

The P-site of the 60S ribosomal subunit signals to Tif6 via Elf1 during ribosomal maturation, suggesting a quasifunctional check of the integrity of the 60S subunit before the first round of translation.
Abstract
Eukaryotic ribosomes are preassembled in the nucleus and mature in the cytoplasm. Release of the antiassociation factor Tif6 by the translocase-like guanosine triphosphatase Efl1 is a critical late maturation step. In this paper, we show that a loop of Rpl10 that embraces the P-site transfer ribonucleic acid was required for release of Tif6, 90 Å away. Mutations in this P-site loop blocked 60S maturation but were suppressed by mutations in Tif6 or Efl1. Molecular dynamics simulations of the mutant Efl1 proteins suggest that they promote a conformation change in Efl1 equivalent to changes that elongation factor G and eEF2 undergo during translocation. These results identify molecular signaling from the P-site to Tif6 via Efl1, suggesting that the integrity of the P-site is interrogated during maturation. We propose that Efl1 promotes a functional check of the integrity of the 60S subunit before its first round of translation.
Introduction
Accurate translation is crucial for proper protein function and, consequently, for the viability of all cellular processes. The complexity of ribosome structure would appear to present an extreme challenge to a cell to ensure the correct assembly and function of the ribosome. Because defects in assembly would likely lead to reduced function and fidelity of the ribosome, strategies must have evolved to ensure the proper function of newly assembled ribosomes. However, the mechanisms that cells use to monitor the correct assembly of their ribosomes are largely unknown.
Ribosomes are comprised of two subunits, the large (60S in eukaryotes) and small (40S) subunits (LSUs and SSUs, respectively) that display a division of labor in translation: the LSU carries out peptidyl transferase activity, whereas the SSU utilizes tRNAs to decode mRNAs. Eukaryotic ribosomes are largely preassembled in the nucleus, requiring >200 trans-acting factors (Henras et al., 2008). The premature subunits are then exported to the cytoplasm, where they undergo final maturation steps before becoming translationally competent. Maturation of the pre-60S subunit involves the recycling of export factors, the removal of placeholder proteins, and the assembly of several critical r-proteins (Zemp and Kutay, 2007; Panse and Johnson, 2010).
We have recently established the order of events of the cytoplasmic maturation pathway of the LSU (Lo et al., 2010). Two different ATPases carry out one series of protein exchanges, leading to the release of the export receptor Arx1 (Lo et al., 2010). The ribosome stalk, which is critical for recruiting and activating translation factors (Mohr et al., 2002), is assembled separately (Kemmler et al., 2009; Lo et al., 2009). These two series of events are prerequisite for the function of the GTPase Efl1, which together with Sdo1 releases the shuttling protein Tif6 (Bécam et al., 2001; Senger et al., 2001; Menne et al., 2007). Tif6 binds to the intersubunit bridge B6, making contacts with the sarcin-ricin loop (SRL), Rpl23, and Rpl24, thereby blocking 40S joining (Gartmann et al., 2010). Efl1 is homologous to the translation elongation factor eEF2 (elongation factor G [EF-G] in prokaryotes; Senger et al., 2001), whereas Sdo1 is orthologous to the human Shwachman–Bodian–Diamond syndrome protein (Shammas et al., 2005; Luz et al., 2009), mutations in which cause Shwachman–Bodian–Diamond syndrome, an autosomal recessive bone marrow failure disease (Boocock et al., 2003). In the last known step, which depends on the prior release of Tif6, the export adaptor Nmd3 is released from the LSU by the GTPase Lsg1 (Hedges et al., 2005; West et al., 2005).
eEF2 and EF-G promote mRNA–tRNA translocation during translation. After peptidyl transfer, the peptidyl tRNA rapidly shifts to the hybrid A/P position through a natural ratchetlike motion of the subunits (Agirrezabala et al., 2008). During translocation, EF-G is recruited to the GTPase-associated center of the ribosome by the L7/L12 stalk (Mohr et al., 2002). GTP hydrolysis by EF-G (Rodnina et al., 1997) induces a conformational change in the protein (Czworkowski et al., 1994; Agrawal et al., 1999) that drives translocation of the peptidyl tRNA from the A/P position into the P/P position.
We previously suggested that cytoplasmic assembly of the P0/P1/P2 protein stalk (the eukaryotic equivalent of L10/L7/L12) is necessary for recruitment and activation of Efl1 to induce the release of Tif6 (Lo et al., 2010). In this model, Efl1 utilizes the known function of the stalk to recruit and activate GTPases during translation for a biogenesis-specific function. Here, we show that a loop of the LSU protein Rpl10 is also intimately involved in the release of Tif6 from the LSU. This loop, which we will refer to as the P-site loop, extends toward the catalytic center of the ribosome, contacting the acceptor stem of the P-site tRNA (Armache et al., 2010). Mutations in this loop prevent the release of Tif6. Mutations in Efl1 bypass the effects of these P-site loop mutations. These Efl1 mutations are predicted to destabilize domain interfaces and facilitate conformational changes analogous to those that eEF2 undergoes during translocation. Our data suggest that in addition to interrogating the correct assembly of the stalk, Efl1 interrogates the P-site of the ribosome in a more rigorous assessment of the integrity of LSU assembly than previously recognized. The utilization of a translocation-like activity during biogenesis suggests that the newly assembled ribosomal subunit undergoes a test drive before being released into the active pool of ribosomes engaged in translating mRNAs.
Results
Rpl10 is required for the release of Tif6
We previously showed that Rpl10 is required for the release of the nuclear export adapter Nmd3 (Hedges et al., 2005; West et al., 2005). However, this is the last step of a multistep pathway (Lo et al., 2010). Hence, we asked whether Rpl10 is required for any of the upstream events. A galactose-inducible GAL1 promoter was integrated into the RPL10 locus in strains expressing GFP-tagged Mrt4, Arx1, or Tif6. These proteins display nuclear localization at steady state but become cytoplasmic under conditions that prevent their release from the LSU (Lo et al., 2010). To monitor Nmd3, which is predominantly cytoplasmic, a leptomycin B (LMB)–sensitive CRM1-T539C mutant was used. Because Crm1 is the nuclear export receptor for Nmd3, LMB inhibits the export of Nmd3, and it accumulates in the nucleus. As expected, under nonrepressing conditions (galactose), Mrt4, Arx1, and Tif6 localized to the nucleus (Fig. 1 A, left), and Nmd3 could be trapped in the nucleus by the addition of LMB, indicating that it was shuttling (Fig. 1 A, left). In repressing conditions (glucose), Mrt4 and Arx1 remained nuclear (Fig. 1 A, right), whereas Nmd3 became cytoplasmic, as we have previously shown (Hedges et al., 2005; West et al., 2005). Surprisingly, repression of RPL10 also caused mislocalization of Tif6 from the nucleus to the cytoplasm (Fig. 1 A, right), indicating a failure in Tif6 recycling. To distinguish between a defect in reimport of free Tif6 or a failure to release it from cytoplasmic 60S subunits, we monitored the sedimentation of Tif6 in sucrose gradients. Under conditions of RPL10 expression or repression conditions, Tif6 sedimented strictly at the position of free 60S subunits (Fig. 1 B), indicating that Tif6 remains bound to the subunit in the cytoplasm when RPL10 expression is repressed.
Figure 1.

The P-site loop of Rpl10 is required for the release of Tif6 from 60S ribosomal subunits. (A) The localization of Mrt4, Arx1, Tif6, and Nmd3 was examined in the presence (galactose) or absence (glucose) of ongoing Rpl10 expression. AJY2766 (PGAL1-RPL10 TIF6-GFP), AJY2767 (PGAL1-RPL10 ARX1-GFP), and AJY2768 (PGAL1-RPL10 MRT4-GFP) were grown in galactose to mid-log phase; the cultures were split in two, and for one, Rpl10 expression was repressed for 2 h by the addition of glucose. GFP-tagged proteins were visualized by microscopy. AJY1837 (PGAL1-RPL10 NMD3-GFP crm1-T539C) was treated, as previously described, with the addition of LMB after glucose addition. DIC, differential interference contrast. (B) Sucrose gradient sedimentation of Tif6. AJY2766 (PGAL1-RPL10 TIF6-GFP) was cultured as described in A. Crude extracts were prepared and fractionated by sucrose gradient sedimentation. The position of Tif6 in gradients was monitored by Western blotting using anti-GFP antibody. Anti-Rpl8 was used to monitor the position of 60S subunits. Numbers in parentheses indicate the predicted molecular mass in kilodaltons of the protein being detected. Sizes (kD) and positions (−) of nearest molecular mass markers are indicated. (C) The Rpl10 P-site loop is required for release of Tif6 from 60S subunits. The GFP-tagged strains described in A were transformed with a vector (pAJ1777) expressing mutant RPL10 deleted of the P-site loop (rpl10-Δ102-112). GFP fluorescence of the tagged proteins was monitored under conditions of Rpl10 expression (galactose) or repression (glucose), as in A. Bars, 5 µm.
