

Abstract
A direct, highly enantioselective alkylation of arylacetic acids via enediolates using a readily available chiral lithium amide as a stereodirecting reagent has been developed. This approach circumvents the traditional attachment and removal of chiral auxiliaries used currently for this type of transformation. The protocol is operationally simple, and the chiral reagent is readily recoverable.
The enantioselective alkylation of carboxylic acids at the α-position is a transformation of fundamental importance in chemical synthesis. The chiral-auxiliary-based methodology has remained dominant in this area, especially for the installation of unactivated primary and secondary alkyl groups.1 The extensively utilized oxazolidinone2 and pseudoephedrine3 auxiliaries have set the standards for generality, high stereocontrol, and practicality since their first introduction into enolate alkylation methodology.4,5 The auxiliary-based alkylation of carboxylic acids necessitates a three-stage process: (1) attachment of an appropriate auxiliary, (2) stereoselective alkylation, and (3) hydrolytic removal and recovery of the auxiliary. With the goal of developing an alternative single-step method, we initiated a study of direct enantioselective alkylation of carboxylic acids through the intermediacy of enediolates employing chiral lithium amides as noncovalent stereodirecting agents.6 Our preliminary findings with aryl- and heteroarylacetic acids indicate that high enantioselectivity is realized by this approach following an operationally simple protocol described herein.
As reactive intermediates, enediolates offer the advantages of superior nucleophilicity and geometrical symmetry, eliminating the issue of E/Z selectivity during enolization and confining the problem to facial selectivity in the enediolate alkylation step. The promise of this approach was first recognized more than two decades ago by the groups of Shioiri7 and subsequently Koga.8 Although moderate yields and enantioselectivities were reported, these seminal studies provided a conceptual basis for the use of chiral lithium amide aggregates for stereoselective alkylation of carboxylic acids. In our work, we discovered that application of readily available9dimeric chiral tetramine bases results in a drastic improvement in both yield and enantioselectivity, providing a straightforward protocol for a direct enantioselective alkylation of carboxylic acids even with less reactive secondary alkyl iodides.
A preliminary screening of C2-symmetric tetramines, a representative group of which is shown in Figure 1, revealed that tetramine 2 containing piperidine subunits and a three-carbon linker exerted the highest enantiodirecting power.10,11 A two-step, multikilogram-scale preparation of enantiopure 2 from styrene oxide was developed by chemists at Amgen, making it a readily available reagent.6
Figure 1.

Selected C2-symmetric amines evaluated for the enantioselective allylation of phenylacetic acid (enantioselectivity is shown).
The direct allylation of phenylacetic acid (shown in Scheme 1) was used for initial optimization studies, and some of the key findings are illustrated in Table 1. In all cases, a clean transformation was observed, providing mixtures of the allylation product and the starting material only. The chiral amine was recovered by a simple extraction with aqueous HCl in nearly quantitative yield. The stoichiometry of the chiral amine and n-butyllithium is among the reaction parameters essential for high enantio-selectivity.4,5 In control experiments with 4.0 equiv of n-butyl-lithium relative to phenylacetic acid, the enantioselectivity eroded substantially when less than 1.0 equiv of (R)-2 was employed (entries 1 and 2). Somewhat lower ee's were noted with 2 equiv of the chiral amine (entries 4 and 5). Reproducibly high enantioselectivity was achieved when only a slight excess of the amine (1.03–1.05 equiv) was used (entry 3). With the free amine instead of its bislithium amide as the additive (2.0 equiv of n-BuLi), racemic product is formed while high conversion is maintained (entry 6). During early experimentation, we found that the level of enantioselectivity was dependent on the quality of n-butyllithium, which we attributed to the presence of ionic lithium contaminants (LiOBu-n, LiOH, etc.) in aged reagent bottles. This conjecture was confirmed by the observation of drastically lower enantioselectivity when the allylation reaction was carried out in the presence of lithium n-butoxide or lithium bromide (entries 8 and 9). Among solents evaluated in this protocol (ethyl ether, toluene, 1,2-dimethoxyethane, 2-methyltetrahydrofuran), tetrahydrofuran (THF) proved to be the optimal solvent for high enantioselectivity and high conversion.
Scheme 1.

Outline of the Direct Enantioselective Alkylation of Arylacetic Acids
Table 1.
