

Mutations in mouse intraflagellar transport–A complex genes alter Sonic hedgehog signaling because of their effects on cilia structure and on trafficking of membrane proteins into cilia.
Abstract
Two intraflagellar transport (IFT) complexes, IFT-A and IFT-B, build and maintain primary cilia and are required for activity of the Sonic hedgehog (Shh) pathway. A weak allele of the IFT-A gene, Ift144, caused subtle defects in cilia structure and ectopic activation of the Shh pathway. In contrast, strong loss of IFT-A, caused by either absence of Ift144 or mutations in two IFT-A genes, blocked normal ciliogenesis and decreased Shh signaling. In strong IFT-A mutants, the Shh pathway proteins Gli2, Sufu, and Kif7 localized correctly to cilia tips, suggesting that these pathway components were trafficked by IFT-B. In contrast, the membrane proteins Arl13b, ACIII, and Smo failed to localize to primary cilia in the absence of IFT-A. We propose that the increased Shh activity seen in partial loss-of-function IFT-A mutants may be a result of decreased ciliary ACIII and that the loss of Shh activity in the absence of IFT-A is a result of severe disruptions of cilia structure and membrane protein trafficking.
Introduction
The Shh signaling pathway is critical for development of nearly every organ system in vertebrate embryos and has important roles in tumorigenesis (Ingham et al., 2011). Recent studies have shown that Hedgehog (Hh) signaling in vertebrates, unlike in Drosophila melanogaster, depends on the primary cilium, a microtubule-based organelle that projects from the surface of most interphase cells (Goetz and Anderson, 2010). Two evolutionarily conserved intraflagellar transport (IFT) protein complexes, IFT-A and IFT-B, are required to build and maintain cilia structure (Cole, 2003; Pedersen et al., 2006). IFT-B proteins and Kinesin-II are required for anterograde trafficking from the base to the tip of the cilium, whereas the IFT-A complex is believed to work with the cytoplasmic dynein-2 motor to control retrograde ciliary trafficking (Pedersen and Rosenbaum, 2008).
The relationship between Shh signaling and cilia is best understood in the patterning of cell types in the ventral half of the developing neural tube, where a gradient of Shh activity controls a set of five different neuronal cell types. Shh is first made in the notochord, which lies ventral to the developing neural tube, and different levels of Shh activity specify different neural cell types, with the highest level of Shh activity specifying the floor plate and V3 interneuron progenitors, whereas intermediate levels of Shh activity specify motor neurons (Briscoe, 2009). Two Gli transcription factors, Gli2 and Gli3, implement the responses to Shh, where the activator form of Gli2 is essential for specification of the most ventral cell types in the developing neural tube. We showed that mutations that inactivate mouse IFT-B proteins block ciliogenesis and prevent the specification of Shh-dependent ventral neural cell types (Huangfu et al., 2003). Subsequent studies showed that all the core Shh pathway proteins that mediate the response to ligand are enriched in cilia and change their ciliary localization in response to pathway activation, including the Shh receptor Patched, the membrane protein Smoothened, the Kinesin-4 family member Kif7, the negative regulator Sufu, and the Gli transcription factors (Goetz and Anderson, 2010).
The roles of IFT-A proteins in ciliogenesis appear to be more complex than those of IFT-B proteins. Biochemical studies have identified six evolutionarily conserved proteins in the IFT-A complex in both Chlamydomonas reinhardtii and mammalian cells, IFT122, IFT144, IFT139 (Ttc21b), IFT140, IFT121 (WDR35), IFT43, and IFT144 (WDR19) (Mukhopadhyay et al., 2010; Behal et al., 2012). A role for IFT-A in retrograde trafficking in cilia and flagella was first defined in the alga Chlamydomonas, where temperature-sensitive mutations in two Chlamydomonas IFT-A genes, fla15 (IFT144) and fla17 (IFT139), cause bulged flagella of normal length, associated with decreased rates of retrograde IFT (Iomini et al., 2001, 2009). Null mutations in mouse Ift139a/Ttc21b and Ift122 cause similar phenotypes: null mutant cilia are of approximately normal length but have bulges at the tips of the axoneme, consistent with a defect in retrograde trafficking (Tran et al., 2008; Cortellino et al., 2009; Qin et al., 2011), and slower rates of retrograde trafficking were measured after knockdown of Ift139a (Tran et al., 2008). In contrast to the relatively mild ciliary defects of Ift139a and Ift122 mutants, null mutations in mouse Ift121/Wdr35, another IFT-A gene, lead to the formation of very short cilia (Mill et al., 2011). Caenorhabditis elegans null mutations in the IFT-A gene encoding IFT140 (che-11) allow formation of normal-length cilia that fail to function normally (Qin et al., 2001), whereas dyf-2 mutants, which lack WDR19/IFT144, make very short cilia, similar to those seen in IFT-B mutants (Efimenko et al., 2006). Thus, some mutants reveal a role for IFT-A proteins in retrograde ciliary trafficking, and others suggest that IFT-A has a role in building cilia. Despite the differences among the mouse and worm phenotypes, mutations in all six human IFT-A genes—IFT139a/TTC21b, IFT121/WDR35, IFT43, IFT122, IFT144/WDR19, and IFT140—have recently been shown to cause a set of related human genetic syndromes, including short-rib polydactyly, Jeune asphyxiating thoracic dystrophy, Sensenbrenner syndrome/cranioectodermal dysplasia, and Mainzer-Saldino syndrome (Gilissen et al., 2010; Arts et al., 2011; Bredrup et al., 2011; Davis et al., 2011; Mill et al., 2011: Perrault et al., 2012).
Paralleling the different effects on cilia structure, IFT-A proteins appear to have different roles in mouse Shh signaling than the IFT-B proteins. Whereas IFT-B–null mutants fail to respond to Shh, embryos that lack Ift139a/Ttc21b and Ift122 show ectopic, ligand-independent activation of the Shh pathway in the neural tube. The basis of the inappropriate activation of the Shh pathway in the IFT-A mutants is not clear.
Here, we carry out studies that resolve the apparently disparate phenotypes caused by loss of different mouse IFT-A proteins. We first compare two different ethylnitrosourea (ENU)-induced mutations that disrupt mouse IFT144, a core component of the mammalian IFT-A complex (Mukhopadhyay et al., 2010). We find that a partial loss-of-function Ift144 allele causes ectopic activation of the Shh pathway in the neural tube, similar to the phenotypes seen in mouse Ift122 and Ift139a mutants. In contrast, mutant embryos homozygous for a strong allele of Ift144 appear to have the opposite phenotype: neural cell types that require a high level Shh activity are not specified. We find that the structure of the weak Ift144 mutant cilia is relatively normal, whereas cilia of the null mutant are short and have highly disrupted axonemes. Analysis of double mutants carrying mutations in two different IFT-A genes confirms that strong loss of IFT-A disrupts ciliogenesis and blocks high-level activity of the Shh pathway. The data indicate that IFT-B is sufficient for trafficking of key proteins required for Shh signaling (Gli2, Sufu, and Kif7) into cilia. In contrast, we find that IFT-A is required for localization of a set of membrane-associated proteins to cilia, and we suggest how the loss of these membrane proteins may account for the Shh pathway phenotypes of both weak and strong IFT-A mutants.
Results
Identification of two alleles of mouse Ift144/Wdr19
We identified a recessive mouse mutant, diamondhead (dmhd), in an ENU mutagenesis screen based on altered expression of a transgenic motor neuron reporter gene (Liem et al., 2009). The HB9-GFP transgene is expressed throughout the spinal cord in motor neurons, a cell type that is specified by intermediate levels of Shh pathway activity (Fig. 1 A). In the dmhd mutant, HB9-GFP was not expressed at rostral levels of spinal cord but was strongly expressed in caudal regions. Homozygous dmhd mutant embryos also had a set of defects in external morphology including exencephaly, with an open diamond-shaped forebrain, and left–right randomization of heart looping. The dmhd mutant embryos arrested at midgestation, at approximately embryonic day 10.5 (E10.5).
Figure 1.

Mutations in Ift144 alter Shh-dependent neural patterning. (A) The motor neuron marker HB9-GFP (green) is not expressed in the rostral neural tube of Ift144dmhd embryos (arrow) but is strongly expressed in the caudal spinal cord. WT, wild type. (B) Ift144twt mutants show increased HB9-GFP expression compared with wild-type littermates. (C) The two Ift144 alleles cause contrasting changes in dorsal–ventral neural patterning, shown in transverse sections at three rostral–caudal levels of the E10.5 neural tube. (D) Expression of the Shh target gene Ptch1-lacZ in E10.5 Ift144 mutant and littermate control embryos at lumbar levels. Ift144dmhd mutants lack strong expression Ptch1-lacZ at the ventral midline at all levels. Ptch1-lacZ is ectopically expressed in the mesoderm adjacent to the neural tube in Ift144dmhd mutants (arrows). Ift144twt mutants show dorsally expanded Ptch1-LacZ expression. Bars: (A and B) 1 mm; (C and D) 200 µm.