The P-site loop of Rpl10 is required for the release of Tif6
In a previous mutational analysis of Rpl10 (Hofer et al., 2007), we identified an internal loop (aa 102–112) that is required for the release of Nmd3. Rpl10 (L16 in bacteria) is located in a cleft between the central protuberance and the P0/P1/P2 ribosome stalk (Fig. 2 A). High-resolution crystal structures of the bacterial ribosome show that this loop of L16 extends toward the P-site and, together with L27, embraces the P-site tRNA (Fig. 2 C; Gao et al., 2009; Voorhees et al., 2009). Thus, we refer to this loop of Rpl10 as the P-site loop. Archaeons and eukaryotes lack L27 but have an elongated P-site loop (Fig. 2 C) that may functionally replace L27 (Schmeing et al., 2009; Voorhees et al., 2009). This extended P-site loop has been modeled by cryo-EM of translating eukaryotic ribosomes, where it is seen making contact with the acceptor stem of the P-site tRNA (Fig. 2 C; Armache et al., 2010). Deletion of the P-site loop (rpl10-Δ102-112) is lethal and blocks the recycling of Nmd3 (Hofer et al., 2007). Here, we asked whether the release of Tif6 also depends on the P-site loop of Rpl10. We introduced plasmid-borne rpl10-Δ102–112 into GAL1-RPL10 strains. Upon repression of wild-type (WT) RPL10 so that only rpl10-Δ102-112 continued to be expressed, Mrt4 and Arx1 remained nuclear, whereas Tif6 and Nmd3 became cytoplasmic (Fig. 1 C, right). Furthermore, overexpression of rpl10-Δ102-112 was dominant negative and caused mislocalization of Tif6 to the cytoplasm in WT cells (unpublished data), indicating that the mislocalization was a result of mutant Rpl10. Thus, deletion of the P-site loop had an effect similar to repression of RPL10 in preventing the release of both Tif6 and Nmd3 from 60S subunits. These results implicate the P-site loop, deep in the catalytic heart of the ribosome, in the release of Tif6.
Figure 2.

Mutagenesis of the P-site loop of Rpl10. (A) A composite image of the LSU showing the expected relative positions of L16/Rpl10, P-site tRNA, EF-G/Efl1, and Tif6 was made by docking yeast Tif6 (Protein Data Bank accession no. 2X7N; Gartmann et al., 2010) onto the bacterial 50S subunit with EF-G (PDB accession nos. 2WRI/2WRJ; Gao et al., 2009). The similarities between L16 and Rpl10 and EF-G and Efl1 suggest that we can use the bacterial proteins as proxies for the eukaryotic structures. (B) Ribbon diagram of the molecular linkage between the P-site loop of Rpl10 and Tif6, derived from A. (C) Comparison of the P-site tRNA interactions of L16 and Rpl10. (top) Bacterial L16, L27, and P-site tRNA (adapted from PDB accession nos. 2WRI/2WRJ; Gao et al., 2009). (bottom) Yeast Rpl10 (blue) and P-site tRNA (purple; PDB accession nos. 3IZC/3IZB/3IZE/3IZF; Armache et al., 2010). Residues of the P-site loop that were targeted for mutation (M102 through A106) are shown in black. Isolated mutations are listed. (D) Growth assay of the rpl10 P-site loop mutants. 10-fold serial dilutions of AJY1437 (rpl10Δ::KanMX) containing either WT (pAJ2522) or P-site loop mutants as the sole source of Rpl10 were spotted onto yeast extract peptone dextrose and grown for 2 d at 30°C. (E) The rpl10 P-site loop mutants can be separated in two classes based on polysome profiles. Extracts were prepared from AJY1437 containing WT (pAJ2522) or P-site loop mutants and sedimented through 7–47% sucrose gradients.
Mutations in the P-site loop of Rpl10 exhibit two distinct phenotypes
To further investigate the function of the P-site loop of Rpl10, we randomized individual codons 102–106 within the loop and screened for viable but slow-growing mutants. We identified 21 such mutants (Fig. 2 D). Polysome profiles were analyzed to determine the effect of these mutations on ribosome biogenesis or translation. Two distinct classes of mutants emerged from this study. Class I (biogenesis) mutants, including rpl10-M102N, M102G, M102H, S104D, S104F, S104Y, S104W, C105G, A106I, A106P, and A106H, displayed halfmers, i.e., mRNAs with a 40S subunit that has not yet joined with the LSU (Figs. 2 E and S1). rpl10-S104D was the strongest biogenesis mutant. Class II mutants, including rpl10-M102W, M102S, L103V, L103S, L103P, and A106R, lacked halfmer polysomes and exhibited higher free 60S than 40S peaks (Figs. 2 E and S1). rpl10-A106R had the strongest phenotype in this class.
We monitored the localization of Nmd3 and Tif6 in strains containing the P-site loop mutants as the sole copy of RPL10. The fifteen mutants tested all trapped Tif6 and Nmd3 in the cytoplasm to various degrees (Figs. 3 A and S2). In addition, Tif6 as well as Nmd3 were retained on 60S subunits in rpl10-S104D (Fig. 3 B). To confirm these microscopy results that suggest rpl10-S104D arrests the 60S maturation pathway at the point of Tif6 release, we monitored the association of upstream trans-acting factors in WT and the rpl10-S104D mutant. Pre-60S particles were immunoprecipitated from extracts with either Tif6 or Nmd3 as bait, and the presence of Rlp24 and Arx1 was monitored by Western blotting. We have previously shown that both Rlp24 and Arx1 are released before the release of Tif6 (Lo et al., 2010). We observed a depletion of both Rlp24 and Arx1 from the immunoprecipitated particles, with a stronger depletion of Rlp24 from the Tif6-containing particles than from the Nmd3-containing particles (Fig. 3 C). This is consistent with the residence time of Tif6 on pre-60S overlapping that of Rlp24 to a greater extent than does Nmd3, which joins the pre-60S particle later than Tif6. The loss of Rlp24 and Arx1 from particles retaining Tif6 and Nmd3 supports the conclusion that rpl10-S104D arrests 60S maturation immediately before the release of Tif6. Although the class I mutants appear to affect subunit biogenesis specifically (see following paragraph), further work is required to understand the molecular defect in the class II mutants, which may affect translation as well as biogenesis.
Figure 3.

The Rpl10 P-site loop mutants trap Tif6 and Nmd3 on cytoplasmic 60S subunits. (A) Tif6-GFP and Nmd3-GFP localization were monitored in WT and rpl10 mutants. AJY2765 (TIF6-GFP rpl10Δ::KanMX) and AJY1837 (NMD3-GFP CRM1-T539C rpl10Δ::KanMX) containing WT (pAJ2522) or the indicated rpl10 P-site loop mutants were grown to mid-log phase, fixed with formaldehyde, DAPI stained, and visualized by microscopy. For Nmd3-GFP localization, cells were incubated with 0.4 µg/ml LMB for 5 min before fixing. DIC, differential interference contrast. Bar, 5 µm. (B) Sucrose gradient sedimentation of Tif6-GFP and Nmd3. AJY2765 (TIF6-GFP rpl10Δ::KanMX) containing WT (pAJ2522) or rpl10-S104D was cultured as described in Fig. 1 A. Cell extracts were prepared and fractionated by sucrose gradient sedimentation. The position of Tif6 and Nmd3 in gradients was monitored by Western blotting using anti-GFP and anti-Nmd3 antibodies, respectively. Anti-Rpl8 was used to monitor the position of 60S subunits. Protein molecular masses and size standards are given as described in the legend to Fig. 1 B. (C) Rpl10-S104D traps Nmd3 and Tif6 on the late-maturing 60S, resulting in a decrease in earlier factors present on Tif6 and Nmd3-bound subunits. Tif6 and Nmd3 were immunoprecipitated (IP) from the following extracts prepared from WT and mutant rpl10 strains: AJY1437 (rpl10Δ::KanMX) containing either WT RPL10 (pAJ2522) or rpl10-S104D and either Nmd3-13myc (pAJ1002) or Tif6-13myc (pAJ1009). The levels of coimmunoprecipitating Rpl8 (as a reporter for 60S), Rpl10, and trans-acting factors Arx1 and Rlp24 were determined by quantitative Western blotting using an infrared imaging system (Odyssey). The levels of Arx1, Rlp24, and Rpl10, relative to Rpl8, were normalized to that of the equivalent WT Rpl10 control. The arrow indicates Arx1; the upper band is nonspecific. Protein molecular masses and size standards are indicated as in B.