Optimization Studies for Enantioselective Allylation of Phenylacetic Acid with (R)-2
| entry | reaction conditionsa | ee (%) | conversion (%) |
|---|---|---|---|
| 1 | n-BuLi (4.0 equiv), (R)-2 (0.8 equiv) | 67 | 68 |
| 2 | n-BuLi (4.0 equiv), (R)-2 (0.9 equiv) | 67 | 88 |
| 3 | n-BuLi (4.0 equiv), (R)-2 (1.03 equiv) | 93 | 91 |
| 4 | n-BuLi (4.0 equiv), (R)-2 (1.10 equiv) | 80–95 | 85–95 |
| 5 | n-BuLi (4.0 equiv), (R)-2 (2.05 equiv) | 75 | 89 |
| 6 | n-BuLi (2.0 equiv), (R)-2 (1.05 equiv) | 0 | 88 |
| 7 | n-BuLi (4.0 equiv), (R)-2 (1.10 equiv)b | 57 | 77 |
| 8 |
n-BuLi (4.0 equiv), (R)-2 (1.10 equiv), n-BuOLi (1.0 equiv) |
52 | 48 |
| 9 |
n-BuLi (4.0 equiv), (R)-2 (1.10 equiv), LiBr (1.0 equiv) |
9 | 23 |
n-BuLi, PhCH2CO2H (0.70 mmol), and (R)-2 were combined at 0 °C in THF. After 15 min, allyl bromide was added at −78 °C over 10 min.
See the Supporting Information for additional experimental details.
Deprotonation with n-BuLi was carried out at −78 °C, not 0 °C.
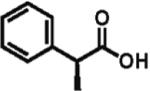
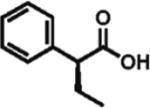
High enantioselectivity is maintained with a broad range of alkyl halides and in fact appears to improve with less reactive, sterically more hindered alkylating agents (Table 2). With more hindered reagents, the high nucleophilicity of enediolates offers a particular advantage. Methylation with iodomethane was complete within seconds and proceeded effectively as a titration, affording (S)-2-methylphenylacetic acid in 83% isolated yield and 88% ee (entry 1). Ethylation of phenylacetic acid was also rapid (10 min), giving the product in 81% yield and 96% ee (entry 2). Higher enantioselectivity was observed with more hindered isobutyl (98% ee), isopropyl (97% ee), and cyclopentyl iodides (96% ee), which required longer reaction times (5–24 h) (entries 5–7). Activated electrophiles such as benzyl bromide and methallyl bromide afforded alkylation products with 92 and 91% ee, respectively (entries 3 and 4). Functionalized alkylating agents are also suitable. For example, alkylation with 4-(tert-butyldimethylsiloxy)-1-iodobutane12 took place in 87% yield and 93% ee (entry 8), while alkylation with 3-bromomethyl-2-fluoropyridine13 occurred in 93% yield and 91% ee (entry 9). High lithium amide-controlled diastereoselectivity was realized with chiral alkyl iodides (entries 10–12). When (S)-1-iodo-2-methylbutane was used,14 highly diastereoselective alkylations (99% dr) were achieved with (R)-2 or (S)-2 (entries 10 and 11). No influence of the alkylating agent stereochemistry on diastereoselectivity was noted. Alkylation with a more functionalized (S)-2,2-dimethyl-4-iodoethyldioxolane15 provided the expected product in 74% yield and 91% dr, while the same alkylation with lithium diisopropylamide (LDA) afforded a ~1:1 mixture of dia-stereomers (entry 12). Importantly, alkylations with valuable functionalized and chiral alkyl halides were performed using a nearly stoichiometric amount of the reagent (1.0–1.2 equiv for entries 8–12). The unreacted halide could be recovered if necessary, as no side reactions such as elimination or amide N-alkylation were observed.
Table 2.
Scope of the Alkylating Agenta
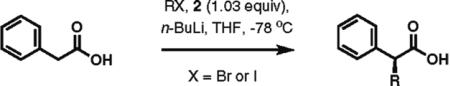
| 1 | 2 | 3 |
 88% ee 83% yield |
 96% ee 81% yield |
 91% ee 80% yield |
| 4 | 5 | 6 |
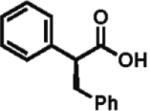 92% ee 85% yield |
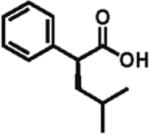 98% ee 82% yield |
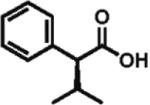 97% ee 85% yield |
| 7 | 8b | 9b |
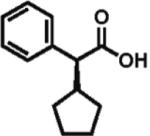 96% ee 76% yield |
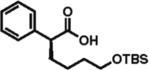 93% ee 87% yield |
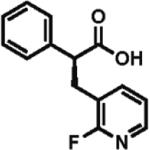 91% ee 93% yield |
| 10c,d | 11c,d | 12c,e |
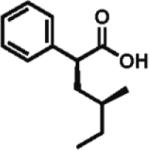 99% d.r. 67% yield |
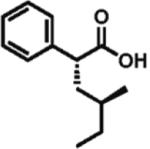 99% d.r. 79% yield |
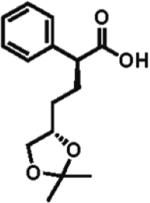 91% d.r. 74% yield |
Enantiomeric excess was determined by HPLC using a chiral stationary phase (see the Supporting Information for details). With the exception of entry 11, all reaction were performed with the R enantiomer of 2.