We used meiotic recombination mapping to localize the dmhd mutation on chromosome 5 (see Materials and methods). Because disruption of primary cilia can cause defects in both left–right asymmetry and Shh signaling, we examined candidate genes in the interval that might affect the formation of primary cilia. We identified a splice site mutation that was predicted to lead to a truncated protein in the gene encoding Wdr19/IFT144 (Fig. 2 A), the homolog of the Chlamydomonas IFT144 and C. elegans Dyf-2, which are required for normal formation of flagella and cilia (Efimenko et al., 2006; Iomini et al., 2009). We were unable to detect wild-type Ift144 transcript by PCR from mutant embryo cDNA and could not detect IFT144 protein with a polyclonal antibody to the N-terminal domain of the protein by immunofluorescence (Fig. 2 B), suggesting that dmhd is a strong loss-of-function or null allele. Consistent with disruption of an IFT protein, cilia structure was strongly disrupted in the dmhd mutant, and only very short cilia were formed (Fig. 2 B).
Figure 2.
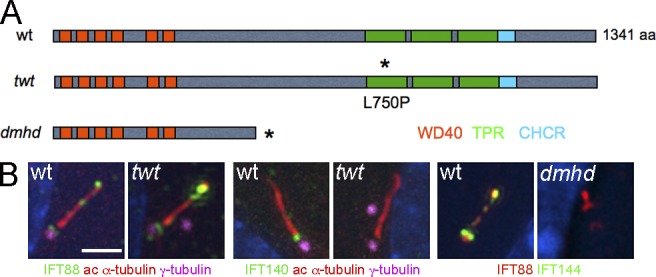
Two mutations in mouse Ift144 affect cilia structure. (A) Schematic drawing of how the mutations affect protein structure. Two isoforms of the 1,341–amino acid WDR19/IFT144 protein have been described; conserved protein–protein interaction motifs are shown in colored boxes. Depending on the isoform, the dmhd splice site mutation would truncate the protein at amino acid 592 (ENSMUSP00000038098) or amino acid 543 (ENSMUSP00000109414). The twt mutation causes a leucine-to-proline substitution in the first tetratrico peptide repeat (TPR; Ashe et al., 2012). Asterisks indicate the position of the sequence changes in the mutant proteins. CHCR, clathrin heavy chain repeat; wt, wild type. (B) Altered cilia protein expression and structure in the mutants. Cilia in twt MEFs are of approximately normal length and width but accumulate higher than normal levels of IFT88 (an IFT-B protein) at cilia tips and lack detectable IFT140 (an IFT-A protein; 0/9 twt cilia were positive for IFT140 compared with 13/14 wild-type cilia). Cilia in dmhd MEFs are short and wide; they accumulate IFT88 but lack any detectable IFT144 protein, consistent with the hypothesis that dmhd is a null allele (0/18 were positive for IFT144). ac, acetylated. Bar, 2 µm.
A second allele of Ift144, twinkletoes (twt), was identified in an independent screen for ENU-induced mutations (Ashe et al., 2012). twt homozygotes survived to the end of gestation, when they showed polydactyly and craniofacial abnormalities. The twt mutation was associated with a missense change in the first tetratricopeptide repeat of IFT144, but the mutation did not affect the amount of protein made (Fig. S1 B; Ashe et al., 2012). The twt mutants showed increased HB9-GFP expression throughout the spinal neural tube (Fig. 1 B). In contrast to the very abnormal dmhd cilia, twt mutant cilia appeared nearly normal in structure, although IFT-140, another IFT-A protein, was not detectable in the twt mutant cilia (Fig. 2 B). dmhd/twt compound heterozygotes died at E12–13 with exencephaly and polydactyly, confirming that the two mutations disrupted the same gene. Because twt homozygotes survived longer than dmhd/twt embryos and because of the stronger disruption in cilia structure in dmhd than in twt, we concluded that twt is a hypomorphic allele of the gene. Therefore, we refer to the dmhd mutation as Ift144dmhd and twt as Ift144twt.
Weak and strong alleles of Ift144 have opposing effects on specification of Shh-dependent ventral neural cell types
To define how IFT144 affects Shh signaling, we examined neural tube patterning in the mutants. Neural patterning in Ift144dmhd embryos was unlike that of other previously characterized IFT mutants. Null mutations in two other IFT-A genes, Ift122 and Ift139a, cause a ventralization of the neural tube, in which the most ventral cell types that require the highest level of Shh (the floor plate and V3 interneuron progenitors) are expanded as a result of elevated ectopic activity of the Shh pathway (Tran et al., 2008; Qin et al., 2011). In contrast, Ift144dmhd mutants lacked the cell types that require high levels of Shh activity, the FoxA2+ floor plate and Nkx2.2+ V3 progenitors, throughout the spinal cord (Figs. 1 C and S1 A). In anterior (cervical) regions, Ift144dmhd embryo mutants lacked all Shh-dependent cell types, but motor neurons, which require intermediate levels of Shh activity, were present in the posterior (lumbar) neural tube, where they expanded to more dorsal positions than seen in wild type (Figs. 1 C and S1 A).
Whereas the ventral neural cell types that require high levels of Shh activity were absent in Ift144dmhd at all rostrocaudal positions, Ift144twt showed an opposing phenotype: the floor plate, V3 progenitor, and motor neuron domains were dorsally expanded in the caudal neural tube (Fig. 1 C). This ectopic specification of Shh-dependent ventral neural cell types in the Ift144twt neural tube was similar to that caused by null mutations in two IFT-A genes, Ift139aalien and Ift122sopb (Tran et al., 2008; Cortellino et al., 2009; Qin et al., 2011).
To test whether the changes in neural patterning were a result of changes in activity of the Shh pathway, we examined expression of Ptch1, a direct transcriptional target of the Shh pathway, using a Ptch1-lacZ reporter (Goodrich et al., 1997). As predicted by the loss of ventral neural cell types, Ptch1-lacZ was reduced in the Ift144dmhd neural tube anterior to the forelimbs (Fig. S1, C and D). In the caudal neural tube, Ptch1-lacZ was expressed ectopically in dorsal regions, although the strong Ptch1-lacZ expression at the ventral midline was absent (Figs. 1 D and S1 D). Ptch1-lacZ was ectopically expressed in the dorsal neural tube of Ift144twt embryos, consistent with the dorsal expansion of ventral neural cell types (Figs. 1 D and S1 E). Thus, the two alleles of Ift144 had contrasting effects on activity of the Shh pathway in the neural tube: Ift144twt increased pathway activity, and Ift144dmhd decreased pathway activity but led to an increased number of cells with an intermediate level of activity in the caudal neural tube.
Other IFT proteins have been shown to act at a step in the Shh pathway between the membrane proteins Patched and Smoothened and the Gli transcription factors. Because the Ift144dmhd phenotype was different than that of other IFT mutants, we used double-mutant analysis to identify the step in the Shh pathway affected by the mutation. Analysis of neural patterning in double mutants showed that Ift144dmhd acted genetically downstream of the two membrane proteins in the pathway, Patched and Smoothened (Fig. S2, A and B), similar to other IFT-B and IFT-A genes (Huangfu et al., 2003: Huangfu and Anderson, 2005; Qin et al., 2011). Double-mutant analysis showed that the neural patterning phenotype of Ift144dmhd was the result of changes in the activity of the Gli2 and Gli3 transcription factors (Fig. S2, C–L), as in other IFT mutants.
The expansion of the motor neuron domain in Ift144dmhd depends on the presence of cilia
IFT144 could, in principle, exert its effect on Shh signaling through either a ciliary or nonciliary role of the protein. To test whether cilia were required for the specification of motor neurons in Ift144dmhd embryos, we generated double mutants that lacked both IFT144 and the IFT-B complex protein IFT172. Embryos homozygous for the Ift172wim mutation lack cilia and fail to specify Shh-dependent cell types in the neural tube, including motor neurons (Huangfu et al., 2003). The Ift144dmhd Ift172wim double mutants showed the same loss of ventral neural cell types seen in Ift172wim single mutants (Fig. S3, A–D). Thus, the changes in the activity of the Shh pathway in Ift144dmhd embryos must be a result of changes in cilia structure or ciliary trafficking caused by loss of this IFT-A protein.
Weak and strong alleles of Ift144 have distinct effects on cilia structure
Each of the neural progenitor cells that respond to Shh has a single primary cilium that projects from its apical surface into the central lumen of the neural tube. When cells of the neural tube were examined en face by scanning electron microscopy, wild-type cilia on E10.5 neural progenitors were ∼1 µm in length, with shorter cilia emanating from ciliary pockets (Fig. 3 A), consistent with the appearance of primary cilia in other cell types (Sorokin, 1962; Molla-Herman et al., 2010; Rohatgi and Snell, 2010). In contrast, the primary cilia on Ift144dmhd neural epithelial cells were short and bulbous (Fig. 3 B and Table S1). The neural cilia in Ift144twt were approximately normal in length (Fig. 3 C and Table S1), whereas Ift144dmhd/twt compound heterozygote neural cilia were shorter and wider than wild type (Fig. 3 D and Table S1). The vast majority of Ift144twt and Ift144dmhd/twt cilia lacked the bulges near the tips that have been described in other IFT-A mutants.
Figure 3.