Mutations in TIF6 suppress the biogenesis class of rpl10 P-site loop mutants
As shown in the previous section, rpl10 loop mutants are defective for the release of Tif6 and its recycling to the nucleus. EFL1, encoding an eEF2-like GTPase, is also required for efficient release of Tif6 from the subunit (Bécam et al., 2001; Senger et al., 2001). Dominant mutations in Tif6 that weaken its affinity for the ribosome suppress the severe growth defect of efl1 mutants (Bécam et al., 2001; Senger et al., 2001; Menne et al., 2007). We asked whether one such mutant, TIF6-V192F, could suppress the effects of the rpl10 P-site loop mutations. Indeed, TIF6-V192F suppressed the growth defect of the biogenesis mutants (rpl10-S104D and C105G) but not class II mutants (rpl10-A106R and L103S; Fig. 4 A). This improved growth was reflected in improved polysome profiles of the rpl10 biogenesis mutants, indicated by the loss of halfmers and increased polysome levels (Fig. 4 B). Furthermore, Tif6-V192F recycled to the nucleus in the rpl10 biogenesis mutants rpl10-S104D and C105G, which it suppressed, but not in the class II mutants rpl10-A106R and L103S (Fig. 4 C).
Figure 4.

TIF6-V192F suppresses class I rpl10 P-site loop mutants. (A) The rpl10 deletion strain (AJY1437) containing WT (pAJ2522) or mutant RPL10 and either vector (pRS413), WT TIF6 (pAJ2543), or TIF6-V192F (pAJ2544) was grown in selective media, and 10-fold serial dilutions were spotted onto plates and incubated for 2 d at 30°C. (B) Extracts were prepared from the rpl10 deletion strain (AJY1437) containing rpl10 P-site loop mutants and either vector (pRS413) or TIF6-V192F (pAJ2544) and sedimented through 7–47% sucrose density gradients. A260 was monitored along the gradient. (C) The rpl10 deletion strain (AJY1437) containing WT (pAJ2522) or mutant RPL10 and vector harboring either WT TIF6-GFP (pAJ1004) or TIF6-V192F-GFP (pAJ2654) was grown in selective media. Cells were fixed with formaldehyde and stained with DAPI before visualization. DIC, differential interference contrast. Bar, 5 µm.
Mutations in the P-site loop of Rpl10 prevent the release of both Tif6 and Nmd3. Because the release of Tif6 is required for the subsequent release of Nmd3 (Lo et al., 2010), P-site loop mutations may only indirectly impinge on the release of Nmd3. To determine whether this is the case, we monitored the localization of Nmd3 in rpl10 P-site loop mutants containing vector, WT TIF6, or the suppressing allele TIF6-V192F. These strains also contained the LMB-sensitive crm1-T539C mutation. In the biogenesis mutant rpl10-S104D, Nmd3 recycled to the nucleus only in the presence of TIF6-V192F (Fig. S3 A). Because the recycling of Nmd3 was restored by mutant Tif6, the effect of rpl10-S104D on Nmd3 recycling is likely an indirect consequence of blocking Tif6 release. TIF6-V192F did not suppress the mislocalization of Nmd3 in rpl10-A106R (Fig. S3 A), giving further support to the idea that this mutant is defective for a pathway distinct from Tif6 release. These results demonstrate that the integrity of the P-site loop of Rpl10 is critical for the release of Tif6.
Thus far, our results suggest that Rpl10 affects Nmd3 indirectly through the release of Tif6. To test the possibility that Rpl10 also directly affects Nmd3 release, we looked for allele specificity between RPL10, TIF6, and NMD3. The temperature-sensitive rpl10-G161D mutant is suppressed by NMD3-I112T,I362T (Hedges et al., 2005). Thus, we introduced WT copies of TIF6 or NMD3 or the suppressing alleles TIF6-V192F or NMD3-I112T,I362T into rpl10-G161D, rpl10-S104D, and rpl10-A106R strains. We observed striking allele specificity: rpl10-G161D was suppressed only by NMD3-I112T,I362T, whereas rpl10-S104D was suppressed only by TIF6-V192F. rpl10-A106R was not suppressed by either TIF6 or NMD3 alleles (Fig. S3 B). Furthermore, rpl10-G161D trapped Nmd3 in the cytoplasm but not Tif6 (Fig. S3). These results show that the P-site loop of Rpl10 specifically affects the release of Tif6, thereby blocking the release of Nmd3, whereas other features of Rpl10 affect the release of Nmd3 independently from Tif6.
Mutations in EFL1 suppress rpl10 P-site loop mutants
The functional interaction between Rpl10 and Tif6 suggests molecular signaling from the P-site to Tif6 during maturation of the 60S subunit. The GTPase Efl1 is closely related to the translocation factor eEF2 and is required for the release of Tif6 (Bécam et al., 2001). We reasoned that if Efl1 were involved in this signaling, then we should be able to find dominant mutations in Efl1 that would be constitutively activated for the release of Tif6, regardless of the status of Rpl10. We randomly mutagenized EFL1 and screened for mutants that suppressed the growth defect of the biogenesis mutant rpl10-S104D, identifying 14 such efl1 mutants (Figs. 5 A [left] and S4). These mutants also suppressed rpl10-C105G but not rpl10-A106R or rpl10-L103S (Fig. 5 A [right] and not depicted), showing a specificity of suppression similar to TIF6-V192F. Suppression was also observed in polysome profiles; the halfmer phenotype of rpl10-S104D was alleviated, and the level of polysomes was increased (see overlay in Fig. 5 B). These EFL1 alleles did not improve the polysome profile of rpl10-A106R (see overlay in Fig. 5 B). The level of suppression of the growth defect of rpl10-S104D by the mutant EFL1-F250S,A669G was comparable with that by TIF6-V192F and restored growth to near WT levels (Fig. 5 C). All of the suppressing efl1 mutants were functional, as they complemented an efl1-null mutant (unpublished data). In addition, mutations in the Walker A motif, required for GTP binding, abrogated the suppressing activity of these mutants (unpublished data), indicating that GTPase activity was required for suppression.
Figure 5.

Mutations in EFL1 suppress rpl10-S104D. (A) AJY1437 (rpl10Δ::KanMX) with rpl10-S104D and either vector (pRS416), WT (pAJ2543), or suppressing alleles of TIF6 (pAJ2544) or WT (pAJ2545) or suppressing allele of EFL1 was grown in selective media. Serial dilutions were spotted on plates and grown for 2 d at 30°C. (B) AJY1437 (rpl10Δ::KanMX) with rpl10-S104D or rpl10-A106R with vector (pRS413), WT (pAJ2545), or mutant EFL1 was grown in selective media to mid-log phase, incubated with 50 µg/ml cycloheximide for 10 min, and harvested on ice. Crude extracts were fractionated by sedimentation through 7–47% sucrose gradients. A260 was monitored along the gradient. On the far right, for comparison, profiles for rpl10-S104D (top) or rpl10-A106R (bottom) with WT EFL1 or EFL1-N193S were overlayed. (C) AJY1437 (rpl10Δ::KanMX) with RPL10 WT or rpl10-S104D and vector (pRS413) or suppressing alleles of EFL1 or TIF6 (pAJ2544) was grown in selective media. 10-fold serial dilutions were spotted on plates and grown for 2 d at 30°C. (D) AJY2770 (PGAL1-RPL10 TIF6-3xHA) containing rpl10-S104D and either empty vector (pRS413) or a suppressing allele of EFL1 (EFL1-F250S,A669G) was grown to mid-log phase in galactose, and expression of genomic RPL10 was repressed by addition of glucose for 4 h, revealing the rpl10-S104D phenotype. Localization of TIF6-3xHA was monitored by indirect immunofluorescence using an anti-HA antibody (see Materials and methods). DIC, differential interference contrast. Bar, 5 µm.
Next, we examined the localization of Tif6 in an rpl10-S104D mutant suppressed by mutant EFL1. Because the GFP tag on Tif6 masked the suppression by the EFL1 mutants (unpublished data), we integrated the smaller 3xHA tag into the TIF6 locus and monitored Tif6 localization via indirect immunofluorescence. In the absence of the suppressing EFL1 mutant, Tif6 was trapped in the cytoplasm (Fig. 5 D), whereas in the presence of a suppressing allele of EFL1, Tif6 was redistributed to the nucleus (Fig. 5 D), indicating that the EFL1 suppressors promote the release of Tif6 even in the presence of mutations in the P-site loop of Rpl10. These data argue for a role of the Rpl10 P-site loop in Efl1-mediated release of Tif6.
Modeling Efl1
To understand the effect of the suppressing mutations on the structure of Efl1, we built atomic models of Efl1 by homology to eEF2 in two different conformations, apo eEF2 and sordarin-bound eEF2 (Jørgensen et al., 2003). The antifungal translation inhibitor sordarin traps eEF2 in an extended conformation that is an intermediate during translocation (Jørgensen et al., 2003; Taylor et al., 2007). We will refer to the corresponding conformation of Efl1 as extended. Fig. 6 A displays the apo Efl1 model, with the residues represented by red sticks highlighting the positions of the suppressing mutations. The extended conformation is very different from the apo conformation (Fig. 6 B), as eEF2 (and bacterial EF-G) undergoes large conformational changes that drive translocation. In the orientation shown in Fig. 6 B, domains I, II, and G’ pivot down relative to domain III, and domains IV and V pivot up, around domain III (compare Fig. 6 B with Fig. 6 C). One key element in these conformational changes is the interface of domains III and V at the center of Efl1 (and eEF2). Indeed, the III:V domain interface is surrounded by domains I, II, and G’ from one side and by domain IV from the other (Fig. 6, A and B).