1.2 equiv of the alkylating agent was used.
1.0 equiv of the alkylating agent was used.
1.3:1 dr was obtained with LDA.
1:1 dr was obtained with LDA.
The scope of aryl- and heteroarylacetic acids was evaluated using enantioselective alkylation with iodoethane as a reference reaction (Table 3, entries 1–16). Uniformly, the reactions were characterized by a clean alkylation process, and in most cases the yields closely corresponded to the conversions, exceeding 90% based on recovered starting material. Substitution at the para position with fluorine, chlorine, or bromine had virtually no effect on the enantioselectivity, which maintained high values in the range 93–96% ee (entries 1–3).16 A slight decrease in enantioselectivity to 89% was observed during ethylation of 4-methoxyphenylacetic acid (entry 4). Methyl and methoxy groups at the ortho and meta positions had no significant effect on enantioselectivity and conversion (entries 5–8). Alkylations of disubsti tuted 3,4-dichlorophenylacetic acid occurred in 92% ee (entry 9), while those of 1- and 2-naphthylacetic acids gave products in 92 and 96% ee with notably higher conversion, which appeared to be general for this type of substrate (entries 10, 11, 21, and 24).
Table 3.
Enantioselective Alkylation: Carboxylic Acid Scopea
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
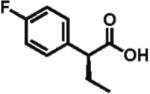 96% ee 84% yield |
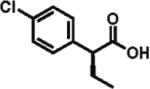 96% ee 81% yield |
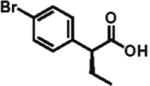 93% ee 65% yield |
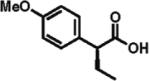 89% ee 71% yield |
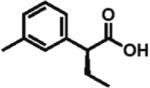 96% ee 83% yield |
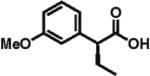 93% ee 82% yield |
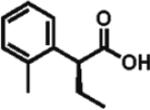 94% ee 82% yield |
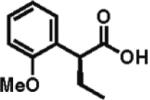 93% ee 87% yield |
| 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
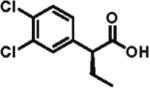 92% ee 82% yield |
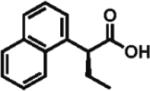 92% ee 89% yield |
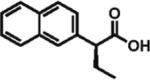 96% ee 88% yield |
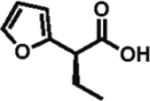 97% ee 60% yield |
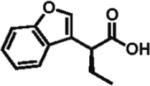 92% ee 62% yield |
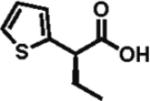 90% ee 85% yield |
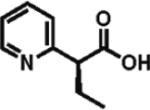 0% ee 12% yield |
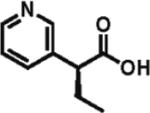 95% ee 61% yieldb |
| 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 |
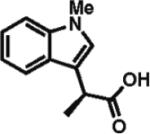 84% ee 71% yield |
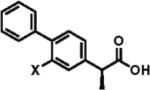 X=H, 91% ee, 87% X=F, 85% ee, 73% |
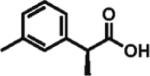 92% ee 87% yield |
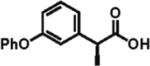 88% ee 87% yield |
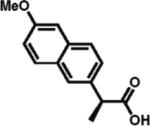 94% ee 96% yield |
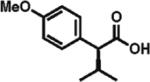 97% ee 78% yield |
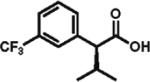 91% ee 70% yield |
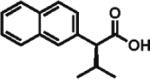 96% ee 89% yield |
Enantiomeric excess was determined by HPLC using a chiral stationary phase (see the Supporting Information for details).
Isolated yield of the corresponding alcohol over two steps after reduction of the acid with LiAlH4.