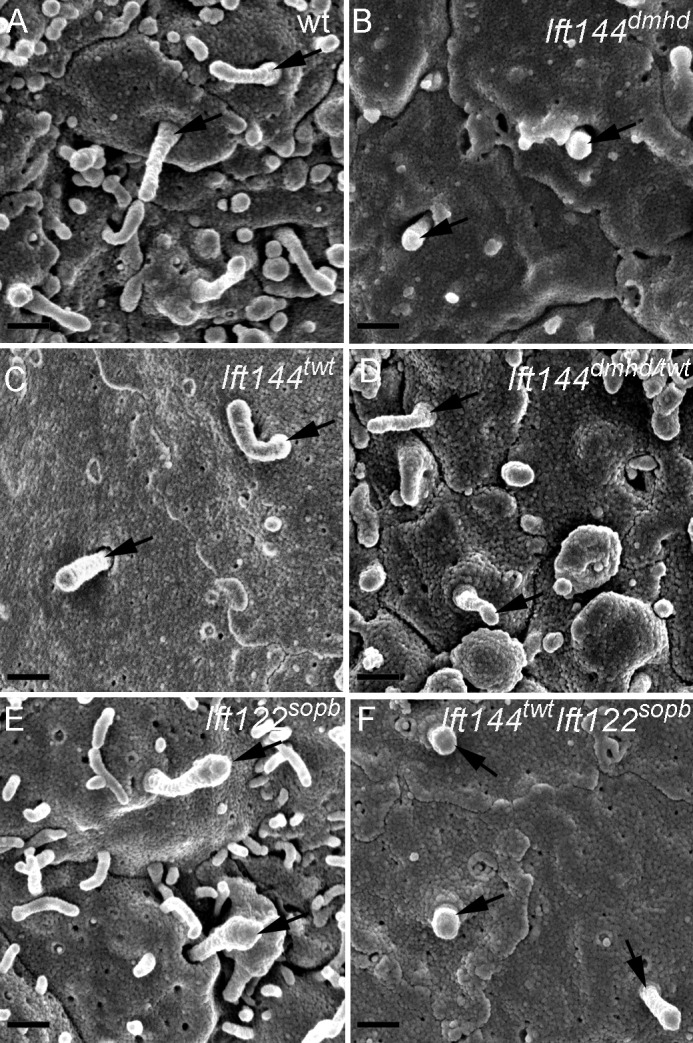
Abnormal morphology of primary cilia in Ift144 mutants. (A–F) Scanning electron micrographs show cilia (arrows) on the lumenal face of the neural tube at E10.5. (A) Most wild-type (wt) cells have a single cilium that projects from the apical surface. (B) Ift144dmhd mutant cilia are shorter, rounder, and more bulged than wild type. (C and D) Ift144twt cilia (C) have normal morphology, whereas Ift144dmhd/twt (D) cilia are shorter than wild type, but few cilia are bulged. (E) Neural tube cilia in embryos that carry a null allele of Ift122 (Ift122sopb) are of approximately normal length but have bulges near the tip. (F) Ift144twt Ift122sopb double-mutant cilia are very short, similar to those of the Ift144dmhd. Bars, 400 nm.
To characterize the aspects of ciliogenesis that require IFT144, we analyzed cilia and basal bodies in neuroepithelial cells by transmission EM (TEM). Cross-sections showed that wild-type basal bodies had the typical triplet microtubule structure, and cilia had the characteristic 9 + 0 doublet microtubule organization (Fig. 4 A). Basal bodies of Ift144dmhd, Ift144twt, and Ift144dmhd/twt cilia appeared to be of normal size and structure and were present close to the apical surface (Fig. 4, B–D). Ift144twt and Ift144dmhd/twt cilia were similar in structure to wild type in both longitudinal sections and cross-sections, although there was a slightly greater distance between the plasma membrane and the microtubule axoneme, which could account for the greater cilia width seen by scanning electron microscopy (Fig. 4 [B and C] and Table S1). Despite the normal structure of the basal body, the majority of Ift144dmhd cilia analyzed lacked any visible microtubules in longitudinal or transverse sections (Figs. 4 D and S4). Instead, the cilia compartment contained disorganized electron-dense material. This phenotype suggests that IFT144 is required for building the cilium rather than functioning exclusively in retrograde trafficking.
Figure 4.

Ultrastructure of wild-type and mutant neural tube cilia. (A–F) Longitudinal sections (left) and cross-sections of cilia (top right) and basal bodies (bb; bottom right) from the neural tube in wild type (WT) and mutants. (A) Wild-type cilia have a 9 + 0 doublet microtubule organization in the axoneme and triplet microtubules in the basal body. (B and C) The microtubules in Ift144twt (B) and Ift144dmhd/twt (C) mutant cilia appear normal, whereas the cilia are slightly wider than wild type. (D) Ift144dmhd mutant cilia lack axonemal microtubules and contain vesicle-like structures and have a normal-appearing basal body. (E) Longitudinal sections show that neural tube Dync2h1mmi cilia are swollen and filled with arrays of electron-dense particles that resemble trains of IFT particles. A few singlet microtubules can be seen in cross-sections of Dync2h1mmi cilia (arrow). (F) The cilia of Ift144twt Dync2h1mmi double mutants are less swollen than the Dync2h1mmi single mutants and do not accumulate IFT trains but show a more severe phenotype than Ift144twt cilia. Cilia are from E9.5 (wild type and Ift144dmhd) and E10.5 (Ift144twt, Ift144dmhd/twt, Dync2h1mmi, and Ift144twt Dync2h1mmi) embryos. Bars, 200 nm.
The phenotype of IFT-A double mutants confirms that the IFT-A complex is required for ciliogenesis and for high-level responses to Shh
The Ift144dmhd mutation caused a much more severe disruption of cilia architecture than null mutations in the mouse IFT-A genes Ift122 or Ift139 (Tran et al., 2008; Qin et al., 2011). To test whether this represented a unique property of the Ift144dmhd allele or was a result of a more crucial role of IFT144 in the IFT-A complex, we analyzed double mutants that were homozygous for both the weak allele Ift144twt and the null allele Ift122sopb. Whereas neural cilia in both Ift144twt and Ift122sopb embryos were of nearly normal length (Fig. 3, C and E), the Ift144twt Ift122sopb double-mutant cilia were less than half the length of wild type and more bulbous than either single mutant (Fig. 3 F and Table S1). The Ift144twt Ift122sopb double-mutant cilia were similar in size and shape to those present in Ift144dmhd embryos.
Whereas Ift144twt and Ift122sopb single mutants showed expansion of Shh-dependent ventral neural cell types, neural patterning in Ift144twt Ift122sopb double mutants was similar to that seen in Ift144dmhd embryos. Like Ift144dmhd, the Ift144twt Ift122sopb double mutants lacked all ventral cell types in spinal cord regions anterior to the forelimbs and lacked floor plate and most Nkx2.2+ V3 progenitors cells at more posterior positions (Figs. 5 [A and B] and S1 A). Also like Ift144dmhd, motor neurons in the double mutant spanned the midline and were expanded dorsally in the caudal neural tube (Fig. 5 B). We conclude that the Ift144twt Ift122sopb double-mutant and Ift144dmhd single-mutant phenotypes in cilia formation and neural patterning are the result of stronger disruption of the IFT-A complex than seen in Ift122-null single mutants.
Figure 5.

Neural patterning in Ift144twt Ift122sopb double mutants resembles that in Ift144dmhd. (A) Based on expression of HB9-GFP, Ift144twt Ift122sopb double-mutant embryos lack motor neurons in the anterior spinal cord (arrow) but retain them in caudal regions. The double mutants are exencephalic and have laterality defects, similar to Ift144dmhd. WT, wild type. (B) Cross-sections through Ift144twt, Ift122sopb, and Ift144twt Ift122sopb double-mutant embryos at E10.5, marked by the expression of HB9-GFP and FoxA2. Ift144twt/Ift122sopb double mutants lack FoxA2+ floor plate cells at all levels, lack cervical motor neurons, and show dorsally expanded motor neurons at lumbar levels, like Ift144dmhd. Bars: (A) 1 mm; (B) 200 µm.
IFT-A and cytoplasmic dynein-2 have distinct roles in cilia structure
Because previous studies have shown that both IFT-A proteins and cytoplasmic dynein-2 are required for normal retrograde ciliary trafficking, we compared the structure of Ift144 mutant cilia with that of mutants homozygous for a strong allele of the gene encoding the heavy chain of the retrograde dynein motor Dync2h1mmi (Liem et al., 2009; Ocbina et al., 2011). TEM sections showed that Dync2h1mmi cilia were filled with electron-dense particles between the axoneme and the ciliary membrane (Fig. 4 E), similar to the accumulation of particles in Chlamydomonas flagella that lack cytoplasmic dynein-2 (Pazour et al., 1999). In cross-sections, it was apparent that Dync2h1mmi cilia lacked most axonemal microtubules, and most of the remaining microtubules were singlets rather than doublets. The particles in the Dync2h1mmi cilia appeared to be highly organized and were regularly spaced at ∼40-nm intervals, very different than the unstructured content of the Ift144dmhd cilium.
As both IFT-A and cytoplasmic dynein-2 are important for retrograde ciliary trafficking, we analyzed the structure of cilia of Ift144twt Dync2h1mmi double mutants. The double-mutant cilia lacked the regular repeating electron-dense particles and were less swollen than Dync2h1 single mutants, and, in the best cases, a reasonably normal-appearing axoneme was restored (Fig. 4 F). Thus, the accumulation of trains of IFT particles in Dync2h1mmi cilia was relieved by the Ift144twt mutation. The partial rescue of cilia structure was paralleled by a partial rescue of Shh signaling, as Ift144twt Dync2h1mmi double mutants specified the most ventral neural cell type, the floor plate, which was never seen in Dync2h1mmi single mutants (Fig. S3, E–J).