Figure 6.

Efl1 models in the apo and translocational conformations. (A) Efl1 model in the apo conformation. Residues in red highlight the positions of single-residue mutations that suppress rpl10-S104D. (B) A superimposition on domain III of Efl1 in two conformations, the apo (in cyan) and the translocational (in yellow) conformations. The orange arrows indicate the direction of movement of domains I, G’, IV, and V, relative to domain III, in the translocational conformation. The inserts unique to Efl1 are not displayed. (C) Zoom on the hydrophobic core of domains III and V of Efl1 in the apo conformation. (D) Zoom on the hydrophobic core of domains III and V of Efl1 in the translocational conformation. (C and D) Residues in red sticks are the inner residues of the hydrophobic core and designate the positions of the single-residue mutations. Residues in orange lines are additional hydrophobic residues surrounding the inner residues of the hydrophobic core.
One cluster of mutations in EFL1 that suppresses the P-site loop mutant rpl10-S104D (I678T in domain III and W973R, V1021, S1028G, E1029G, L1043S, and F1045S in domain V) mapped to the III:V interface. These mutations correspond closely in position to the sordarin binding site in eEF2 (Jørgensen et al., 2003). Similarly, fusidic acid traps bacterial EF-G on the ribosome in an extended conformation (Agrawal et al., 1998; Spiegel et al., 2007). Crystal structures reveal that fusidic acid binds in the interface of domains II and III (Gao et al., 2009). Another cluster of mutations in EFL1 (S411P, T657R, L668P, and A669G) mapped to domain II at the interface with domain III. An additional mutation in this region arose from a 36-bp DNA inversion, altering aa 664–675, which comprise the linker between domains II and III. Although these mutations map to the II–III interface, their position is offset from the site where fusidic acid binds in EF-G. Notably, all of these mutations map to dynamic domain interfaces that are involved in the conformational change that eEF2 and EF-G undergo during translocation. Two other mutations, N193S and F250S (originally isolated in combination with A669G), mapped to domain I of Efl1.
efl1 suppressor mutations disrupt domain interfaces that promote conformational changes in eEF2
According to our model of Efl1, the III:V domain interface is firmly sealed and forms an organized hydrophobic core in the apo conformation (Fig. 6 C) but becomes disrupted in the extended conformation (Fig. 6 D). Interestingly, 7 of the 14 suppressing mutations that we identified by random mutagenesis are located in the hydrophobic core of the III:V interface, and 5 of these affect residues constituting the heart of the hydrophobic core. The localization of this cluster of suppressing mutations to the hydrophobic core of the III:V interface suggests an important role for this particular structure in Efl1 function.
We propose that mutations in the III:V domain interface destabilize this hydrophobic core and result in the loss of the characteristic interaction between domains III and V in the apo conformation. On the structural level, mutations that disrupt the hydrophobic core of the III:V interface could result in displacing the equilibrium between conformations of Efl1 toward the extended conformation. If such a conformational change normally requires proper signaling from the P-site, these mutations may predispose Efl1 to undergo a conformational change that triggers the release of Tif6, regardless of the integrity of the P-site.
To investigate our hypothesis that the suppressing mutations in Efl1 destabilize the hydrophobic core and thus could promote a conformational change, we studied the effect of these mutations on the stability of the hydrophobic core by molecular dynamics (MD) simulations in explicit solvent. We first built a model of the hydrophobic core of Efl1 that comprised domains III and V and part of domain IV of WT Efl1 in the apo conformation. Then, we introduced the five individual mutations that affect the innermost residues of the hydrophobic core at the interface between domains III and V.
Simulation of the WT system
The stability of the hydrophobic core in the WT simulation, as well as in the mutant simulations, was assessed by monitoring the dynamics and the interactions of water molecules surrounding the hydrophobic core, in other words by monitoring the structure of the hydration sphere of the hydrophobic core of the domain III:V interface. The analysis of the structure of this hydration sphere in the WT simulation (Fig. 7 B) shows that the integrity of the hydrophobic core is maintained, as no water molecules penetrate into it. Furthermore, the visualization of the hydration sphere reveals the presence of a narrow tunnel of structured water molecules that are drawn into the hydrophobic core through interaction with the hydroxyl group of Tyr 976 (Fig. 7 B, right). These structured water molecules could play a role in the equilibrium of conformations in Efl1. The root-mean-square (RMS) of the WT structure indicates that the III:V interface is relatively stable, as the RMS vibrates around 3 Å during the simulation time (Fig. 7 A, red line). To highlight the destabilization of the III:V interface, RMS values were calculated only over domains III and V for all the simulations.
Figure 7.

MD simulations of the WT and mutant domain III:V interface. (A) RMSs of Efl1 hydrophobic core in the WT and mutant simulations. RMSs were calculated over domains III and V. Simulations were run for 10 ns. The time courses shown begin at 1 ns of simulation, after equilibration. (B, left) The hydration sphere surrounding the hydrophobic core during the simulation (for the WT simulation). (right) Zoom on the hydration tunnel passing around TYR976 residue in the hydrophobic core. (C) The hydration sphere surrounding the hydrophobic core during the simulation (for the I678T mutant simulation). The yellow dashed circle highlights the water molecules penetrating into the III:V domain interface in the hydrophobic core, leading to its partial opening. (D, left) The hydration sphere surrounding the hydrophobic core during the simulation (for the V1021A simulation). (right) Zoom on the region where water molecules penetrate the interface between the broken β sheet and the α helices in domain III. The hydration spheres were realized by superimposing a representative number of frames, extracted from the simulation trajectories (see Materials and methods for more details).
Simulations of the mutant systems
A similar analysis of all the mutant systems reveals the destabilization of the hydrophobic core, most significantly in the mutants I678T, V1021A, and L1043S. In the I678T mutant, one observes the penetration of water molecules between domains III and V (Fig. 7 C, dashed yellow circle) and the partial opening of the III:V domain interface. The unstable character of the hydrophobic core in the I678T mutant is further indicated by the RMS (Fig. 7 A, green line) as it oscillates between 3 and 5 Å. Similar instability of the III:V domain interface and penetration of water molecules are observed in the L1043S mutant (unpublished data), leading to the partial opening of the III:V domain interface. It is interesting to note that this mutant presents the highest and the most unstable RMS of all the simulations (Fig. 7 A, blue line) as it increases gradually from 3 to 6 Å during the trajectory.
V1021A also induces the penetration of water molecules into the III:V domain interface and its partial opening (Fig. 7 D). In this mutant, the destabilization of the hydrophobic core could be caused by the disruption of an intradomain III interface, part of the hydrophobic core, which can be observed by water molecules penetrating the space between the α helices and the β sheet in domain III (Fig. 7 D, right). The III:V domain interface RMS oscillates between 4 and 5 Å, indicating a lower stability compared with the WT simulation (Fig. 7 A, gray line).
F1045S and W973R mutants display an intermediate stability between that of WT and mutants I678T, V1021A, and L1043S. Here again, water molecules penetrate the III:V domain interface. The disruption is caused by the replacement of two big hydrophobic nonpolar residues by polar or charged ones, threonine and arginine, which bring additional water molecules to the hydrophobic core (unpublished data). The RMS of these two mutants, although low in mean, exhibits a less stable profile than the WT (Fig. 7 A, orange and purple lines).
None of the mutant simulations displays the same pattern of structured water molecules similar to the tunnel observed in the WT simulation, and, in general, their hydration patterns are significantly different from the WT. In summary, our MD simulations of the interface between domains III and V show that the substitution of certain hydrophobic residues in the heart of this hydrophobic interface, by alanine or by polar residues, disrupts the hydrophobic core, allowing water to partially penetrate and destabilize the III:V domain interface. As discussed earlier, the organization of the III:V domain interface in Efl1 is distinct in both the apo and the extended conformations. The disruption and the opening of the hydrophobic core may shift the equilibrium from the apo to the extended conformation. To test the suggestion from MD that the domain III:V interface is destabilized in the mutants, we assayed the sensitivity of WT and the F1045S mutant of Efl1 to limited proteolysis by trypsin. Indeed, the mutant showed an altered cleavage pattern (Fig. 8), indicating altered accessibility to cleavage sites and, hence, altered conformation. Thus, our MD study supports a model in which a shift in the equilibrium of Efl1 conformation toward the extended conformation promotes suppression of defects in the P-site loop of Rpl10. Although we did not carry out MD simulations for the mutations that cluster at the II–III domain interface, we imagine that they similarly disrupt that interface to promote the extended conformation.
Figure 8.
