolved on an 8% non-denaturing polyacrylamide gel (see Materials and Methods). (Top) A representative gel image of three independent experimental EMSAs. The bottom graph shows the relative DNA binding activity (compared to APE1 without DA, GSH, or DTT treatment) with means±SD (n=3). NE, no enzyme and DNA-only control; S, unbound substrate; C, APE1/AP–DNA complexes.
S-Glutathionylation of Cys99 is responsible for APE1 endonuclease inhibition
To determine which cysteine residue(s) is involved in the S-glutathionylation-dependent inactivation of APE1, we initially examined the endonuclease activity of glutathionylated APE1 proteins that harbored only the following cysteine residues: 65/93, 65/99, or 65/93/99; all other cysteine residues were substituted with Ala (see Materials and Methods). Cys65, Cys93, and Cys99 (depicted in Fig. 4a, three-dimensional APE1 protein rendering) fall within the N-terminal portion of APE1 thought to be critical for the redox regulatory role of the protein (residues 1–127). We reasoned that cysteine residues within the Ref-1 domain of APE1 would be the most probable sites for redox regulation by S-glutathionylation. For these studies, the wild-type, 65/93, 65/99, and 65/93/99 APE1 proteins were expressed as N-terminal hexa-His-SUMO fusion proteins in bacteria and, following cell lysis, were bound to a nickel column before treatment with the SUMO protease (Ulp1) to release partially purified full-length untagged protein (see Materials and Methods for further details).
Fig. 4.

Identification of S-glutathionylated cysteine residue in APE1. (a) The three-dimensional structure of the human APE1 protein is shown as a magenta ribbon rendering with the seven cysteine residues depicted as van der Waals spheres. Substrate DNA is shown as a stick rendering with the abasic site containing strand in green and the complementary strand in purple. The rendering was generated from coordinate file 1DEW (Protein Data Bank accession number) by using the PyMOL Molecular Graphics System (Version 1.3, Schrödinger, LLC). Inhibitory effect of S-glutathionylation on AP endonuclease activity was measured using APE1 proteins retaining all seven cysteine residues (WT) or two or three of the N-terminal cysteine residues (Cys65, Cys93, and Cys99). The other cysteine residues not designated are substituted with Ala. Treatment parameters are indicated, and S=substrate and P=product. The data are representative of at least three independent experiments. APE1 cysteine mutants were generated by a single amino acid substitution (to Ala) and tested for AP endonuclease activity. Each cysteine mutant protein was treated with 1 mM diamide plus 1 mM GSH where indicated (+). The data are representative of at least three independent experiments.
As expected, wild-type APE1 lost AP endonuclease activity after treatment with 1 mM diamide/1 mM GSH, and its activity was restored when DTT was added (Fig. 4b, lanes 4 and 5). The APE1 mutant protein that retained only Cys65 and Cys93 but was mutated at Cys99 (and all other cysteine residues) to Ala (i.e., Cys65/Cys93 or 65/93) was notably not affected by oxidation (lane 7) or glutathionylation (lane 9). Conversely, the APE1 mutant protein forms that retained Cys99 (i.e., 65/99 and 65/93/99) showed a similar AP endonuclease inhibitory pattern (lanes 14 and 19) as wild type. These data indicate that inactivation of APE1 endonuclease activity requires Cys99 and, therefore, that this residue is likely S-glutathionylated.
To determine whether S-glutathionylation of any of the cysteine residues not within the Ref-1 domain affects APE1 endonuclease activity, we employed a set of single, site-specific mutant proteins. Human APE1 has a total of seven cysteine residues at positions 65, 93, 99, 138, 208, 296, and 310 (three-dimensional rendering shown in Fig. 4a). Each of these residues was specifically converted to alanine independently, and the different APE1 mutants were purified as glutathione S-transferase (GST) fusion proteins (see Material and Methods), with the C65A, C93A, and C99A mutants serving as controls in this experiment. As shown in Fig. 4c, only the C99A protein retained substantial AP site incision activity following diamide/GSH treatment, consistent with this residue being essential for the S-glutathionylation-mediated inactivation of APE1 endonuclease function.