IFT-A is required for recruitment of specific cilia components, but not IFT88, to cilia
The TEM analysis showed that Ift144dmhd cells have short, abnormal cilia. However, standard molecular markers for cilia were not detectable in the mutant cilia. Arl13b, a palmitoylated membrane-associated protein of the ARL family of small GTPases (Caspary et al., 2007; Cevik et al., 2010; Larkins et al., 2011), is localized to all cilia in the wild-type embryos and mouse embryo fibroblasts (MEFs). Arl13b was present in primary cilia of Ift144twt and Ift122sopb embryos and MEFs (Fig. 6, A and B). In contrast, Arl13b was not detectable in cilia in the neural tube of Ift144dmhd single mutants or in Ift144twt Ift122sopb double mutants (Fig. 6 A) or in primary MEFs derived from Ift144dmhd or Ift144twt Ift122sopb embryos (Fig. 6 B). Acetylated α-tubulin, another standard marker for cilia, was easily detected in MEF cilia in wild-type, Ift144twt, and Ift122sopb embryos but was not detected in most Ift144dmhd mutant or Ift144twt Ift122sopb double-mutant MEF cilia (Fig. 6 C). A similar pattern of expression of acetylated α-tubulin was observed in cilia on mesenchymal cells near the notochord in wild-type and mutant embryos (Fig. S5). Another ciliary marker, the membrane protein adenylyl cyclase III (ACIII), was present in neural cilia in wild type but appeared to be present in far fewer cilia in the IFT-A mutants (Fig. 6 A). Because of the density of neural cilia, it was difficult to quantitate the effect on ACIII localization in the neural tube, but the decreased localization of ACIII to mutant cilia could be confirmed in MEFs. ACIII localized to wild-type MEF cilia but was not detected in Ift144dmhd mutant or Ift144twt Ift122sopb double-mutant MEFs (Fig. 6 D). ACIII was detected in Ift144twt and Ift122sopb MEFs but at significantly lower levels than in wild-type cilia (Fig. 6 D). A stronger effect on ACIII localization was seen in mesenchymal cells in the embryo, where ACIII was present in wild-type mesenchymal cells, but ACIII was reduced in mesenchymal cells of the weak IFT-A mutants, and little or no ACIII was detected in strong IFT-A mutants (Fig. S5 and Table S2).
Figure 6.

A subset of cilia markers is not detected in IFT-A mutant cilia. (A) Neural tube cilia. Arl13b is expressed in the cilia that project into the lumen of the E10.5 neural tube in wild-type (wt), Ift144twt, and Ift122sopb embryos. Arl13b cannot be detected in Ift144dmhd or Ift144twt Ift122sopb double-mutant cilia. IFT88 localizes to cilia in all genotypes analyzed. ACIII is present in neural cilia in wild type but is present in fewer neural cilia in the IFT-A mutants. Bars: (top row) 200 µm; (bottom rows) 10 µm. (B–D) MEF cilia. The basal body is marked by γ-tubulin and the axoneme by IFT88. (B) Arl13b marks the ciliary axoneme in wild-type (35/35), Ift144twt (27/32), and Ift122sopb (12/13) mutant MEFs but is not detectable in Ift144dmhd (0/9) and greatly reduced or Ift144twt Ift122sopb (1/12) mutant cilia. IFT88 accumulates in the cilia of Ift144dmhd or Ift144twt Ift122sopb mutants. (C) Like Arl13b, acetylated α-tubulin (ac α-tub) is detected in the primary cilia of wild-type (12/12), Ift144twt (17/17 cilia scored were positive for Arl13b), and Ift122sopb (8/9) mutant MEFs but is detected in only a small fraction of Ift144dmhd (1/13) or Ift144twt Ift122sopb (4/14) mutant cilia. (D) ACIII is strongly expressed in wild-type MEF cilia (12/13) but is not detectable in either Ift144dmhd (0/8) or Ift144twt Ift122sopb cilia (0/12). Ift144twt MEF cilia showed ACIII staining (13/13), but the intensity of staining was reduced to 62 ± 14% of wild type. Only 30% of Ift122sopb (6/21) showed ACIII staining, and, in the ACIII+ cilia, the intensity was 22 ±10% of wild type. Bars, 2 µm.
However, it was possible to visualize Ift144dmhd cilia by immunofluorescent staining, as the mutant cilia showed strong expression of the IFT-B protein IFT88. IFT88 was localized to neural cilia that point into the lumen of the neural tube in all genotypes analyzed: wild type, Ift144dmhd, Ift144twt, Ift122sopb, and Ift144twt Ift122sopb (Fig. 6 A). Thus, we infer that IFT-A is not required for targeting or entry of IFT88 into cilia. In wild-type MEFs, IFT88 was enriched at the transition zone at the base of the cilium and in a punctate pattern along the ciliary axoneme (Figs. 2 B and 6 [B–D]). In Ift144dmhd and Ift144twt Ift122sopb MEF cilia, IFT88 extended distal to the basal bodies (marked by γ-tubulin) and often appeared to be continuous from the transition zone into the short mutant axoneme, in contrast to its more punctate localization in wild type, suggesting that this IFT-B protein accumulates in cilia in the absence of IFT-A.
IFT-A is required for recruitment of Smo, but not Gli2, Sufu, and Kif7, to cilia
To determine how the changes in cilia structure and composition in the mutants affected the activity of the Shh pathway, we analyzed the localization of Shh pathway proteins in cilia of MEFs derived from mutant embryos. Smo was not detected in the cilium of any of the genotypes analyzed in the absence of Shh pathway activation. In wild-type, Ift144twt, and Ift122sopb MEF cilia, Smo moved into cilia when the Shh pathway was activated with the small molecule Smoothened agonist (SAG; Fig. 7 A). In contrast, no Smo was detected in the cilium of Ift144dmhd mutant or Ift144twt Ift122sopb double-mutant MEFs when the pathway was activated with SAG (Fig. 7 A).
Figure 7.

Localization of Shh pathway proteins in IFT-A mutant MEF cilia. (A and B) γ-Tubulin marks the basal body; IFT88 marks the ciliary axoneme. (A) Smo is present in the cilia of wild-type (wt), Ift144twt, and Ift122sopb MEFs in cells where the Shh pathway is activated by SAG (>90% of cilia are Smo+; n > 10). Smo movement into the cilium in response to activation of the Shh pathway by SAG is abolished in Ift144dmhd (0/9 cilia were Smo+) and greatly reduced Ift144twt Ift122sopb (1/17). (B) Gli2 is two- to threefold enriched at wild-type MEF cilia tips in response to SAG (n > 14 for each condition). The level of Gli2 is slightly elevated at the tips of the Ift144twt and Ift122sopb mutant cilia in the absence of SAG and is further enriched in the presence SAG (n > 16 for each condition). The level of Gli2 is elevated at the tips of the Ift144dmhd or Ift144twt Ift122sopb mutant cilia in the absence of SAG and does not increase further in the presence SAG (n > 9 for each condition). Bar, 2 µm.
In wild-type cells, Gli2 localizes to cilia tips in the absence of ligand, and its concentration at cilia tips increases in response to pathway activation (Fig. 7 B; Chen et al., 2009; Endoh-Yamagami et al., 2009). In the absence of pathway activation, the amount of Gli2 at cilia tips appeared to be elevated in all of the IFT-A mutants examined (Fig. 7 B).
To confirm that the localization of Shh pathway proteins in MEFs reflected their behavior in the embryo, we examined the distribution of the proteins in mesenchymal cells surrounding the neural tube in wild-type and mutant embryos. In cells near the notochord, a source of Shh, Smo was present along the ciliary axoneme of wild-type cells (Fig. S5). In mesenchymal cells in Ift144dmhd embryos, no Smo was detected in the axoneme, as in MEFs. As in MEFs, Gli2 was present at the tips of wild-type and Ift144dmhd mesenchymal cilia (Fig. S5). Sufu and Kif7, essential components of the Shh signaling pathway that localize to the tips of wild-type cilia, were also present at the tips of Ift144dmhd mesenchymal cilia. Mesenchymal cells in Ift144twt Ift122sopb showed a similar pattern of Shh pathway protein localization as Ift144dmhd MEFs: Gli2, Sufu, and Kif7 were present at the tips of double-mutant cilia, but no Smo was detected in the cilium (Fig. S5). Thus, despite the gross structural defects, Gli2, Sufu, and Kif7 traffic into Ift144dmhd and Ift144twt Ift122sopb cilia, but Smo does not move into these cilia in response to pathway activation.