Heteroarylacetic acids also proved to be substrates, showing high enantioselectivities for a diverse range of substrates. 2-Furylacetic acid was ethylated in higher enantioselectivity (97%; entry 12), but the conversion was ~65%. A similar level of enantioselectivity was observed with 3-benzofuranacetic acid and 2-thiopheneacetic acid, and conversion was restored to typical values with the latter substrate (entries 13 and 14). Remarkably, R-ethylation of 3-pyridylacetic acid closely matched that of phenylacetic acids in terms of enantioselectivity (95% ee, 61% yield; entry 16), while 2-pyridylacetic acid was unreactive and displayed poor enantioselectivity (entry 15).17
To gain a broader overview of the reaction scope, the alkylating agent was varied to include iodomethane and 2-iodo-propane in the next set of experiments. Direct α-methylation of N-methyl-3-indoleacetic acid, which we anticipated to be among the more challenging processes, took place in good yield (71%) with a reasonably high enantioselectivity of 84% (entry 17). A number of known nonsteroidal anti-inflammatory drugs (NSAIDs) have been prepared by direct enantioselective α-methylation of the corresponding arylacetic acids. Naproxen, flurbiprofen, and fenoprofen were readily produced in 94, 85, and 88% ee and high isolated yields, respectively, (entries 18, 20, and 21). Methylation of 4-biphenylacetic acid proceeded in higher enantioselectivity relative to (2-fluorobiphen-4-yl)acetic acid (entry 18). The exquisitely high enantioselectivity and yield in the methylation of (6-methoxynaphthalen-2-yl)acetic acid to give naproxen was characteristic of naphthaleneacetic acids. With less reactive 2-iodopropane, high enantioselectivity was maintained for 4-methoxyphenyl-, 3-(trifluoromethyl)phenyl-, and 3-naphthaleneacetic acids (97, 91, and 96% ee, respectively; entries 22–24).
A large-scale application of the protocol was illustrated by direct alkylation of 10 g of phenylacetic acid with 2-iodopropane (Scheme 2). High enantioselectivity and yield were maintained on this scale, and the chiral amine was recovered in pure form in 99% yield by a simple extraction with aqueous hydrochloric acid. In fact, both the enantioselectivity and yield improved upon scale-up.
Scheme 2.

Large-Scale Direct Enantioselective Alkylation Experiment
The direct enantioselective alkylation was applied to the synthesis of γ-secretase modulator 1, which has been investigated in preclinical studies for its potential in treatment of Alzheimer's disease (Scheme 3).18 Consecutive bromination of ethyl (3-chloro-4-hydroxyphenyl)acetate and O-alkylation of the resulting substituted phenol with (bromomethyl)cyclopropane afforded 2, which was advanced further by a Suzuki–Miyaura cross-coupling with 4-(trifluoromethyl)phenylboronic acid followed by hydrolysis to give carboxylic acid 3. Direct enantioselective alkylation of 3 with isobutyl iodide using the protocol developed in our studies delivered 1 in 97% ee and 72% isolated yield.
Scheme 3.

Asymmetric Synthesis of γ-Secretase Inhibitor 1 by Enantioselective Alkylation
In closing, an operationally simple protocol for direct enantioselective alkylation of aryl- and heteroarylacetic acids has been developed. This protocol offers a one-step alternative to traditional auxiliary-based methods and utilizes a readily accessible chiral lithium amine that can be recovered with high efficiency by a simple extraction with aqueous acid. Applications on scale and with functionalized substrates and alkylating agents have been demonstrated. Efficient alkylations with unreactive alkyl halides are enabled by the high nucleophilicity of enediolates. Further studies to probe the origin of the stereoselectivity19 and expand the scope of the alkylation process to other types of carboxylic acids are underway.20
Supplementary Material
ACKNOWLEDGMENT
We thank Amgen for the donation of more than 100 g of amine (R)-2 that enabled the research described in this article, and the assistance of Dr. Seb Caille (Amgen, Thousand Oaks, CA) is especially appreciated. Financial support was provided by the National Institutes of Health National Institute of General Medical Sciences (R01 GM077379) and kind gifts from Amgen and Eli Lilly, with additional support from the Tobacco-Related Disease Research Program (predoctoral award to C.E.S.). We are grateful to Dr. Hongjun Zhou for continued assistance with NMR spectroscopy.