S-Glutathionylation of cysteine residues in APE1 as determined by mass spectrometric analysis
To determine directly the extent and sites of S-glutathionylation, we reacted the comparable wild-type, C65/C93, and C65/C99 APE1 samples (see above) with 10 mM diamide and 1 mM GSH and subjected them to global electrospray ionization (ESI)–quadrupole time-of-flight (QTOF) mass spectrometric analysis (Fig. 5). S-Glutathionylation of wild-type APE1 resulted in complete protein modification, yielding two products with mass differences corresponding to the addition of two GSH (Δ mass 610.7 Da) and seven GSH (Δ mass 2136.9 Da), respectively, in a ratio of approximately 2.5:1 based on relative peak heights (Fig. 5a). The formation of a product incorporating two GSH is consistent with results obtained for modification of APE1 by N-ethyl maleimide (MalNEt), in which the two solvent-accessible cysteine residues (Cys99 and Cys138) are preferentially modified.39 The product with seven GSH incorporated indicates that treatment with diamide results in exposure of all seven cysteines. A similar result was obtained for treatment of APE1 with E3330 (a dimethoxy quinone derivative able to inhibit the redox function of APE1) and MalNEt; in this experiment, products with two and seven incorporated MalNEts were obtained over incubation times of several hours with no accumulation of intermediate products.39 The product containing seven MalNEts was found to be a partially unfolded form of APE1 through characterization of hydrogen deuterium exchange monitored by mass spectrometry. By analogy, APE1 modified by seven GSH may also correspond to a partially unfolded form of the enzyme and represent the slightly altered mobility shift species noted above.
Fig. 5.

ESI––QTOF mass spectrometric analysis of wild-type (a), C65/C93 (b), and C65/C99 (c) APE1 proteins. S-Glutathionylation of wild-type (WT), C65/C93, and C65/C99 APE1 proteins (10 μM) was carried out by treatment with 10 mM diamide and 1 mM GSH. The reaction mixtures were subjected to global ESI–QTOF mass spectrometric analysis. Plotted on the x-axis is the deconvoluted mass for observed peaks, and plotted on the y-axis is the peak intensity. In (b), the peak corresponding to the unmodified parent protein is designated as “P”.
Mass spectrometric analysis following S-glutathionylation of the C65/C93 APE1 protein showed the parent ion with no modifications and a comparatively low abundance ion showing the addition of two GSH in a ratio of approximately 29:1 (Fig. 5b). S-Glutathionylation of the C65/C99 APE1 sample resulted in two products corresponding to addition of one (Δ mass 305.24) and two GSH in a 3:1 ratio (Fig. 5c). For wild-type and C65/C99 samples, no unmodified parent ion was observed in the deconvoluted spectra, indicating efficient glutathionylation of these samples and their respective cysteine residues (Fig. 5a and c). In contrast, the C65/C93 sample remained largely unmodified following treatment with GSH and diamide, indicating that Cys99 is necessary for efficient glutathionylation (Fig. 5b). Taken together, these results are consistent with the loss of APE1 endonuclease activity stemming from modification of Cys99.
H2O2 induces S-glutathionylation of APE1 in vitro and in cells
We next determined whether APE1 was S-glutathionylated by a more physiologically relevant agent, H2O2, using an artificial in vitro treatment paradigm. Wild-type APE1 (full-length untagged protein expressed as an N-terminal hexa-His-SUMO fusion; see Materials and Methods) was treated with H2O2 and then reacted with GSH. The modifications of APE1, specifically disulfide bond formation involving APE1 and GSH, were subsequently assessed by liquid chromatography (LC)–tandem mass spectrometry (MS/MS) peptide analysis after digestion with trypsin. In contrast to diamide-induced modification by GSH, which results only in disulfide bonds, treatment with H2O2 will result in not only disulfide bond formation in APE1 but also formation of sulfenic or sulfinic acid production by oxidation of cysteine.40 GSH can react with any of the oxidized forms of cysteine residues in APE1 in this case, but most likely with sulfenic acid forms. As summarized in Table 1, relatively low levels of glutathionylation were detected involving cysteine residues 65, 93, 99, and 138. The low yields of APE1–GSH disulfide bonds (around 1% of the total protein at each residue) may stem from the low concentration of H2O2 used and from the ability of excess GSH to reduce any disulfide bonds formed during the experiment leading to the formation of GSSG. The modification of only the four cysteine residues 65, 93, 99, and 138 is consistent with results obtained for treatment of APE1 with E3330, in which the only disulfide bonds observed were Cys65/Cys93, Cys65/Cys99, Cys93/Cys99, Cys65/Cys138, and Cys93/Cys138.39
Table 1.
LC–MS/MS analysis of S-glutathionylation of APE1 by H2O2
| APE1 peptide disulfide bonded to GSH | Cys residuea |
|---|---|
| ICSWNVDGLR | 65 |
| EEAPDILCLQETK | 93 |
| CSENKLPAELQELPGLSHQYWSAPSDK | 99 |
| QCPLK | 138 |
The residue number for the Cys (in boldface) found in each peptide that was disulfide bonded to GSH.