Discussion
IFT144 is required for anterograde, as well as retrograde, ciliary trafficking
Our results define an allelic series in the mouse Ift144 gene, which shows that partial loss of IFT-A and complete loss of IFT-A have very different effects on cilia structure and on cilia-dependent Shh signaling. Previous experiments have shown that the IFT-A complex in both Chlamydomonas and mouse is important for the normal rate of retrograde ciliary trafficking (Iomini et al., 2001, 2009; Tran et al., 2008). Embryos homozygous for the partial loss-of-function mutation Ift144twt have normal-length neural cilia that show characteristics expected for a slowed rate of retrograde trafficking: the cilia are somewhat wider than wild-type cilia, and they accumulate IFT88 and Gli2. In contrast, embryos homozygous for the apparent null allele Ift144dmhd have a strong defect in ciliogenesis. The loss of microtubules and the absence of Arl13b, ACIII, and Smo in Ift144dmhd mutant cilia suggest that the mutation decreases anterograde trafficking in the cilium.
Additional evidence for a role of IFT144 in anterograde trafficking comes from the analysis of double mutants that lack the dynein motor. Although both IFT-A and cytoplasmic dynein-2 are important for retrograde trafficking in the cilium, our data highlight the different requirements for the two classes of proteins in cilia structure. In Ift144dmhd cilia, a membrane compartment filled with electron-dense material extends distal to the basal body, but the normal microtubule structure of the axoneme is absent, and only a few microtubules are present in the cilium. This contrasts with the structure of Dync2h1mmi cilia, which are filled with a regular array of particles spaced at ∼40-nm intervals. These structures are likely to be trains of IFT particles, based on comparison with the arrays of IFT trains with 40-nm periodicity observed by electron tomography in Chlamydomonas LC8 mutants, which lack a dynein light chain (Pigino et al., 2009). Ift144twt Dync2h1mmi double-mutant cilia do not accumulate the ordered IFT trains seen in Dync2h1mmi. This is consistent with our previous results showing that reduction of either IFT-B or IFT-A partially rescued the cilia phenotype of Dync2h1 mutants because of roles of both IFT-B and IFT-A in anterograde ciliary trafficking (Ocbina et al., 2011). Thus, we conclude that IFT144 is required for normal anterograde trafficking.
The cilia phenotypes of the mouse Ift144 allelic series parallel findings with Chlamydomonas IFT-A mutants. At the permissive temperature, temperature-sensitive mutations in two Chlamydomonas IFT-A genes, fla15 (IFT144) and fla17 (IFT139), cause bulged flagella, associated with slowed rates of retrograde IFT (Iomini et al., 2001, 2009). However, at restrictive temperature, fla15 and fla17 mutant cells appear to be aflagellate, similar to the phenotype of null mutants in Chlamydomonas Ift140 (another IFT-A gene; Cole, 2003). Thus, in both Chlamydomonas and the mouse, partial loss of IFT-A proteins leads to a defect in retrograde trafficking, and complete loss of IFT-A function leads to the formation of very short cilia. Studies on Dyf-2, the C. elegans homolog of IFT144, are also consistent with roles of this protein in both retrograde and anterograde trafficking (Efimenko et al., 2006).
Overlapping functions of mouse IFT-A genes
Our genetic analysis of mouse mutants provides a different perspective on the function of individual IFT-A proteins than seen in biochemical experiments. Those studies suggested that mammalian IFT144/Wdr19 and IFT122 were core components of the mammalian IFT-A complex, whereas IFT139/Ttc21b and IFT121/Wdr35 were peripheral proteins that were not required for the formation of the core complex (Mukhopadhyay et al., 2010). In contrast, the genetic data indicate that loss of IFT144 or IFT121/Wdr35 (Mill et al., 2011) has a stronger effect on IFT-A function than does loss of IFT139a or IFT122.
Double mutants that are homozygous for both a null allele of Ift122 and the weak allele of Ift144 (Ift144twt Ift122sopb) have stronger defects in ciliogenesis than either single mutant and are indistinguishable from Ift144dmhd mutants in both cilia structure and protein localization. Therefore, we conclude that IFT144 and IFT122 partially overlap in function, and, in the absence of IFT122, decreased activity of IFT144 is sufficient to block the activity of the IFT-A complex. This conclusion is consistent with data from Chlamydomonas that some IFT proteins can substitute for each other (Iomini et al., 2009) and reveals functional differences among the IFT-A proteins.
Loss of Shh pathway activity in strong IFT-A mutants is associated with the disruption in cilia structure
Strong disruption of IFT-A, as in Ift144dmhd mutants, blocks the formation of a normal ciliary axoneme and the specification of ventral neural cell types that require high levels of Shh activity (Fig. 8 C). Because of the important roles of cilia in promoting Shh activity, a simple model is that the decreased activity of the Shh pathway in Ift144dmhd is a result of the collapse of cilia structure in the absence of the IFT-A complex and that normal cilia structure is required for cilia-associated Shh pathway proteins to respond correctly to ligand.
Figure 8.
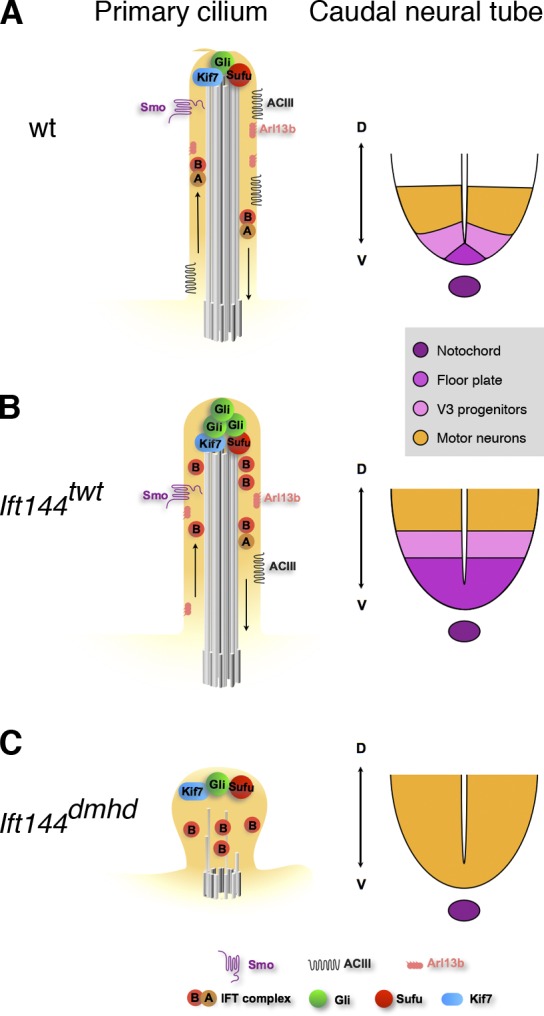
Model for the relationship between the composition of the primary cilium and Shh-dependent patterning of the neural tube in IFT144 mutants. (A) In wild type (wt), IFT is required to maintain the axonemal structure and to mediate trafficking of mammalian Shh pathway proteins in the primary cilium. When Shh ligand is present (illustrated here), Smo moves into cilia, and a complex of proteins required for activation of the pathway, including Kif7, Gli2, and Sufu, is present at cilia tips. Primary cilia are required for the production of the Gli2 transcription activator and therefore to respond to a gradient of Shh activity, which specifies ventral cell types in the developing neural tube, including floor plate, V3 progenitors, and motor neurons. Arl13b and ACIII are present in cilia both in the presence or absence of Shh and can modulate the activity of the pathway. (B) In Ift144twt mutants, cilia appear to have nearly normal morphology but are slightly wider than wild type. IFT-A complex proteins are reduced in cilia, whereas IFT-B particles tend to accumulate in the cilia tips. Smo trafficking into cilia in response to ligand is not affected, but Gli2 is enriched in the cilia regardless of pathway activation, as seen in other IFT-A complex mutants such as Ift122sopb. The level of ACIII in the ciliary membrane is reduced, which may lead to decreased cAMP and decreased activity of PKA, a negative regulator of the pathway. Shh-dependent ventral cell types are dorsally expanded in the neural tube. (C) Ift144dmhd mutant cilia have only rare microtubules and accumulate IFT-B particles, but no IFT-A particles are present. Smo, Arl13b, and ACIII fail to localize to the ciliary membrane, but Kif7, Sufu, and Gli2 accumulate at the tip of the short cilium. In the caudal neural tube, floor plate and V3 progenitors are absent, but motor neurons are formed, demonstrating the presence of an intermediate level of Shh pathway activity. Ift144twt Ift122sopb double mutants are indistinguishable from Ift144dmhd mutants in both cilia structure and neural patterning. D, dorsal; V, ventral.
Despite the loss of high-level responses to Shh, the Ift144dmhd and Ift144twt Ift122sopb embryos show ectopic activation of the pathway in the caudal neural tube, albeit at a lower level than in Ift144twt. As the ectopic motor neurons seen in Ift144dmhd embryos are also present in Ift144dmhd Dync2h1mmi double-mutant embryos (Fig. S3 F), where retrograde trafficking should be completely disrupted, the data argue that the activation of the Shh pathway seen in IFT-A mutants is not the result of defects in retrograde trafficking. Instead, the results suggest that the ligand-independent activation of the Shh pathway is the result of a defect in IFT-A–dependent anterograde trafficking.