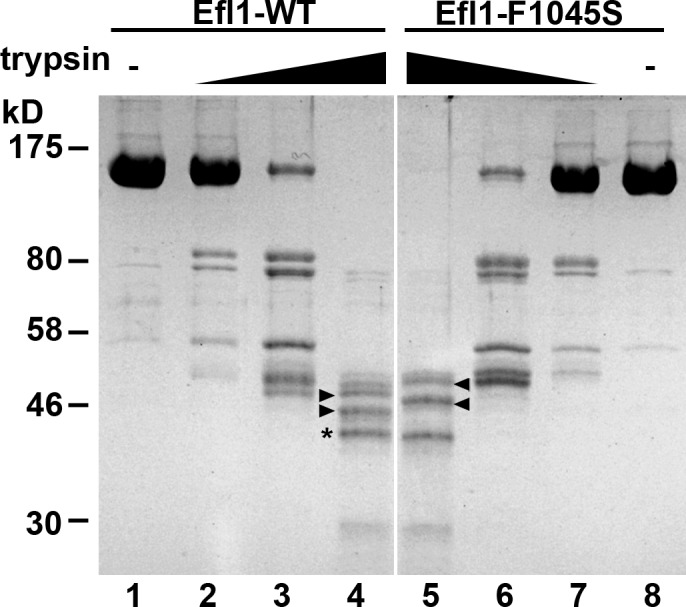
WT and mutant Efl1 show differential sensitivity to proteolysis. 0.5 µg of purified WT Efl1 or Efl1-F1045S was treated with increasing amounts of trypsin: no trypsin, lanes 1 and 8; 0.1 ng trypsin, lanes 2 and 7; 1 ng trypsin, lanes 3 and 6; 10 ng trypsin, lanes 4 and 5. Products were separated by SDS-PAGE and stained with Coomassie blue. Common fragments are indicated with an asterisk, and fragments that differ between WT and mutant are indicated by arrowheads. Positions of molecular mass markers are indicated to the left.
Discussion
Here, we provide compelling evidence that maturation of the large ribosomal subunit involves extensive probing of its functional center. We found that mutations in a loop of ribosomal protein Rpl10, 13 Å from the catalytic center, prevent the release of the trans-acting factor Tif6, 90 Å away. We propose that the integrity of the P-site is communicated to Tif6 through the translocase-like GTPase Efl1. Because Tif6 prevents subunit association, coupling its release to the assembly of the P-site and, indeed, to the entire GTPase-associated center via the GTPase Efl1 affords a mechanism to ensure that only properly assembled subunits enter the active pool of ribosomes.
The P-site loop of Rpl10
Recent high-resolution cryo-EM images of translating eukaryotic ribosomes reveal a loop of Rpl10, which we have termed the P-site loop, extending toward the acceptor stem of the P-site tRNA (Armache et al., 2010). We identified two classes of mutations within the P-site loop of Rpl10. One class of mutations, illustrated by rpl10-S104D, primarily impacts 60S biogenesis, as these mutants display halfmer polysomes and can be suppressed by mutations in the biogenesis factors Tif6 and Efl1. However, the second class of mutations, represented by rpl10-A106R, does not lead to halfmers and cannot be suppressed in a similar fashion. We considered that this class might act through eEF2 to impact translation, analogous to class I acting through Efl1. However, we were unable to identify mutations in eEF2 that would bypass rpl10-A106R. Considering the location of these mutations in the P-site loop, they could affect either peptidyl transferase activity or termination. Additional work will be required to understand the molecular defect of the class II mutants.
Efl1 is a eukaryote-specific factor
Efl1 appears to have evolved from the translation elongation factor eEF2 as a specialized factor required for 60S subunit maturation. Efl1 works in conjunction with Sdo1 to release Tif6 (Bécam et al., 2001; Senger et al., 2001; Menne et al., 2007; Finch et al., 2011). All three of these factors are conserved throughout eukaryotes. However, archaea lack Efl1 but have Sdo1 and Tif6. Assuming that the mechanism of the release of Tif6 is fundamentally conserved, it is likely that archaeal EF-G acts in biogenesis to release Tif6 in addition to its canonical role as an elongation factor. Bacteria have neither Sdo1 nor Tif6. Recently, Finch et al. (2011) showed that Sdo1 couples the GTPase activity of Efl1 to the release of Tif6 from the ribosome.
What distinguishes Efl1 from eEF2? In yeast, the two proteins share 40% sequence identity, which extends throughout the entire protein. However, Efl1 contains several insertions that are not present in eEF2. The most conspicuous difference, an insertion of 160 residues in domain II, can be deleted without any significant impairment in protein function (unpublished data). Presumably, differences in the structures of the two proteins allow Efl1 but not eEF2 to be recruited to pre-60S subunits. However, it remains possible that the two proteins retain some overlap in function. Efl1 is not essential, leaving open the possibility that eEF2 can function in its place, albeit inefficiently. Such functional overlap has been recently reported for the divergent release factors Dom34 and Hbs1, which act primarily on stalled ribosomes but have retained the ability to act on terminating ribosomes, the substrate primarily of eRF1 and eRF3 (Shoemaker et al., 2010).
Test drive of the LSU by Efl1
Translocation of tRNAs by eEF2 (or EF-G) requires a conformational change in the factor that is driven by its GTPase activity. The altered structure extends domain IV of eEF2 or EF-G into the decoding center of the SSU, where it repositions the anticodon stem loop of the peptidyl tRNA. We propose that a similar conformational change in Efl1 drives maturation of the 60S subunit to release Tif6. Supporting this proposed similarity in protein dynamics of translocases and Efl1, deletion of domain IV or mutations in the Walker A motif of Efl1 led to loss of Efl1 function in vivo (unpublished data). Furthermore, as described in this work, we identified mutations in Efl1 that bypassed the biogenesis defect caused by mutations in the P-site loop of Rpl10. These mutations in Efl1 map to dynamic domain interfaces that control the conformational changes of the protein. Indeed, MD simulations of the factor with these mutations on the interface of domains III and V predict that the suppressing mutations at this interface all disrupt the hydrophobic core of the interface. In turn, this perturbation would induce a conformational change similar to the one observed in eEF2 and EF-G during translocation.
Considering Efl1 in the larger context of the 60S biogenesis pathway, we suggest that Efl1 assesses multiple aspects of ribosome assembly in addition to the integrity of the P-site. We recently showed that Efl1 functions immediately after assembly of the stalk, a structure required to recruit and activate GTPases on the ribosome. Thus, recruitment of Efl1 is a marker of stalk assembly. Furthermore, activation of the catalytic activity of the GTPases of translation requires their proper recognition of the SRL, whose presence repositions a critical histidine into the catalytic site of the GTPase (Voorhees et al., 2010). Hence, activation of Efl1 function also is likely to assess the correct folding of the SRL.
Our results suggest that during maturation in the cytoplasm, the LSU undergoes a quasifunctional check for conformational responses that depend on the correct assembly and dynamic properties of the subunit. We propose that the function of Efl1 represents a test drive of the nascent subunit that serves as a quality control check of ribosome assembly. The check involves interactions that, in the fully assembled ribosome, belong to the repertoire of eEF2 during mRNA–tRNA translocation but here are performed by a close homolog of the elongation factor Efl1. A prerequisite for Efl1 function is the assembly of the ribosome stalk, which, upon completion, would recruit Efl1 to the nascent subunit. Efl1 then interrogates the P-site, defined in part by the P-site loop of Rpl10. The recognition of a correctly assembled subunit, including proper folding of the SRL, would trigger a GTPase-dependent conformational change in Efl1 analogous to that of the translocases during translation. In turn, this change would promote the final steps of maturation of the 60S subunit through the release of the antiassociation factor Tif6. Thus, we propose that Efl1 links the release of Tif6 to the correct assembly of the functional center of the LSU.
What occupies the P-site during biogenesis?
Our data point to a molecular connection between Efl1 and the P-site loop of Rpl10. During translation, a tRNA bridges the space between these proteins in the P-site (Fig. 2, A and B). Does a tRNA also occupy the P-site during biogenesis? This would seem to be the ideal substrate for testing the function of the ribosome and could facilitate assembly of the P-site. Alternatively, a protein could be located in the P-site in lieu of a tRNA. The structure of human and archaeal Sdo1 has been solved (Savchenko et al., 2005; Shammas et al., 2005; de Oliveira et al., 2010) and has been likened to that of a tRNA (Ng et al., 2009) or bacterial ribosome recycling factor (Finch et al., 2011). Thus, Sdo1 could act as a tRNA mimic to test the P-site for tRNA binding and perhaps for its ability to support tRNA translocation. In this model, a trivial explanation for the effect of rpl10-S104D on the release of Tif6 is that this mutant is defective for Sdo1 binding. However, two results argue against this idea. First, we do not detect any loss of Sdo1 sediments with rpl10-S104D ribosomes compared with WT (unpublished data). Second, suppression of rpl10-S104D by mutant Efl1 requires the presence of Sdo1. These results implicate Sdo1 in the assessment of the P-site by Efl1.