Lastly, we explored whether APE1 S-glutathionylation occurred in mammalian cells and whether this modification changed during oxidative stress (i.e., following a challenge with the oxidizing agent H2O2). In these experiments, human cervical carcinoma HeLa cells were initially treated with a range of H2O2 concentrations to determine the dose at which cytotoxicity was minimized (<10% cell killing; data not shown). Following exposure for 30 min to a relatively nontoxic dose of H2O2 (100 μM), Western blotting of whole cell extracts with anti-SSG and anti-APE1 antibodies suggested that intracellular APE1 underwent S-glutathionylation (Fig. 6a). Most notably, there appeared to be a slight molecular mass shift in APE1 that was consistent with such a modification. To interrogate this idea further, we monitored S-glutathionylation of APE1 more directly by using biotinylated glutathione. In particular, HeLa cells were treated with BioGEE (biotinylated glutathione ethyl ester), a membrane-permeable analog of glutathione,41 for 1 h prior to H2O2 exposure. Biotinylated proteins were then precipitated using streptavidin agarose beads, followed by washing and elution with DTT (see Materials and Methods). Glutathionylated APE1 was specifically identified and quantified via Western blot analysis with anti-APE1 antibody. Figure 6b shows that 100 μM H2O2 induces a significant increase in S-glutathionylated APE1 protein after a 30-min challenge in HeLa cells. Although we did not see S-glutathionylated APE1 protein in the absence of BioGEE pretreatment (Fig. 6c, “−” lane), there appears to exist a basal level of S-glutathionylated APE1 protein in untreated cells (Fig. 6b, “C” lane, and c, “+” lane). Taken together, these results indicate that APE1 protein is S-glutathionylated within cells at a steady-state level and that APE1 S-glutathionylation is increased after an oxidative challenge.
Fig. 6.
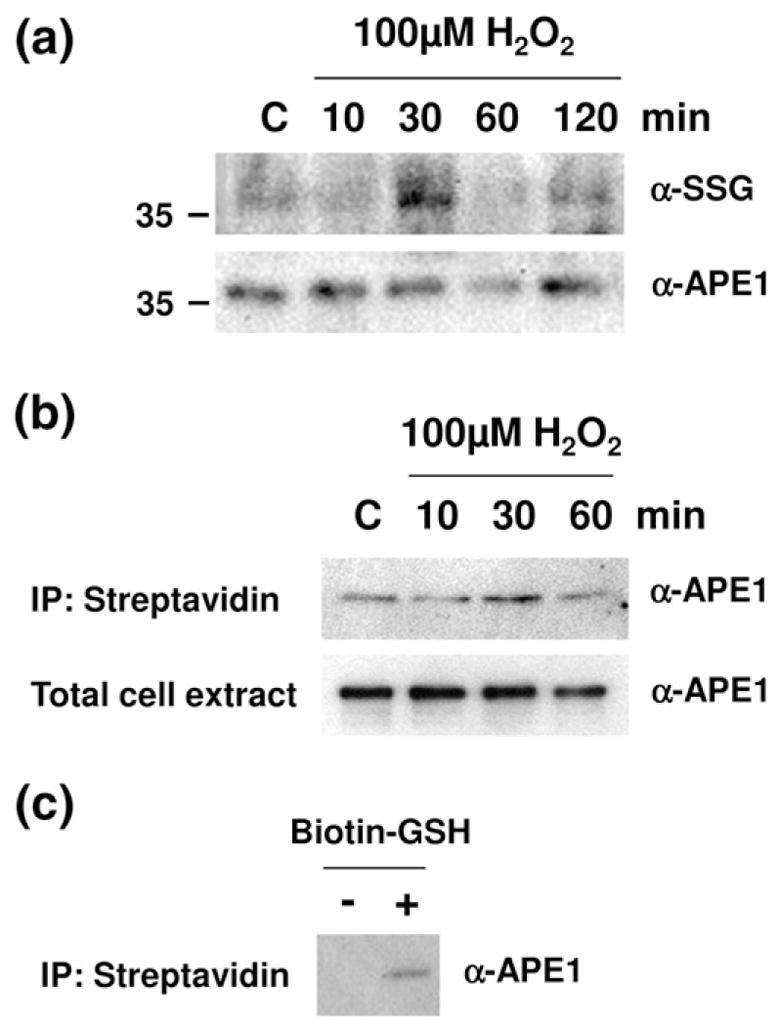
H2O2-induced S-glutathionylation of APE1 in cells of APE1 in cells (a) HeLa cells were exposed to 100 μM H2O2 for 10, 30, 60, or 120 min. An equal amount of total whole cell extract (10 μg) at the designated time point was then subjected to nonreducing SDS-PAGE and probed with antibodies specific to SSG or APE1. Shown is a representative gel (cropped to show only the molecular mass region relevant to APE1) of at least three independent experiments. (b) Cells were preincubated with 100 μM BioGEE for 1 h and subsequently exposed to 100 μM H2O2 for 10, 30, and 60 min. Biotin-bound proteins were extracted using streptavidin agarose and eluted with DTT. The eluent was subjected to nonreducing SDS-PAGE and probed with an antibody specific to APE1. The data are representative of at least three independent experiments. (c) Cells were incubated with or without BioGEE. Biotin-bound proteins were extracted using streptavidin agarose and eluted with DTT. The eluent was subjected to nonreducing SDS-PAGE and probed with an antibody specific to APE1.