Cilia localization of soluble Shh pathway proteins does not depend on IFT-A
Given the absence of most microtubules in the ciliary axoneme in the null allele Ift144dmhd, it is remarkable that the mutants can specify the ventral neural cell types that require intermediate levels of Shh activity and that this intermediate level of pathway activity depends on the presence of the Ift144dmhd cilium. We observed that Gli2, Kif7, and Sufu, which are critical for activity of the Shh pathway, are all present at the tips of the small Ift144dmhd and Ift144twt Ift122sopb double-mutant cilia, which suggests that the IFT-B complex is sufficient for trafficking Gli, Kif7, and Sufu to cilia tips and therefore for the activation of midlevel Shh activity. A critical role for IFT-B in cilia localization of Gli proteins is consistent with the reduction of Gli2 at cilia tips caused by a modest reduction in the level of the IFT-B protein IFT172 (Friedland-Little et al., 2011).
IFT-A–dependent trafficking of membrane proteins into cilia may account for the neural patterning phenotypes of IFT-A mutants
Our analysis indicates that a variety of proteins and protein complexes, including IFT88, Gli2, Sufu, and Kif7, can translocate into cilia that lack a functional IFT-A complex. In contrast, several membrane proteins, including Arl13b, ACIII, and Smo, are not detectable in cilia of either of the strong IFT-A mutants (Ift144dmhd single mutants or Ift144twt Ift122sopb double mutants). A role for the IFT-A–associated protein Tulp3 in trafficking of specific G protein–coupled receptors into cilia was previously defined (Mukhopadhyay et al., 2010), and our findings suggest that IFT-A has a general role in the trafficking of membrane proteins into cilia. Recent work has shown that a membrane-associated protein complex at the transition zone that includes the protein Tectonic1 is important for entry of some membrane proteins (ACIII and Arl13b) but had less effect on others (Smo) into cilia (Garcia-Gonzalo et al., 2011). This raises the possibility that the Tectonic1 and IFT-A complexes act in concert to promote the transport of membrane proteins into cilia.
The membrane proteins affected by loss of IFT-A (Arl13b, Smo, and ACIII) all have documented or plausible roles in Shh signaling and are likely to contribute to the Shh phenotypes of the IFT-A mutants. Mouse mutants that lack Arl13b show strong defects in Shh signaling and neural patterning (Caspary et al., 2007), so the absence of detectable ciliary Arl13b in the strong IFT-A mutants (Ift144dmhd and Ift144twt Ift122sopb) could be responsible for some aspects of the neural patterning phenotypes of these embryos. Indeed, cell types that require high levels of Shh activity (the floor plate and V3 interneurons) fail to be specified, and the motor neuron domain is expanded in the caudal neural tube of Arl13b mutants, as in Ift144dmhd and Ift144twt Ift122sopb embryos. However, there must be additional targets of IFT-A that contribute to the neural patterning defects in the mutant embryos because the Arl13b and IFT-A neural patterning phenotypes are not identical. For example, neural patterning is normal in the rostral neural tube of Arl13b mutant embryos, but ventral neural cell types are not specified in this region of the neural tube in the strong IFT-A mutants; this could be explained if an unidentified membrane protein with an Arl13b-like function depends on IFT-A for its cilia localization in the rostral neural tube. However, even in the caudal neural tube, where the Arl13b and IFT-A phenotypes are similar, double mutants reveal differences between the genotypes. For example, motor neurons are specified in Arl13b Gli2 double mutants but not in Ift144dmhd Gli2 double-mutant embryos. Therefore, we suggest that the IFT-A neural patterning phenotype could be the result of the absence of several different membrane proteins in the cilium, including Arl13b.
The one core component of the Shh pathway that does not traffic into cilia of the strong IFT-A mutants (Ift144dmhd and Ift144twt Ift122sopb) is Smo, a positive regulator of the pathway. However, the absence of Smo in the cilium cannot account for the neural patterning phenotypes in Ift144dmhd embryos, as our double-mutant analysis shows that the Ift144dmhd phenotype is independent of the presence or absence of Smo. Nevertheless, our data provide the first evidence that IFT proteins play a role for Smo movement into cilia and contrast with models that suggest Smo moves into cilia by lateral transport from the plasma membrane (Milenkovic et al., 2009). IFT-A could directly promote Smo movement from the transition zone into the cilium. Alternatively, the strong IFT-A mutations might disrupt axonemal structure in a way that other proteins cannot transport Smo past the transition zone.
The increased Shh pathway activity seen in the weak IFT-A mutants is a long-standing puzzle. The neural patterning phenotype of the hypomorphic mutant Ift144twt is similar to those of null alleles of Ift122 and Ift139a (Fig. 8, A and B; Tran et al., 2008; Qin et al., 2011). All three mutations cause an expansion of Shh-dependent cell types in the neural tube, and the three mutants also cause polydactyly and craniofacial phenotypes (Tran et al., 2008; Qin et al., 2011; Ashe et al., 2012). Mutations in Tulp3, an IFT-A–associated protein, also cause ectopic activation of the Shh pathway in the neural tube, but the structure of Tulp3 cilia appears to be normal (Cameron et al., 2009; Norman et al., 2009; Patterson et al., 2009), and it has been suggested that the Tulp3 neural patterning phenotype reflects its role in the anterograde trafficking of a membrane-associated negative regulator of the pathway (Mukhopadhyay et al., 2010).
Our findings suggest a possible explanation for gain of Shh activity in the weak IFT-A mutants. Although ACIII is generally considered a cilia marker, ACIII and other adenylyl cyclases (Bishop et al., 2007; Choi et al., 2011) that are localized to cilia are likely to be important for Shh signaling. cAMP-dependent protein kinase is a strong negative regulator of Shh signaling (Tuson et al., 2011). It is reasonable to propose that the adenylyl cyclases in the cilium are the source of the cAMP that activates PKA (Barzi et al., 2010). In the absence of ciliary cAMP, we predict that PKA activity would be decreased, which would lead to ligand-independent activation of the Shh pathway. Therefore, we hypothesize that the gain of Shh activity seen in the weak IFT-A mutants may be the result of decreased levels of adenylyl cyclase in the cilia. The activation of intermediate, but not high, levels of Shh pathway activity in the strong IFT-A mutants (Fig. 8 C) could be a result of the combined loss of ACIII and Arl13b in the mutant cilia or a result of the absence of those proteins in combination with the disrupted cilia structure.
IFT-A and human genetic disease
Mutations in all six IFT-A genes—IFT139a/TTC21b, IFT121/WDR35, IFT43, IFT122, IFT144, and IFT140—cause human ciliopathies (Gilissen et al., 2010; Arts et al., 2011; Bredrup et al., 2011; Davis et al., 2011; Mill et al., 2011; Perrault et al., 2012), whereas disease-causing mutations have been identified in only a single IFT-B gene (Beales et al., 2007). The higher frequency of disease-causing IFT-A mutations is likely to reflect the overlap in function we have observed among IFT genes, so that even null mutations cause relatively mild phenotypes that are compatible with postnatal life. As the Ift144twt mutant recapitulates many of the skeletal and craniofacial anomalies seen in these diseases (Ashe et al., 2012), our findings suggest that the complex phenotypes seen in IFT-A syndromes are likely to be a result of the loss of specific membrane proteins in the mutant cilia.
Materials and methods
Mouse strains and phenotypic analysis
A screen for recessive ENU-induced mutations (García-García et al., 2005) was performed by mutagenizing C57BL6/J (The Jackson Laboratory) male mice and out-crossing to FVB/NJ (The Jackson Laboratory) females homozygous for the HB9-GFP transgene, which expresses EGFP in spinal motor neurons (Liem et al., 2009). The dmhd mutation was identified based on decreased GFP expression in whole E10.5 N3 embryos that were potentially homozygous for newly induced mutations. For analysis of neural patterning, embryos were processed as previously described (Caspary et al., 2007): embryos were fixed in 4% PFA in PBS for 40 min on ice, cryoprotected with 30% sucrose, embedded in Tissue-Tek (Sakura), and cryosectioned at 12 µm. The sections were then stained for expression of region-specific neural antigens. All sections were counterstained with DAPI (blue in all figures) to stain nuclei. The following mutant mouse strains were used for analysis: Ptch1tm1Mps (a gift from M. Scott, Stanford University; Stanford, CA; Goodrich et al., 1997), Smobnb (a null ENU-induced allele; Caspary et al., 2002), Ift172wim (an ENU-induced allele; Huangfu et al., 2003), Gli2 (a gift from A. Joyner, Sloan-Kettering Institute, New York, NY; Matise et al., 1998), Gli3Xt (a gift from A. Joyner; Hui and Joyner, 1993), Dync2h1mmi (an ENU-induced allele; Liem et al., 2009), IFT144twt (an ENU-induced allele; Ashe et al., 2012), and Ift122sopb (a null ENU-induced allele provided by J.T. Eggenschwiler, Princeton University, Princeton, NJ; Qin et al., 2011).