Materials and methods
Strains, plasmids, and media
Cells were grown at 30°C in rich media (yeast extract and peptone) or appropriate synthetic drop-out medium with 2% glucose or 1% galactose as the carbon source. Strains, plasmids, and primers used in this study are listed in Tables 1, 2, and 3, respectively. AJY2766, 2767, and 2768 were made by amplifying genomic DNA from AJY2104 (Hofer et al., 2007) with oligonucleotides AJO645/646 and transforming the PCR product into Tif6-GFP, Arx1-GFP, and Mrt4-GFP strains (Huh et al., 2003) and selecting for G418R colonies. AJY2765 was made by integrating TIF6-GFP::HIS3 (Huh et al., 2003) into AJY1437 (rpl10Δ::KanMX), derived from sporulating the heterozygous diploid (Research Genetics) containing pAJ392. AJY2770 was made by integrating the PCR product from oligonucleotide F2CORE/R1CORE and template pFA6a-HA-K1URA3 (Sung et al., 2008) into AJY2766. pAJ2522 was constructed by amplifying BY4741 genomic DNA with AJO491/268. The product was digested with SalI and BamHI and ligated into the same sites of pAJ1197 (Hofer et al., 2007). pAJ2543 was constructed by amplifying BY4741 genomic DNA with AJO534/454, the product digested with EagI and SalI and ligated into pRS413. pAJ2544 was constructed by digesting pAJ2240 (A. Warren, University of Cambridge, Cambridge, England, UK) with SstI and XhoI, and the fragment was ligated into pRS413. pAJ2545 was constructed by amplifying BY4741 genomic DNA with AJO1352/1353, the product digested with Sst1 and XhoI and ligated into pRS413. pAJ2652 and pAJ2653 were constructed by digesting pAJ538 and pAJ1315, respectively (Hedges et al., 2005), with EagI and XhoI, and the fragments were ligated into pRS416. pAJ2654 was constructed by fusion PCR amplifying pAJ2240 with AJO1384/1369 and Tif6-GFP genomic DNA (Huh et al., 2003) with AJO1367/1368 (PCR2). PCR products were combined and reamplified with AJO1384/1367, digested with XhoI and MluI, and ligated into pAJ2240. pAJ2665 was constructed by digesting pAJ1004 with BstEII, and the fragment was ligated into pAJ2240.
Table 1.
Strains used in this study
| Strain no. | Genotype | Source |
| AJY1437 | MATα rpl10::KanMX lysΔ0 met15Δ0 his3Δ0 leu2Δ0 ura3Δ0 pAJ392 | This study |
| AJY1657 | MATa rpl10-G161D ura3 leu2 | Hedges et al., 2005 |
| AJY1837 | MATα rpl10Δ::KanMX NMD3-GFP::KanMX CRM1-T539C pDEQ2#5 | Hedges et al., 2005 |
| AJY2104 | MATα KanMX::GAL::RPL10 ade2 ade3 ura3 leu2 | Hofer et al., 2007 |
| AJY2765 | MATa rpl10Δ::KanMX TIF6-GFP::HIS3 LYS2 met15Δ0 his3Δ1 leu2Δ0 ura3Δ0 pAJ392 | This study |
| AJY2766 | MATα KanMX::GAL::RPL10 TIF6-GFP::HIS3 ade2 ade3 ura3 leu2 | This study |
| AJY2767 | MATa KanMX:: GAL::RPL10 ARX1-GFP::HIS3 his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | This study |
| AJY2768 | MATα KanMX:: GAL::RPL10 MRT4-GFP::HIS3 his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | This study |
| AJY2770 | MATα KanMX::GAL::RPL10 TIF6-6HA::URA3 ade2 ade3 ura3 leu2 | This study |
| W303 | MATα ade2-1 can1-100 his3-11, leu2-3,112 trp1-1 ura3-1 SSD1-d | This study |
Table 2.
Plasmids used in this study
| Plasmid | Description | Source |
| pAJ392 | RPL10 URA3 CEN | This study |
| pAJ538 | NMD3-13myc LEU2 CEN | Ho et al., 2000 |
| pAJ758 | nmd3-ILL-AAA-GFP URA3 CEN | Hedges et al., 2005 |
| pAJ1002 | Nmd3-13myc URA3 CEN | This study |
| pAJ1004 | TIF6-GFP URA3 CEN | This study |
| pAJ1009 | TIF6-GFP URA3 CEN | This study |
| pAJ1197 | RPL10-13myc Leu2 CEN | Hofer et al., 2007 |
| pAJ1315 | NMD3-I112T,I362T-13myc LEU2 CEN | Hedges et al., 2005 |
| pAJ1777 | rpl10-102-112Δ-13myc LEU2 CEN | Hofer et al., 2007 |
| pAJ2147 | PGPD-MBP-HIS6-TEV-EFL1 LEU2 2µ | This study |
| pAJ2240 | TIF6-V192F URA3 CEN | A. Warner |
| pAJ2522 | RPL10 LEU2 CEN | This study |
| pAJ2543 | TIF6 HIS3 CEN | This study |
| pAJ2544 | TIF6-V192F HIS3 CEN | This study |
| pAJ2545 | EFL1 HIS3 CEN | This study |
| pAJ2652 | NMD3 URA3 CEN | This study |
| pAJ2653 | NMD3-I112T,I362T URA3 CEN | This study |
| pAJ2654 | TIF6-V192F-GFP URA3 CEN | This study |
| pAJ2665 | TIF6 URA3 CEN | This study |
| pDEQ2#5 | PGal1::RPL10 URA3 CEN | Eisinger et al., 1997 |
Table 3.
Primers used in this study
| Oligonucleotide | Sequence |
| AJO268 | 5′-CGCGGATCCTACCCAACATGCTGAAC-3′ |
| AJO454 | 5′-GCTGTCGACTCTTTCGCATACAACTG-3′ |
| AJO491 | 5′-GTGCCATGGCTAGAAGACCAGCT-3′ |
| AJO534 | 5′-CTGCCCGGGCGGCCGTTTAAACCCATATTCCTTTG-3′ |
| AJO645 | 5′-CGTGAGCTCTTGTATCTCTTCACCGAA-3′ |
| AJO646 | 5′-CCGTGGATCCTAGCTTGAGCAGCAAAGTA-3′ |
| AJO932 | 5′-CCGTGGGAGCTCATTTGTCGGTGC-3′ |
| AJO933 | 5′-CGACAAATGAGCTCCCACGGTTAACG-3′ |
| AJO1320 | 5′-GTCTTACGTATCAACAAGNNNTTGTCTTGTGCCGGTGCGGATAGATTG-3′ |
| AJO1321 | 5′-GTCTTACGTATCAACAAGATGNNNTCTTGTGCCGGTGCGGATAGATTG-3′ |
| AJO1322 | 5′-GTCTTACGTATCAACAAGATGTTGNNNTGTGCCGGTGCGGATAGATTG-3′ |
| AJO1323 | 5′-GTCTTACGTATCAACAAGATGTTGTCTNNNGCCGGTGCGGATAGATTG-3′ |
| AJO1324 | 5′-GTCTTACGTATCAACAAGATGTTGTCTTGTNNNGGTGCGGATAGATTGCAAC-3′ |
| AJO1352 | 5′-CGCCCTCGAGAATGAAAGATAATGAACAGC-3′ |
| AJO1353 | 5′-CGCCGAGCTCGAAAGAATTTTAGTCAGCGC-3′ |
| AJO1367 | 5′-GCCTCTCGAGCTAGCATTCTGGGCCTCCATGTCGC-3′ |
| AJO1368 | 5′-CCGCTTGCAAGATGCCCAACC-3′ |
| AJO1369 | 5′-GGTTGGGCATCTTGCAAGCGG-3′ |
| AJO1384 | 5′-TGCTGGTACGCGTATCATCGG-3′ |
| F2CORE | 5′-TCGATGAATTCGAGCTCGTT-3′ |
| R1CORE | 5′-GGTCGACGGATCCCCGGGTT-3′ |
Mutagenesis of Rpl10
The P-site loop of Rpl10 was amplified with five different forward primers (AJO1320–1324), each containing a single randomized codon, and a common reverse primer (AJO268). The PCR products were cloned as SnaBI to BamHI fragments into pAJ1777, and the resulting pools of vectors were transformed into the RPL10 shuffle strain AJY1437. Slow-growing mutants were identified on 5-FOA–containing medium and sequenced.
Mutagenesis of Efl1
The ORF of EFL1 was randomly mutagenized by PCR using Taq DNA polymerase WT EFL1 (pAJ2545) as a template with oligonucleotides AJO1352 and 1353. The PCR product was cotransformed with gapped (StuI cut) pAJ2545 into AJY1437, in which WT RPL10 had been replaced with rpl10-S104D. Fast-growing colonies were selected, and EFL1-containing plasmids were extracted and sequenced.
Polysome profiles
All steps were performed at 4°C. Extracts of log-phase cells were prepared in 10 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 6 mM β-mercaptoethanol, 150 mM KCl, 50 µg/ml cycloheximide, 1 mM PMSF, 1 µM leupeptin, and 1 µM pepstatin by glass bead lysis. Extracts were clarified by centrifugation at 15,000 g for 10 min, and nine A260 units were layered onto 7–47% sucrose gradients in extraction buffer. After centrifugation, gradients were monitored continuously at 254 nm as fractions were collected. Samples were precipitated with TCA and analyzed by SDS-PAGE and Western blotting.