Discussion
In addition to the many well-known posttranslational modifications, alterations involving redox are recognized as prominent mechanisms for modulating protein function.42,43 Many proteins are specific targets of redox regulation due to the presence of reactive cysteine residues in their primary structure. Among them, S-glutathionylation has been identified as a major form of redox modification. Modulation of protein function by glutathionylation of cysteine residues spans a wide variety of biological processes, including energy metabolism, signal transduction, redox homeostasis, calcium homeostasis, ion channel activity, cytoskeletal assembly, and protein stability.33,34 In this study, we demonstrate that human APE1 undergoes S-glutathionylation in vitro and in cells. Among the seven cysteine residues in APE1, glutathionylation of Cys99 was found to lead to inactivation of the endonuclease function of this repair enzyme, suggesting a novel link between APE1 redox modification and DNA repair function.
The position of Cys99 within the three-dimensional structure of APE1 is likely important in the mechanism by which S-glutathionylation impairs its AP endonuclease activity. In particular, in the crystal structures of APE1 bound to substrate DNA (1DEW and 1DE8), the Cys99 residue is in close proximity to the bound substrate (Fig. 4a). Glutathionylation of Cys99 would be expected to alter the local structure of the protein and thereby affect the positioning of the substrate on the enzyme and/or the affinity with which the substrate is bound. This idea is consistent with a previous report in which Cys99 of human APE1 was shown to be a residue that ensures optimum APE1 activity.44 In particular, Mantha et al. found reduced affinity of the Cys99Ser mutant for AP-site-containing DNA in the absence of Mg2+.44 Moreover, we report herein both an increase in KM and a reduced protein/DNA complex stability in EMSAs following diamide/GSH treatment. Thus, the inhibitory effect of Cys99 S-glutathionylation on AP endonuclease activity appears to be mediated by altered substrate interactions with the enzyme.
Our data herein reveal a novel mechanism involving posttranslational modification that may modulate APE1 repair and redox regulatory functions. However, despite the potential significance of APE1 S-glutathionylation, future studies will need to explore more comprehensively the biological contribution(s) of this modification. In particular, why would it be advantageous for APE1 nuclease activity to be susceptible to redox inactivation? Although this feature may simply reflect the harmful nature of oxidative stress, it is possible that APE1 uses this modification as a mechanism to switch between redox regulatory and repair functions (a so-called molecular switch), depending on the status of the cell. Moreover, as shown for S-nitrosation of APE1,28 S-glutathionylation may provide a means of regulating the intracellular distribution of the protein. It will be important, in this regard, to explore the extent of APE1 S-glutathionylation in cells exhibiting high cytoplasmic staining, such as seen in certain cancers.45–47 Finally, given that our studies reveal that human APE1 is naturally S-glutathionylated and that the extent of this modification increases following H2O2 treatment in HeLa cells, it will be interesting to determine whether APE1 S-glutathionylation varies depending on the cell type, stress level, nuclear microenvironment, and GSH abundance. In closing, we postulate that redox modification of cysteine residues within APE1 provides a mechanism for regulating the different functions of the protein.
Materials and Methods
Reagents
Diamide, DTT, GSH, GSSG, and H2O2 were purchased from Sigma (St. Louis, MO). Streptavidin immobilized on 4% agarose beads was from EMD Bioscience (Gibbstown, NJ). BioGEE (Invitrogen, Carlsbad, CA), molecular weight standards for SDS-PAGE, protease inhibitors, and all cell culture reagents were from Invitrogen. Anti-SSG antibody was from ViroGen (cat# 101-A; Watertown, MA), and anti-APE1 antibody was from Novus Biologicals (cat# NB 100-116; Littleton, CO). Bradford protein assay reagent was purchased from Bio-Rad (Hercules, CA).
APE1 proteins
Recombinant native (untagged) human APE1 protein was expressed in and subsequently purified to near homogeneity from bacteria using a two-step ion-exchange chromatography procedure that has been previously described.48,49 APE1 proteins harboring cysteine residues 65/93, 65/99, or 65/93/99, with all other cysteine residues substituted with Ala (as well as a comparable wild-type protein), were expressed as N-terminal His-SUMO-fusion proteins and purified as previously described.39 In brief, full-length untagged wild-type APE1 protein and 65/93, 65/99, and 65/93/99 mutant APE1 proteins were eluted from a Ni-NTA column following on-column cleavage with the Ulp1 SUMO-specific protease and subsequently subjected to S-Sepharose and size-exclusion chromatographic purification steps.