Cell culture and antibodies
MEFs were derived from E10.5 embryos using standard protocols, as previously described (Ocbina and Anderson, 2008). Confluent MEF cultures were serum starved for 48 h to induce ciliogenesis and treated with 100 nM SAG (EMD) for 24 h to activate the Shh pathway. Cultures were fixed for 10 min in 2% PFA followed by 5 min in 100% methanol. The Gli2 antibody (raised in guinea pig) was a gift from J.T. Eggenschwiler. The IFT88 and IFT140 antibodies (both raised in rabbits) were gifts from B. Yoder (University of Alabama, Birmingham, AL) and G. Pazour (University of Massachusetts Medical School, Worcester, MA), respectively. The Smo antibody (rabbit) was previously described (Ocbina et al., 2011). The rabbit Kif7 polyclonal antibody (Pocono Rabbit Farm and Laboratory, Inc.) was raised to a Kif7 peptide (amino acids 362–600) fused to a His tag expressed in bacteria. The following additional antibodies were used: rabbit anti-Arl13b (Caspary et al., 2007), mouse anti-Nkx2.2 (Developmental Studies Hybridoma Bank), rabbit anti-FoxA2 (Abcam), mouse anti-WDR19/IFT144 antibody (immunofluorescence; Abnova), rabbit anti-WDR19/IFT144 antibody (Western blot; GeneTex Inc.), mouse anti–γ-tubulin (Sigma-Aldrich), mouse antiacetylated α-tubulin (Sigma-Aldrich), rabbit anti-IFT88 (Proteintech Group, Inc.), and rabbit anti-ACIII (Santa Cruz Biotechnology, Inc.). Immunofluorescence staining was performed with Alexa Fluor (488, 594, and 633)–conjugated secondary antibodies (Invitrogen). Double antibody staining with rabbit antibodies was performed in series using Dylight 488-Fab fragment goat anti–rabbit H+L (Jackson ImmunoResearch Laboratories, Inc.).
Image acquisition
Immunofluorescent imaging of cilia was performed using an image restoration system (DeltaVision; Applied Precision) with a wide-field microscope (IX70; Olympus). Imaging was performed using a 100× 1.4 NA objective at room temperature mounted in VECTASHIELD (Vector Laboratories) and captured with a cooled charge-coupled device camera (CoolSNAP QE; Photometrics). Images were acquired using SoftWorks, and datasets were processed using the Volocity software package to produce an extended-view snapshot (PerkinElmer). To improve clarity, some images were adjusted using the curves tool in Photoshop (Adobe); in each case, the entire image was adjusted identically, and all panels in a figure were adjusted with identical settings. Quantification of proteins within cilia was performed using MetaMorph software (Molecular Devices) and measuring the sum of intensities in DeltaVision z stacks. Neural tube sections were analyzed with a microscope (Axioplan2; Carl Zeiss) using a 10× 0.5 NA lens at room temperature in VECTASHIELD. Images were acquired with a wide-field monochrome camera (AxioCam MR) using AxioVision software (v4.6; Carl Zeiss).
Identification of the dmhd mutation
The dmhd mutation was mapped to a segment of chromosome 5 based on the identification of regions homozygous for C57BL6/J polymorphisms in DNA from mutant embryos on a genome-wide single-nucleotide polymorphism panel (Moran et al., 2006). The mutation mapped between two single-nucleotide polymorphisms, CEL-5_34263494 and rs6409508 (Ensembl build M37). A splice site mutation was identified in the Wdr19 gene: a T to C substitution at the second nucleotide of intron 16–17 (Ensembl transcript ENSMUST00000041892). No wild-type transcript was detected by PCR from mutant embryo cDNA, indicating that this mutation completely blocks normal splicing. The Ift144dmhd mutation disrupts a Kpn1 restriction site present in the wild-type sequence, creating a restriction fragment–length polymorphism that was used for genotyping. PCR amplification from genomic DNA (primers 5′-TGATGCGACCTATGAGATTCC-3′ and 5′-CCAGCCAAAATAACCTTGGA-3′) generated a 287-bp PCR product containing the restriction fragment–length polymorphism. Kpn1 digestion cleaves the wild-type, but not the dmhd mutant, amplicon.
EM
E9.5 or 10.5 embryos were fixed with 2% PFA and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. For scanning electron microscopy, embryos were dissected to expose the lumen of the neural tube in 0.1 M cacodylate buffer and dehydrated in ethanol (Huangfu et al., 2003). Scanning electron microscopy was performed using a field emission microscope (Supra 25; Carl Zeiss), and images were acquired with SmartSEM (Carl Zeiss). For TEM, samples were postfixed with 1% osmium tetroxide followed by 1% uranyl acetate, dehydrated through a graded series of ethanol, and embedded in LX112 resin (Ladd Research Industries). Ultrathin sections were cut on an ultramicrotome (Reichert Ultracut; Leica), stained with uranyl acetate followed by lead citrate, and viewed on a transmission electron microscope (1200 EX; JEOL Ltd.) at 80 kV.
Online supplemental material
Fig. S1 shows the molecular and phenotypic characterization of the Ift144twt and Ift144dmhd mutants. Fig. S2 shows double-mutant analysis that places Ift144 in the genetic pathway downstream of Ptch1 and Smo and upstream of Gli2 and Gli3. Fig. S3 shows that the Ift144dmhd phenotype depends on cilia but is independent of Dync2h1. Fig. S4 shows the range of structural defects of Ift144dmhd neural cilia seen by TEM. Fig. S5 shows the localization of Shh pathway and ciliary components in IFT-A mutant embryonic mesenchymal cells. Table S1 shows cilia dimensions in wild type and mutants. Table S2 shows cilia localization of Shh pathway proteins in embryonic mesenchymal cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201110049/DC1.
Supplementary Material
Acknowledgments
We thank Leslie Gunther-Cummins (Albert Einstein College of Medicine, Bronx, NY) for assistance with TEM, Carlo Iomini (Mount Sinai Hospital, New York, NY) for helpful discussions, Jonathan Eggenschwiler for antibodies and the Ift122sopb mice, and Brad Yoder and Greg Pazour for antibodies. We thank Sarah Goetz for helpful comments on the manuscript. We thank Nina Lampen for help with scanning electron microscopy and the Memorial Sloan-Kettering Cancer Center core facilities and Tao Tong of the Rockefeller University Bio-Imaging Resource Center for help with imaging.
This work was supported by National Institutes of Health grant no. NS044385 to K.V. Anderson, National Health and Medical Research Council of Australia project grant no. 631493 to C. Wicking, and National Institutes of Health grant no. HD43430 to D. Beier.
Footnotes
Abbreviations used in this paper:
- ENU
- ethylnitrosourea
- IFT
- intraflagellar transport
- MEF
- mouse embryo fibroblast
- TEM
- transmission EM
References
- Arts H.H., Bongers E.M., Mans D.A., van Beersum S.E., Oud M.M., Bolat E., Spruijt L., Cornelissen E.A., Schuurs-Hoeijmakers J.H., de Leeuw N., et al. 2011. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J. Med. Genet. 48:390–395 10.1136/jmg.2011.088864 [DOI] [PubMed] [Google Scholar]
- Ashe A., Butterfield N.C., Town L., Courtney A.D., Cooper A.N., Ferguson C., Barry R., Olsson F., Liem K.F., Jr., Parton R.G., et al. 2012. Mutations in mouse Ift144 model the craniofacial, limb and rib defects in skeletal ciliopathies. Hum. Mol. Genet. 21:1808–1823 10.1093/hmg/ddr613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai C.B., Stephen D., Joyner A.L. 2004. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell. 6:103–115 10.1016/S1534-5807(03)00394-0 [DOI] [PubMed] [Google Scholar]
- Barzi M., Berenguer J., Menendez A., Alvarez-Rodriguez R., Pons S. 2010. Sonic-hedgehog-mediated proliferation requires the localization of PKA to the cilium base. J. Cell Sci. 123:62–69 10.1242/jcs.060020 [DOI] [PubMed] [Google Scholar]
- Beales P.L., Bland E., Tobin J.L., Bacchelli C., Tuysuz B., Hill J., Rix S., Pearson C.G., Kai M., Hartley J., et al. 2007. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39:727–729 10.1038/ng2038 [DOI] [PubMed] [Google Scholar]
- Behal R.H., Miller M.S., Qin H., Lucker B.F., Jones A., Cole D.G. 2012. Subunit interactions and organization of the Chlamydomonas reinhardtii intraflagellar transport complex A proteins. J. Biol. Chem. 287:11689–11703 10.1074/jbc.M111.287102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop G.A., Berbari N.F., Lewis J., Mykytyn K. 2007. Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol. 505:562–571 10.1002/cne.21510 [DOI] [PubMed] [Google Scholar]
- Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbø M., Filhol E., Bole-Feysot C., et al. 2011. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 89:634–643 10.1016/j.ajhg.2011.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J. 2009. Making a grade: Sonic Hedgehog signalling and the control of neural cell fate. EMBO J. 28:457–465 10.1038/emboj.2009.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D.A., Pennimpede T., Petkovich M. 2009. Tulp3 is a critical repressor of mouse hedgehog signaling. Dev. Dyn. 238:1140–1149 10.1002/dvdy.21926 [DOI] [PubMed] [Google Scholar]
- Caspary T., García-García M.J., Huangfu D., Eggenschwiler J.T., Wyler M.R., Rakeman A.S., Alcorn H.L., Anderson K.V. 2002. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr. Biol. 12:1628–1632 10.1016/S0960-9822(02)01147-8 [DOI] [PubMed] [Google Scholar]
- Caspary T., Larkins C.E., Anderson K.V. 2007. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 12:767–778 10.1016/j.devcel.2007.03.004 [DOI] [PubMed] [Google Scholar]