Immunoprecipitation
Immunoprecipitation was performed as previously described using anti–c-myc monoclonal antibody 9e10 (Covance). Proteins were separated by SDS-PAGE and transferred to nitrocellulose for Western blotting using α-Nmd3, α-Tif6 (provided by F. Fasiolo, Institute de Biologie Moléculaire et Cellulaire, Strasbourg, France), α-Arx1, α-Rlp24 (provided by M. Fromont-Racine, Institut Pasteur, Paris, France), α-Rpl10, or α-Rpl8p antibodies. The levels of coimmunoprecipitating 60S subunits reporter Rpl8, Rpl10, and trans-acting factors Arx1 and Rlp24 were determined by quantitative Western blotting using an infrared imaging system (Odyssey; LI-COR Biosciences). The signals for Arx1, Rlp24, and Rpl10 relative to the corresponding Rpl8 signals were normalized to those of the equivalent WT control.
Microscopy
For direct fluorescence, cells were grown to mid-log phase, fixed with formaldehyde, treated with DAPI, washed, and resuspended in PBS. As indicated, cells were treated with 0.4 µg/ml LMB before fixation. For indirect immunofluorescence, cells were grown and fixed as previously described. Cells were permeabilized in cold methanol followed by washing in acetone. Anti-HA antibody (HA.11; Covance) was diluted 3,000 fold in PBS plus 0.1% BSA. Cy3-conjugated donkey anti–mouse antibody (Jackson ImmunoResearch Laboratories, Inc.) was used at a 300-fold dilution. After antibody application, cells were incubated for 1 min in 1 µg/ml DAPI and mounted in Aqua-Poly/Mount (Polysciences, Inc.). Images were captured at ambient temperature using a microscope (E800; Nikon) fitted with a Plan-Apochromat 100×/1.4 objective and a camera (CoolSNAP ES; Photometrics) controlled by NIS-Elements AR 2.10 software (Nikon). Images were prepared using Photoshop CS5 (Adobe).
Protein purification and limited trypsin digestion
Efl1 WT and Efl1-F1045S were expressed as maltose-binding protein (MBP) fusion proteins in yeast using vector pAJ2147. MBP was linked to Efl1 with a HIS6 tag and a tobacco etch virus (TEV) protease site. All steps were performed at 0–4°C. Extracts were made by glass bead lysis in extraction buffer (50 mM NaPO4, pH 8, 500 mM NaCl, 1 mM EDTA, 5 mM β-mercaptoethanol, and 10% glycerol and containing leupeptin and pepstatin at 1 µM each and 1 mM PMSF). Extracts were clarified by centrifugation at 50,000 g for 20 min and loaded onto amylose resin. After washing in extraction buffer lacking EDTA, protein was eluted in the same buffer containing 20 mM maltose. Eluted protein was loaded onto Ni–nitrilotriacetic acid, washed, and eluted in the same buffer containing 250 mM imidazole. Eluted proteins were cleaved with TEV protease during dialysis in 20 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM β-mercaptoethanol, and 10% glycerol. After dialysis, the sample was passed over Ni–nitrilotriacetic acid to remove residual uncleaved fusion protein, cleaved MBP, and TEV protease. Cleavage with TEV protease leaves three additional aa on the amino terminus of purified Efl1. Trypsin digests were performed in dialysis buffer for 30 min at 16°C.
Efl1 models
Models of Saccharomyces cerevisiae Efl1 in both conformations, apo and translocational, were realized by homology to the S. cerevisiae eEF2 crystal structures in these same conformations (Protein Data Bank accession nos. 1N0V and 1N0U, respectively) based on a multiple alignments (see supplemental pdf) realized using T-Coffee (Notredame et al., 2000). SWISS-MODEL (Arnold et al., 2006) was used to build the homology models. Efl1 inserts were modeled by homology using Phyre (Kelley and Sternberg, 2009). When no homologs were found, some inserts were modeled completely or partly de novo also using SWISS-MODEL and SYMPRED for the secondary structure prediction (Simossis and Heringa, 2004). The models were fine tuned manually using PyMOL and SWISS-PdbViewer (Guex and Peitsch, 1997).
MD simulations
MD simulation systems were prepared using VMD (Humphrey et al., 1996). Each system is a reduced model of Efl1 in the apo conformation. For the WT simulation, the reduced model was hydrated by ∼13,000 TIP3P water molecules. The system was neutralized by potassium cations K+, and an excess of KCl was added to ∼0.2 M. All the simulated systems were prepared using CHARMM force field parameters (combined CHARMM all-hydrogen topology file for CHARMM22 proteins and CHARMM27 lipids; Taylor et al., 2007). The WT system was relaxed by 4,000 minimization steps in NAMD (Phillips et al., 2005). The mutated systems were prepared by making the appropriate aa substitutions in the minimized WT system and then reminimized after substitution. MD simulations were conducted using NAMD for a total simulation time of 10 ns per simulation. RMS calculations were realized using PyMOL after superimposition of a representative set of the simulation frames (1/25) on domains III and V. Figures were rendered using PyMOL (molecular graphics system; version 1.3; Schrödinger, LLC) and UCSF Chimera (Pettersen et al., 2004) and KaleidaGraph (Synergy Software).
Hydration sphere of hydrophobic core
This type of analysis is inspired by a previous work on nucleic acids (Auffinger and Hashem, 2007). Hydration was visualized as follows: a representative number of frames was superimposed on domain III and V; solvent ions were removed; only the closest 350 waters to the center of the hydrophobic core were retained; and a certain number of water molecules was removed to view the internal shape and size of the hydration spheres, as seen in Fig. 7. Operations were realized using the Ptraj program from the AmberTool 1.4 package (The Amber Molecular Dynamics Package) and PyMOL.
Online supplemental material
Fig. S1 shows that mutation of the P-site loop of Rpl10 yields two classes of mutants. Fig. S2 shows that the Rpl10 P-site loop mutants trap Tif6 and Nmd3 in the cytoplasm to various degrees. Fig. S3 shows that Rpl10 is independently involved in release of Tif6 and Nmd3 from the LSU. Fig. S4 shows that mutations in EFL1 suppress rpl10-S104D. A pdf file listing the multiple sequence alignment of Efl1 proteins with eEF2 and EF-G is provided online. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201112131/DC1.
Supplementary Material
Acknowledgments
We thank A. Warren for plasmids, M. Fromont-Racine for anti-Rlp24, and F. Fasiolo for anti-Tif6 antibodies.
This work was supported by National Institutes of Health grant GM53655 to A.W. Johnson and by Howard Hughes Medical Institute and National Institutes of Health grant R01 GM29169 to J. Frank.