The single-cysteine mutant APE1 proteins tagged at the N-terminus with GST were prepared as described previously.9 In brief, GST-C65A-hApe1, GST-C93A-hApe1, GST-C99A-hApe1, GST-C138A-hApe1, GST-C208A-hApe1, GST-C296A-hApe1, and GST-C310A-hApe1 pGEX-4T plasmid constructs were generated by site-directed mutagenesis (QuikChange kit; Stratagene, Wilmington, DE) and confirmed by DNA sequencing. For protein production, the plasmids were transformed into BL21 (λDE3) Escherichia coli (Stratagene), and selected colonies were inoculated and grown in 50-mL Luria broth (LB) medium containing 100 μg/mL ampicillin until the OD600 reached 0.5. At that point, expression was induced by the addition of isopropyl-β-D-1-thiogalactopyranoside to a final concentration of 1 mM and conducted overnight at 22 °C. The cells were harvested by centrifugation and resuspended in 2-mL phosphate-buffered saline (PBS) solution. Cells were then lysed by sonication, followed by centrifugation at 14,000g for 20 min at 4 °C. The clarified cell extracts were loaded onto a Glutathione Sepharose 4B column, and the bound GST-hApe1 fusion protein was isolated using the standard glutathione affinity purification protocol (GE Healthcare, Piscataway, NJ). Following three consecutive 1-mL elution steps, each fraction was examined for GST-hApe1 protein by SDS-PAGE and Coomassie blue staining. The fraction containing the highest amount of protein was dialyzed against 50 mM Hepes (pH 7.5), 50 mM KCl, and 10% glycerol overnight at 4 °C. Protein concentrations were determined by using the standard Bradford method, and aliquots were stored at −80 °C until needed.
S-Glutathionylation and deglutathionylation of APE1
Purified APE1 (0.2 μg) protein was treated with diamide (1–5 mM) and GSH (1–10 mM) in degassed buffer A [40 mM Tris–HCl (pH 8.0), 140 mM NaCl, and 0.2 mM ethylenediaminetetraacetic acid (EDTA)] at room temperature for 90 min. After the reaction, where indicated, DTT (5–10 mM) was subsequently added for 30 min at room temperature to induce deglutathionylation. APE1 protein was then analyzed using one of the methods outlined below. For S-glutathionylation by disulfide exchange with GSSG, recombinant APE1 protein was incubated for 90 min with 10 mM GSSG at room temperature in degassed buffer A.
Electrophoresis and Western blot analysis
Equal amounts of protein were separated on a 10% nonreducing SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. Blots were incubated with anti-SSG or anti-APE1 antibody (see above). Immunoreactive bands were detected with horseradish-peroxidase-conjugated secondary antibodies and enhanced chemiluminescence plus reagents (GE Healthcare).
AP endonuclease and EMSA
The incision activity of APE1 was measured as previously described.50 In brief, a 5′-32P-end-labeled oligonucleotide duplex substrate was formed by using 18F NMR (5′-GTC ACC GTG FTA CGA CTC-3′; where F is the abasic site analog tetrahydrofuran) and 18G NMR (5′-GAG TCG TAG CAC GGT GAC-3′). Incision reactions were performed at 37 °C for 5 min with 400 pg of APE1 (with or without diamide/GSH treatment; see above) and 0.5 pmol of AP–DNA substrate in 10-μL INH buffer [50 mM Hepes (pH 7.5), 100 mM KCl, and 1 mM MgCl2]. For the enzyme kinetic studies (carried out in triplicate), 100 pg of APE1 (280 pM) was used with final substrate concentrations of 5, 10, 25, 50, and 100 nM. The incision reaction products were resolved on a denaturing urea-polyacrylamide gel and visualized by standard phosphorimager analysis.
EMSAs were conducted essentially as described previously.51 In brief, purified APE1 was treated as above with 1 mM diamide, 1 mM GSH, and/or 1 mM DTT. Two nanograms of the protein was then incubated with 100 fmol of AP–DNA in 50 mM Hepes–KOH (pH 7.5), 50 mM KCl, 10% glycerol, 0.01% Triton X-100, 100 μg/mL bovine serum albumin, and 4 mM EDTA at a final volume of 10 μL for 5 min on ice. Binding reactions were resolved on an 8% non-denaturing polyacrylamide gel and analyzed following electrophoresis using a phosphorimager.