- Cevik S., Hori Y., Kaplan O.I., Kida K., Toivenon T., Foley-Fisher C., Cottell D., Katada T., Kontani K., Blacque O.E. 2010. Joubert syndrome Arl13b functions at ciliary membranes and stabilizes protein transport in Caenorhabditis elegans. J. Cell Biol. 188:953–969 10.1083/jcb.200908133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.H., Wilson C.W., Li Y.J., Law K.K., Lu C.S., Gacayan R., Zhang X., Hui C.C., Chuang P.T. 2009. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23:1910–1928 10.1101/gad.1794109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.H., Suzuki A., Hajarnis S., Ma Z., Chapin H.C., Caplan M.J., Pontoglio M., Somlo S., Igarashi P. 2011. Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc. Natl. Acad. Sci. USA. 108:10679–10684 10.1073/pnas.1016214108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole D.G. 2003. The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic. 4:435–442 10.1034/j.1600-0854.2003.t01-1-00103.x [DOI] [PubMed] [Google Scholar]
- Cortellino S., Wang C., Wang B., Bassi M.R., Caretti E., Champeval D., Calmont A., Jarnik M., Burch J., Zaret K.S., et al. 2009. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev. Biol. 325:225–237 10.1016/j.ydbio.2008.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E.E., Zhang Q., Liu Q., Diplas B.H., Davey L.M., Hartley J., Stoetzel C., Szymanska K., Ramaswami G., Logan C.V., et al. 2011. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43:189–196 10.1038/ng.756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efimenko E., Blacque O.E., Ou G., Haycraft C.J., Yoder B.K., Scholey J.M., Leroux M.R., Swoboda P. 2006. Caenorhabditis elegans DYF-2, an orthologue of human WDR19, is a component of the intraflagellar transport machinery in sensory cilia. Mol. Biol. Cell. 17:4801–4811 10.1091/mbc.E06-04-0260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endoh-Yamagami S., Evangelista M., Wilson D., Wen X., Theunissen J.W., Phamluong K., Davis M., Scales S.J., Solloway M.J., de Sauvage F.J., Peterson A.S. 2009. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 19:1320–1326 10.1016/j.cub.2009.06.046 [DOI] [PubMed] [Google Scholar]
- Friedland-Little J.M., Hoffmann A.D., Ocbina P.J., Peterson M.A., Bosman J.D., Chen Y., Cheng S.Y., Anderson K.V., Moskowitz I.P. 2011. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum. Mol. Genet. 20:3725–3737 10.1093/hmg/ddr241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-García M.J., Eggenschwiler J.T., Caspary T., Alcorn H.L., Wyler M.R., Huangfu D., Rakeman A.S., Lee J.D., Feinberg E.H., Timmer J.R., Anderson K.V. 2005. Analysis of mouse embryonic patterning and morphogenesis by forward genetics. Proc. Natl. Acad. Sci. USA. 102:5913–5919 10.1073/pnas.0501071102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J., et al. 2011. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 43:776–784 10.1038/ng.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen C., Arts H.H., Hoischen A., Spruijt L., Mans D.A., Arts P., van Lier B., Steehouwer M., van Reeuwijk J., Kant S.G., et al. 2010. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am. J. Hum. Genet. 87:418–423 10.1016/j.ajhg.2010.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz S.C., Anderson K.V. 2010. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 11:331–344 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L.V., Milenović L., Higgins K.M., Scott M.P. 1997. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 277:1109–1113 10.1126/science.277.5329.1109 [DOI] [PubMed] [Google Scholar]
- Huangfu D., Anderson K.V. 2005. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA. 102:11325–11330 10.1073/pnas.0505328102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. 2003. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 426:83–87 10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- Hui C.C., Joyner A.L. 1993. A mouse model of greig cephalopolysyndactyly syndrome: The extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3:241–246 10.1038/ng0393-241 [DOI] [PubMed] [Google Scholar]
- Ingham P.W., Nakano Y., Seger C. 2011. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 12:393–406 10.1038/nrg2984 [DOI] [PubMed] [Google Scholar]
- Iomini C., Babaev-Khaimov V., Sassaroli M., Piperno G. 2001. Protein particles in Chlamydomonas flagella undergo a transport cycle consisting of four phases. J. Cell Biol. 153:13–24 10.1083/jcb.153.1.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iomini C., Li L., Esparza J.M., Dutcher S.K. 2009. Retrograde intraflagellar transport mutants identify complex A proteins with multiple genetic interactions in Chlamydomonas reinhardtii. Genetics. 183:885–896 10.1534/genetics.109.101915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkins C.E., Aviles G.D., East M.P., Kahn R.A., Caspary T. 2011. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol. Biol. Cell. 22:4694–4703 10.1091/mbc.E10-12-0994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem K.F., Jr., He M., Ocbina P.J., Anderson K.V. 2009. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA. 106:13377–13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise M.P., Epstein D.J., Park H.L., Platt K.A., Joyner A.L. 1998. Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development. 125:2759–2770 [DOI] [PubMed] [Google Scholar]
- Milenkovic L., Scott M.P., Rohatgi R. 2009. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J. Cell Biol. 187:365–374 10.1083/jcb.200907126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill P., Lockhart P.J., Fitzpatrick E., Mountford H.S., Hall E.A., Reijns M.A., Keighren M., Bahlo M., Bromhead C.J., Budd P., et al. 2011. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am. J. Hum. Genet. 88:508–515 10.1016/j.ajhg.2011.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla-Herman A., Ghossoub R., Blisnick T., Meunier A., Serres C., Silbermann F., Emmerson C., Romeo K., Bourdoncle P., Schmitt A., et al. 2010. The ciliary pocket: An endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci. 123:1785–1795 10.1242/jcs.059519 [DOI] [PubMed] [Google Scholar]
- Moran J.L., Bolton A.D., Tran P.V., Brown A., Dwyer N.D., Manning D.K., Bjork B.C., Li C., Montgomery K., Siepka S.M., et al. 2006. Utilization of a whole genome SNP panel for efficient genetic mapping in the mouse. Genome Res. 16:436–440 10.1101/gr.4563306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Wen X., Chih B., Nelson C.D., Lane W.S., Scales S.J., Jackson P.K. 2010. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 24:2180–2193 10.1101/gad.1966210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman R.X., Ko H.W., Huang V., Eun C.M., Abler L.L., Zhang Z., Sun X., Eggenschwiler J.T. 2009. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum. Mol. Genet. 18:1740–1754 10.1093/hmg/ddp113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina P.J., Anderson K.V. 2008. Intraflagellar transport, cilia, and mammalian Hedgehog signaling: Analysis in mouse embryonic fibroblasts. Dev. Dyn. 237:2030–2038 10.1002/dvdy.21551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocbina P.J., Eggenschwiler J.T., Moskowitz I., Anderson K.V. 2011. Complex interactions between genes controlling trafficking in primary cilia. Nat. Genet. 43:547–553 10.1038/ng.832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson V.L., Damrau C., Paudyal A., Reeve B., Grimes D.T., Stewart M.E., Williams D.J., Siggers P., Greenfield A., Murdoch J.N. 2009. Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: Identification of Tulp3 as a novel negative regulator of the Sonic hedgehog pathway. Hum. Mol. Genet. 18:1719–1739 10.1093/hmg/ddp075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour G.J., Dickert B.L., Witman G.B. 1999. The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J. Cell Biol. 144:473–481 10.1083/jcb.144.3.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen L.B., Rosenbaum J.L. 2008. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 85:23–61 10.1016/S0070-2153(08)00802-8 [DOI] [PubMed] [Google Scholar]
- Pedersen L.B., Geimer S., Rosenbaum J.L. 2006. Dissecting the molecular mechanisms of intraflagellar transport in chlamydomonas. Curr. Biol. 16:450–459 10.1016/j.cub.2006.02.020 [DOI] [PubMed] [Google Scholar]
- Perrault I., Saunier S., Hanein S., Filhol E., Bizet A.A., Collins F., Salih M.A., Gerber S., Delphin N., Bigot K., et al. 2012. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am. J. Hum. Genet. 90:864–870 10.1016/j.ajhg.2012.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G., Geimer S., Lanzavecchia S., Paccagnini E., Cantele F., Diener D.R., Rosenbaum J.L., Lupetti P. 2009. Electron-tomographic analysis of intraflagellar transport particle trains in situ. J. Cell Biol. 187:135–148 10.1083/jcb.200905103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Rosenbaum J.L., Barr M.M. 2001. An autosomal recessive polycystic kidney disease gene homolog is involved in intraflagellar transport in C. elegans ciliated sensory neurons. Curr. Biol. 11:457–461 10.1016/S0960-9822(01)00122-1 [DOI] [PubMed] [Google Scholar]
- Qin J., Lin Y., Norman R.X., Ko H.W., Eggenschwiler J.T. 2011. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc. Natl. Acad. Sci. USA. 108:1456–1461 10.1073/pnas.1011410108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Snell W.J. 2010. The ciliary membrane. Curr. Opin. Cell Biol. 22:541–546 10.1016/j.ceb.2010.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin S. 1962. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 15:363–377 10.1083/jcb.15.2.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran P.V., Haycraft C.J., Besschetnova T.Y., Turbe-Doan A., Stottmann R.W., Herron B.J., Chesebro A.L., Qiu H., Scherz P.J., Shah J.V., et al. 2008. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 40:403–410 10.1038/ng.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuson M., He M., Anderson K.V. 2011. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development. 138:4921–4930 10.1242/dev.070805 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.