Footnotes
Abbreviations used in this paper:
- EF-G
- elongation factor G
- LMB
- leptomycin B
- LSU
- large subunit
- MBP
- maltose-binding protein
- MD
- molecular dynamics
- RMS
- root-mean-square
- SRL
- sarcin-ricin loop
- SSU
- small subunit
- TEV
- tobacco etch virus
- WT
- wild type
References
- Agirrezabala X., Lei J., Brunelle J.L., Ortiz-Meoz R.F., Green R., Frank J. 2008. Visualization of the hybrid state of tRNA binding promoted by spontaneous ratcheting of the ribosome. Mol. Cell. 32:190–197 10.1016/j.molcel.2008.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R.K., Penczek P., Grassucci R.A., Frank J. 1998. Visualization of elongation factor G on the Escherichia coli 70S ribosome: The mechanism of translocation. Proc. Natl. Acad. Sci. USA. 95:6134–6138 10.1073/pnas.95.11.6134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R.K., Heagle A.B., Penczek P., Grassucci R.A., Frank J. 1999. EF-G-dependent GTP hydrolysis induces translocation accompanied by large conformational changes in the 70S ribosome. Nat. Struct. Biol. 6:643–647 10.1038/10695 [DOI] [PubMed] [Google Scholar]
- Armache J.P., Jarasch A., Anger A.M., Villa E., Becker T., Bhushan S., Jossinet F., Habeck M., Dindar G., Franckenberg S., et al. 2010. Localization of eukaryote-specific ribosomal proteins in a 5.5-Å cryo-EM map of the 80S eukaryotic ribosome. Proc. Natl. Acad. Sci. USA. 107:19754–19759 10.1073/pnas.1010005107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K., Bordoli L., Kopp J., Schwede T. 2006. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 22:195–201 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- Auffinger P., Hashem Y. 2007. SwS: A solvation web service for nucleic acids. Bioinformatics. 23:1035–1037 10.1093/bioinformatics/btm067 [DOI] [PubMed] [Google Scholar]
- Bécam A.M., Nasr F., Racki W.J., Zagulski M., Herbert C.J. 2001. Ria1p (Ynl163c), a protein similar to elongation factors 2, is involved in the biogenesis of the 60S subunit of the ribosome in Saccharomyces cerevisiae. Mol. Genet. Genomics. 266:454–462 10.1007/s004380100548 [DOI] [PubMed] [Google Scholar]
- Boocock G.R., Morrison J.A., Popovic M., Richards N., Ellis L., Durie P.R., Rommens J.M. 2003. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 33:97–101 10.1038/ng1062 [DOI] [PubMed] [Google Scholar]
- Czworkowski J., Wang J., Steitz T.A., Moore P.B. 1994. The crystal structure of elongation factor G complexed with GDP, at 2.7 A resolution. EMBO J. 13:3661–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira J.F., Sforça M.L., Blumenschein T.M., Goldfeder M.B., Guimarães B.G., Oliveira C.C., Zanchin N.I., Zeri A.C. 2010. Structure, dynamics, and RNA interaction analysis of the human SBDS protein. J. Mol. Biol. 396:1053–1069 10.1016/j.jmb.2009.12.039 [DOI] [PubMed] [Google Scholar]
- Eisinger D.P., Dick F.A., Trumpower B.L. 1997. Qsr1p, a 60S ribosomal subunit protein, is required for joining of 40S and 60S subunits. Mol. Cell. Biol. 17:5136–5145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch A.J., Hilcenko C., Basse N., Drynan L.F., Goyenechea B., Menne T.F., González Fernández A., Simpson P., D’Santos C.S., Arends M.J., et al. 2011. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 25:917–929 10.1101/gad.623011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.G., Selmer M., Dunham C.M., Weixlbaumer A., Kelley A.C., Ramakrishnan V. 2009. The structure of the ribosome with elongation factor G trapped in the posttranslocational state. Science. 326:694–699 10.1126/science.1179709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartmann M., Blau M., Armache J.P., Mielke T., Topf M., Beckmann R. 2010. Mechanism of eIF6-mediated inhibition of ribosomal subunit joining. J. Biol. Chem. 285:14848–14851 10.1074/jbc.C109.096057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N., Peitsch M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 18:2714–2723 10.1002/elps.1150181505 [DOI] [PubMed] [Google Scholar]
- Hedges J., West M., Johnson A.W. 2005. Release of the export adapter, Nmd3p, from the 60S ribosomal subunit requires Rpl10p and the cytoplasmic GTPase Lsg1p. EMBO J. 24:567–579 10.1038/sj.emboj.7600547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henras A.K., Soudet J., Gérus M., Lebaron S., Caizergues-Ferrer M., Mougin A., Henry Y. 2008. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell. Mol. Life Sci. 65:2334–2359 10.1007/s00018-008-8027-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J.H., Kallstrom G., Johnson A.W. 2000. Nmd3p is a Crm1p-dependent adapter protein for nuclear export of the large ribosomal subunit. J. Cell Biol. 151:1057–1066 10.1083/jcb.151.5.1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer A., Bussiere C., Johnson A.W. 2007. Mutational analysis of the ribosomal protein Rpl10 from yeast. J. Biol. Chem. 282:32630–32639 10.1074/jbc.M705057200 [DOI] [PubMed] [Google Scholar]
- Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K. 2003. Global analysis of protein localization in budding yeast. Nature. 425:686–691 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. 1996. VMD: Visual molecular dynamics. J. Mol. Graph. 14:33–38: 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- Jørgensen R., Ortiz P.A., Carr-Schmid A., Nissen P., Kinzy T.G., Andersen G.R. 2003. Two crystal structures demonstrate large conformational changes in the eukaryotic ribosomal translocase. Nat. Struct. Biol. 10:379–385 10.1038/nsb923 [DOI] [PubMed] [Google Scholar]
- Kelley L.A., Sternberg M.J. 2009. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc. 4:363–371 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- Kemmler S., Occhipinti L., Veisu M., Panse V.G. 2009. Yvh1 is required for a late maturation step in the 60S biogenesis pathway. J. Cell Biol. 186:863–880 10.1083/jcb.200904111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K.Y., Li Z., Wang F., Marcotte E.M., Johnson A.W. 2009. Ribosome stalk assembly requires the dual-specificity phosphatase Yvh1 for the exchange of Mrt4 with P0. J. Cell Biol. 186:849–862 10.1083/jcb.200904110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo K.Y., Li Z., Bussiere C., Bresson S., Marcotte E.M., Johnson A.W. 2010. Defining the pathway of cytoplasmic maturation of the 60S ribosomal subunit. Mol. Cell. 39:196–208 10.1016/j.molcel.2010.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz J.S., Georg R.C., Gomes C.H., Machado-Santelli G.M., Oliveira C.C. 2009. Sdo1p, the yeast orthologue of Shwachman-Bodian-Diamond syndrome protein, binds RNA and interacts with nuclear rRNA-processing factors. Yeast. 26:287–298 10.1002/yea.1668 [DOI] [PubMed] [Google Scholar]
- Menne T.F., Goyenechea B., Sánchez-Puig N., Wong C.C., Tonkin L.M., Ancliff P.J., Brost R.L., Costanzo M., Boone C., Warren A.J. 2007. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 39:486–495 10.1038/ng1994 [DOI] [PubMed] [Google Scholar]
- Mohr D., Wintermeyer W., Rodnina M.V. 2002. GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry. 41:12520–12528 10.1021/bi026301y [DOI] [PubMed] [Google Scholar]
- Ng C.L., Waterman D.G., Koonin E.V., Walters A.D., Chong J.P., Isupov M.N., Lebedev A.A., Bunka D.H., Stockley P.G., Ortiz-Lombardía M., Antson A.A. 2009. Conformational flexibility and molecular interactions of an archaeal homologue of the Shwachman-Bodian-Diamond syndrome protein. BMC Struct. Biol. 9:32 10.1186/1472-6807-9-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C., Higgins D.G., Heringa J. 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- Panse V.G., Johnson A.W. 2010. Maturation of eukaryotic ribosomes: Acquisition of functionality. Trends Biochem. Sci. 35:260–266 10.1016/j.tibs.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina M.V., Savelsbergh A., Katunin V.I., Wintermeyer W. 1997. Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature. 385:37–41 10.1038/385037a0 [DOI] [PubMed] [Google Scholar]
- Savchenko A., Krogan N., Cort J.R., Evdokimova E., Lew J.M., Yee A.A., Sánchez-Pulido L., Andrade M.A., Bochkarev A., Watson J.D., et al. 2005. The Shwachman-Bodian-Diamond syndrome protein family is involved in RNA metabolism. J. Biol. Chem. 280:19213–19220 10.1074/jbc.M414421200 [DOI] [PubMed] [Google Scholar]
- Schmeing T.M., Voorhees R.M., Kelley A.C., Gao Y.G., Murphy F.V., IV, Weir J.R., Ramakrishnan V. 2009. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science. 326:688–694 10.1126/science.1179700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger B., Lafontaine D.L., Graindorge J.S., Gadal O., Camasses A., Sanni A., Garnier J.M., Breitenbach M., Hurt E., Fasiolo F. 2001. The nucle(ol)ar Tif6p and Efl1p are required for a late cytoplasmic step of ribosome synthesis. Mol. Cell. 8:1363–1373 10.1016/S1097-2765(01)00403-8 [DOI] [PubMed] [Google Scholar]
- Shammas C., Menne T.F., Hilcenko C., Michell S.R., Goyenechea B., Boocock G.R., Durie P.R., Rommens J.M., Warren A.J. 2005. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman-Diamond Syndrome. J. Biol. Chem. 280:19221–19229 10.1074/jbc.M414656200 [DOI] [PubMed] [Google Scholar]
- Shoemaker C.J., Eyler D.E., Green R. 2010. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 330:369–372 10.1126/science.1192430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simossis V.A., Heringa J. 2004. The influence of gapped positions in multiple sequence alignments on secondary structure prediction methods. Comput. Biol. Chem. 28:351–366 10.1016/j.compbiolchem.2004.09.005 [DOI] [PubMed] [Google Scholar]
- Spiegel P.C., Ermolenko D.N., Noller H.F. 2007. Elongation factor G stabilizes the hybrid-state conformation of the 70S ribosome. RNA. 13:1473–1482 10.1261/rna.601507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung M.K., Ha C.W., Huh W.K. 2008. A vector system for efficient and economical switching of C-terminal epitope tags in Saccharomyces cerevisiae. Yeast. 25:301–311 10.1002/yea.1588 [DOI] [PubMed] [Google Scholar]
- Taylor D.J., Nilsson J., Merrill A.R., Andersen G.R., Nissen P., Frank J. 2007. Structures of modified eEF2 80S ribosome complexes reveal the role of GTP hydrolysis in translocation. EMBO J. 26:2421–2431 10.1038/sj.emboj.7601677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees R.M., Weixlbaumer A., Loakes D., Kelley A.C., Ramakrishnan V. 2009. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 16:528–533 10.1038/nsmb.1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhees R.M., Schmeing T.M., Kelley A.C., Ramakrishnan V. 2010. The mechanism for activation of GTP hydrolysis on the ribosome. Science. 330:835–838 10.1126/science.1194460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M., Hedges J.B., Chen A., Johnson A.W. 2005. Defining the order in which Nmd3p and Rpl10p load onto nascent 60S ribosomal subunits. Mol. Cell. Biol. 25:3802–3813 10.1128/MCB.25.9.3802-3813.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemp I., Kutay U. 2007. Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett. 581:2783–2793 10.1016/j.febslet.2007.05.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.