LC–MS/MS analysis
To induce disulfide bond formation, we treated a 50-μL solution of full-length APE1 (10 μM) with H2O2 (1 mM) for 2 h in 10 mM Hepes (pH 7.5). The buffer was then exchanged, and the sample was reacted with GSH (100 μM) for 30 min at room temperature and was quenched with 1 mM MalNEt prior to digestion to prevent disulfide bond scrambling. The sample was diluted 10-fold with water to give a final APE1 concentration of 1 μM and then digested using a protein:trypsin ratio of 50:1 at 37 °C for 4 h. Native digestion conditions were similar to those successfully used for other problems involving purified protein samples.52,53 Triplicate LC–MS/MS measurements of 5 μL of the digestion solution were made following reversed-phase capillary LC separations with an Eksigent NanoLC-1D pump (Eksigent Technologies, Inc., Dublin, CA). The reversed-phase capillary column (0.075 mm×150 mm) was packed in-house by using a PicoFrit™ tip (New Objective, Inc., Woburn, MA) with C18 particles (Magic, 5 μm, 120 Å; Michrom Bioresources, Inc., Auburn, CA). The mobile phase consisted of water containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B). Immediately after sample loading, the mobile phase was held at 98% solvent A for 12 min. A linear gradient was used, whereby the amount of solvent B was increased from 2% to 60% over 60 min, then to 80% over 10 min at 260 nL/min, followed by a 12-min re-equilibration step by 100% solvent A. The flow was directed by PicoView Nanospray Source (PV550; New Objective, Inc.) to the LTQ Orbitrap (Thermo Fisher Scientific, West Palm Beach, FL). The spray voltage was 1.8–2.2 kV, and the capillary voltage was 27 V. The LTQ Orbitrap was operated in standard data-dependent MS/MS acquisition mode controlled by its Xcalibur 2.0.7 software. A full mass spectral scan was followed by six product ion (MS/MS) scans of the six most abundant ions. The mass spectra of the peptides were acquired at high mass resolving power (60,000 for ions of m/z 400) with the FT analyzer over the range of m/z 350–2000. The six most abundant precursor ions were dynamically selected in the order of highest-to-lowest signal intensity (minimal intensity of 1000 counts) and were subjected to collision-induced dissociation. Precursor activation was performed with an isolation width of 2 Da and an activation time of 30 ms. The normalized collision energy was 35% of the maximum available. The automatic gain control target value was regulated at 1×106 for the FT analyzer and at 3×104 for the ion trap with a maximum injection time of 1000 ms for the FT analyzer and of 200 ms for the ion trap. The instrument was externally calibrated by using a standard calibration mixture of caffeine, the peptide MRFA, and Ultramark 1621 (Thermo Fisher Scientific).
LC–MS/MS data were searched with Mascot 2.2 (Matrix Science, Boston, MA) against the NCBI database to identify covalent modifications.54–56 Parameters used in Mascot were as follows: enzyme, trypsin; maximum missed cleavage, 3; fixed modifications, oxidation on methionines; variable modifications, GSH on cysteines; peptide mass tolerance, 10 ppm with one C13 peak; peptide charge, +1 to +3; product mass tolerance, 0.6 Da; and instrument type, default (searching for all types of b and y ions).
ESI–QTOF analysis
Glutathionylation for APE1 samples treated with diamide and GSH was characterized by global QTOF mass spectrometry. Full-length APE1 samples including C65/C93, C65/C99, and wild-type APE1 at a concentration of 10 μM were reacted with 1 mM GSH and 10 mM diamide for 2 h at room temperature. Samples were analyzed using ESI mass spectrometry following Agilent 1200 series capillary HPLC with a flow rate of 50 μL/min consisting of 70% H2O and 30% acetonitrile, both with 0.1% formic acid. Mass spectra were measured using an Agilent 6520 QTOF operating in time-of-flight mode with ESI. The sample was nebulized at 10 psi and dried with 325 °C N2 gas flowing at 5 L/min. The capillary voltage was 4000 V, and a mass region of m/z 300–1700 was scanned. Mass spectra of the sample were averaged and deconvoluted using the MassHunter Bioconfirm software supplied by the manufacturer.
Cell culture and preparation of total cell lysates
Human cervical cancer HeLa cells (ATCC CCL-185) were maintained in a high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were grown at 37 °C in an atmosphere of 5% CO2 and 95% air. To generate whole cell extracts, we rinsed cells three times with ice-cold PBS and lysed them in RIPA buffer [50 mM Tris–Cl (pH 7.4), 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 μg/mL each of aprotinin and leupeptin, and 1 mM Na3VO4]. After centrifugation at 12,000g for 30 min, the supernatant was collected, and the protein concentration was determined by the Bradford method.
Purification of S-glutathionylated proteins using BioGEE in cells
BioGEE was added to the medium 1 h prior to the addition of 100 μM H2O2. Soluble proteins covalently bound to biotin were extracted in batches by using streptavidin agarose. The agarose beads were washed three times with RIPA buffer. Proteins bound to streptavidin via a disulfide bond were then eluted from the beads by incubation for 30 min with PBS/EDTA/SDS containing 10 mM DTT. Proteins in the eluent were resolved by SDS-PAGE and detected by Western blotting.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging, as well as by grants from the National Institutes of Health (CA114571) to M.M.G. and from the National Center for Research Resources (2P41RR000954) to M.L.G.
Abbreviations used
- AP
apurinic/apyrimidinic
- APE1
AP endonuclease 1
- EDTA
ethylenediaminetetraacetic acid
- EMSA
electrophoretic mobility shift assay
- ESI
electrospray ionization
- GSH
reduced glutathione
- GSSG
glutathione disulfide
- GST
glutathione S-transferase
- LC
liquid chromatography
- MalNEt
N-ethyl maleimide
- MS/MS
tandem mass spectrometry
- PBS
phosphate-buffered saline
- QTOF
quadrupole time-of-flight
- redox
reduction–oxidation
- Ref-1
redox effector factor 1
References
- 1.Wilson DM, 3rd, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 2.Demple B, Sung JS. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair (Amst) 2005;4:1442–1449. doi: 10.1016/j.dnarep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36:425–435. doi: 10.1016/j.ctrv.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos–Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seiffert M, Cant C, Chen Z, Rappold I, Brugger W, Kanz L, et al. Human signal-regulatory protein is expressed on normal, but not on subsets of leukemic myeloid cells and mediates cellular adhesion involving its counterreceptor CD47. Blood. 1999;94:3633–3643. [PubMed] [Google Scholar]
- 6.Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-κB. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- 7.Ueno M, Masutani H, Arai RJ, Yamauchi A, Hirota K, Sakai T, et al. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem. 1999;274:35809–35815. doi: 10.1074/jbc.274.50.35809. [DOI] [PubMed] [Google Scholar]
- 8.Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res. 2007;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res. 2007;13:260–267. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 12.Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Fishel ML, He Y, Reed AM, Chin-Sinex H, Hutchins GD, Mendonca MS, Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair. 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelley MR, Fishel ML. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem. 2008;8:417–425. doi: 10.2174/187152008784220294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, et al. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005;33:4379–4394. doi: 10.1093/nar/gki751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrison JF, Rinne ML, Kelley MR, Druzhyna NM, Wilson GL, Ledoux SP. Altering DNA base excision repair: use of nuclear and mitochondrial-targeted N-methylpurine DNA glycosylase to sensitize astroglia to chemotherapeutic agents. Glia. 2007;55:1416–1425. doi: 10.1002/glia.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 18.Yacoub A, Kelley MR, Deutsch WA. The DNA repair activity of human redox/repair protein APE/Ref-1 is inactivated by phosphorylation. Cancer Res. 1997;57:5457–5459. [PubMed] [Google Scholar]
- 19.Fritz G, Kaina B. Phosphorylation of the DNA repair protein APE/REF-1 by CKII affects redox regulation of AP-1. Oncogene. 1999;18:1033–1040. doi: 10.1038/sj.onc.1202394. [DOI] [PubMed] [Google Scholar]
- 20.Hsieh MM, Hegde V, Kelley MR, Deutsch WA. Activation of APE/Ref-1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res. 2001;29:3116–3122. doi: 10.1093/nar/29.14.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang E, Qu D, Zhang Y, Venderova K, Haque ME, Rousseaux MW, et al. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol. 2010;12:563–571. doi: 10.1038/ncb2058. [DOI] [PubMed] [Google Scholar]
- 22.Bhakat KK, Izumi T, Yang SH, Hazra TK, Mitra S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003;22:6299–6309. doi: 10.1093/emboj/cdg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Busso CS, Iwakuma T, Izumi T. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53–MDM2 signaling pathway. Oncogene. 2009;28:1616–1625. doi: 10.1038/onc.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meisenberg C, Tait PS, Dianova II, Wright K, Edelmann MJ, Ternette N, et al. Ubiquitin ligase UBR3 regulates cellular levels of the essential DNA repair protein APE1 and is required for genome stability. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr744. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Busso CS, Lake MW, Izumi T. Posttranslational modification of mammalian AP endonuclease (APE1) Cell Mol Life Sci. 2010;67:3609–3620. doi: 10.1007/s00018-010-0487-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghezzi P, Bonetto V, Fratelli M. Thiol-disulfide balance: from the concept of oxidative stress to that of redox regulation. Antioxid Redox Signal. 2005;7:964–972. doi: 10.1089/ars.2005.7.964. [DOI] [PubMed] [Google Scholar]
- 27.Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35:2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miersch S, Mutus B. Protein S-nitrosation: biochemistry and characterization of protein thiol–NO interactions as cellular signals. Clin Biochem. 2005;38:777–791. doi: 10.1016/j.clinbiochem.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Sanchez LM, Muntane J, de la Mata M, Rodriguez-Ariza A. Unraveling the S-nitro-soproteome: tools and strategies. Proteomics. 2009;9:808–818. doi: 10.1002/pmic.200800546. [DOI] [PubMed] [Google Scholar]
- 31.Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A. S-Glutathionylation in protein redox regulation. Free Radical Biol Med. 2007;43:883–898. doi: 10.1016/j.freeradbiomed.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Shelton MD, Chock PB, Mieyal JJ. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 33.Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. FEBS J. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- 34.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Bhakat KK, Mantha AK, Mitra S. Transcriptional regulatory functions of mammalian AP-endonuclease (APE1/Ref-1), an essential multi-functional protein. Antioxid Redox Signal. 2009;11:621–638. doi: 10.1089/ars.2008.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kosower NS, Kosower EM. Diamide: an oxidant probe for thiols. Methods Enzymol. 1995;251:123–133. doi: 10.1016/0076-6879(95)51116-4. [DOI] [PubMed] [Google Scholar]
- 37.Demasi M, Piassa Filho GM, Castro LM, Ferreira JC, Rioli V, Ferro ES. Oligomerization of the cysteinyl-rich oligopeptidase EP24.15 is triggered by S-glutathionylation. Free Radical Biol Med. 2008;44:1180–1190. doi: 10.1016/j.freeradbiomed.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 38.Kelley MR, Parsons SH. Redox regulation of the DNA repair function of the human AP endonuclease Ape1/Ref-1. Antioxid Redox Signal. 2001;3:671–683. doi: 10.1089/15230860152543014. [DOI] [PubMed] [Google Scholar]
- 39.Su D, Delaplane S, Luo M, Rempel DL, Vu B, Kelley MR, et al. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme. Biochemistry. 2010 doi: 10.1021/bi101248s. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-α induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- 42.Biswas S, Chida AS, Rahman I. Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol. 2006;71:551–564. doi: 10.1016/j.bcp.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 43.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mantha AK, Oezguen N, Bhakat KK, Izumi T, Braun W, Mitra S. Unusual role of a cysteine residue in substrate binding and activity of human AP-endonuclease 1. J Mol Biol. 2008;379:28–37. doi: 10.1016/j.jmb.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puglisi F, Barbone F, Tell G, Aprile G, Pertoldi B, Raiti C, et al. Prognostic role of Ape/Ref-1 subcellular expression in stage I–III breast carcinomas. Oncol Rep. 2002;9:11–17. [PubMed] [Google Scholar]
- 46.Di Maso V, Avellini C, Croce LS, Rosso N, Quadrifoglio F, Cesaratto L, et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: possible prognostic significance. Mol Med. 2007;13:89–96. doi: 10.2119/2006-00084.DiMaso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheng Q, Zhang Y, Wang R, Zhang J, Chen B, Wang J, et al. Prognostic significance of APE1 cytoplasmic localization in human epithelial ovarian cancer. Med Oncol. 2011 doi: 10.1007/s12032-011-9931-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 48.Erzberger JP, Barsky D, Scharer OD, Colvin ME, Wilson DM., 3rd Elements in abasic site recognition by the major human and Escherichia coli apurinic/apyrimidinic endonucleases. Nucleic Acids Res. 1998;26:2771–2778. doi: 10.1093/nar/26.11.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen LH, Barsky D, Erzberger JP, Wilson DM., 3rd Mapping the protein–DNA interface and the metal-binding site of the major human apurinic/apyrimidinic endonuclease. J Mol Biol. 2000;298:447–459. doi: 10.1006/jmbi.2000.3653. [DOI] [PubMed] [Google Scholar]
- 50.Wilson DM., 3rd Properties of and substrate determinants for the exonuclease activity of human apurinic endonuclease Ape1. J Mol Biol. 2003;330:1027–1037. doi: 10.1016/s0022-2836(03)00712-5. [DOI] [PubMed] [Google Scholar]
- 51.Wilson DM, 3rd, Takeshita M, Demple B. Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res. 1997;25:933–939. doi: 10.1093/nar/25.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gau BC, Chen H, Zhang Y, Gross ML. Sulfate radical anion as a new reagent for fast photochemical oxidation of proteins. Anal Chem. 2010;82:7821–7827. doi: 10.1021/ac101760y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hambly DM, Gross ML. Laser flash photochemical oxidation to locate heme binding and conformational changes in myoglobin. Int J Mass Spectrom. 2007;259:124–129. [Google Scholar]
- 54.Xu H, Freitas MA. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu H, Yang L, Freitas MA. A robust linear regression based algorithm for automated evaluation of peptide identifications from shotgun proteomics by use of reversed-phase liquid chromatography retention time. BMC Bioinformatics. 2008;9:347. doi: 10.1186/1471-2105-9-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu H, Zhang L, Freitas MA. Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J Proteome Res. 2008;7:138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]