Footnotes
Supporting Information. Detailed experimental procedures, characterization data, copies of 1H and 13C NMR spectra, and HPLC traces for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Evans DA, Helmchen G, Rüping M. In: Asymmetric Synthesis—The Essentials. Christmann M, Bräse S, editors. Wiley-VCH; Weinheim, Germany: 2006. p. 3. [Google Scholar]; (b) Caine D. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 3. Pergamon Press; New York: 1991. pp. 1–63. [Google Scholar]
- (2).(a) Evans DA, Ennis MD, Mathre DJ. J. Am. Chem. Soc. 1982;104:1737–1739. [Google Scholar]; (b) Evans DA, Urpi F, Somers TC, Clark JS, Bilodeau MT. J. Am. Chem. Soc. 1990;112:8215–8216. [Google Scholar]
- (3).(a) Myers AG, Yang BH, Chen H, Gleason JL. J. Am. Chem. Soc. 1994;116:9361–9362. [Google Scholar]; (b) Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496–6511. [Google Scholar]
- (4).(a) Meyers AI, Knaus G, Kamata K, Ford ME. J. Am. Chem. Soc. 1976;98:567–576. [Google Scholar]; (b) Sonnet P, Heath RR. J. Org. Chem. 1980;45:3137–3139. [Google Scholar]; (c) Evans DA, Takacs JM. Tetrahedron Lett. 1980;21:4233–4266. [Google Scholar]; (d) Schmierer R, Grotemeier G, Helmchen G, Selim A. Angew. Chem., Int. Ed. Engl. 1981;20:207–208. [Google Scholar]; (e) Kawanami Y, Ito Y, Kitagawa T, Taniguchi Y, Katsuki T, Yamaguchi M. Tetrahedron Lett. 1984;25:857–860. [Google Scholar]; (f) Oppolzer W, Moretti R, Thomi S. Tetrahedron Lett. 1989;30:5603–5606. [Google Scholar]; (g) Abiko A, Moriya O, Filla SA, Masamune S. Angew. Chem., Int. Ed. Engl. 1995;34:793–795. [Google Scholar]; (h) Lin J, Chan WH, Lee AWM, Wong WY. Tetrahedron. 1999;55:13983–13998. [Google Scholar]
- (5).For selected recent applications in synthesis, see: Levin S, Nani RR, Reisman SE. J. Am. Chem. Soc. 2011;133:774–776. doi: 10.1021/ja110192b.. Nicolas L, Anderl T, Sasse F, Steinmetz H, Jansen R, Höfle G, Laschat S, Taylor RE. Angew. Chem., Int. Ed. 2011;50:938–941. doi: 10.1002/anie.201005530.. Hung K.-y., Harris PWR, Brimble MA. J. Org. Chem. 2010;75:8728–8731. doi: 10.1021/jo102038q.. Sun H, Abbott JR, Roush WR. Org. Lett. 2011;13:2734–2737. doi: 10.1021/ol200834p.. Fuwa H, Nakajima M, Shi J, Takeda Y, Saito T, Sasaki M. Org. Lett. 2011;13:1106–1109. doi: 10.1021/ol1031409..
- (6).For structure and reactivity of enediolates, see: Hauser CR, Chambers WJ. J. Am. Chem. Soc. 1956;78:4942–4944.. Creger PL. J. Am. Chem. Soc. 1967;89:2500–2501.. Pfeffer PE, Silbert LS, Chirinko JM. J. Org. Chem. 1972;37:451–458.. Brun EM, Gil S, Mestres R, Parra M. Tetrahedron. 1998;54:15305–15320.. Wang DZ, Kim Y-J, Streitwieser A. J. Am. Chem. Soc. 2000;122:10754–10760.. Streitwieser A, Husemann M, Kim Y-J. J. Org. Chem. 2003;68:7937–7942. doi: 10.1021/jo034377+.. For selected examples of chiral lithium amides as stereodirecting agents, see:Imai M, Hagihara A, Kawasaki H, Manabe K, Koga K. J. Am. Chem. Soc. 1994;116:8829–8830.. Shirai R, Tanaka M, Koga K. J. Am. Chem. Soc. 1986;108:543–545. doi: 10.1021/ja00263a051.. Simpkins NS. Pure Appl. Chem. 1996;68:691–694.. Qin Y.-c., Stivala CE, Zakarian A. Angew. Chem., Int. Ed. 2007;46:7466–7469. doi: 10.1002/anie.200702142.. Gu Z, Herrmann AT, Stivala CE, Zakarian A. Synlett. 2010:1717–1722.. Stivala CE, Zakarian A. Org. Lett. 2009;11:839–842. doi: 10.1021/ol8027797.. Stivala CE, Zakarian A. J. Am. Chem. Soc. 2008;130:3774–3776. doi: 10.1021/ja800435j..
- (7).Ando A, Shioiri T. Chem. Commun. 1987:656–658. [Google Scholar]
- (8).Matsuo J, Koga K. Chem. Pharm. Bull. 1997;45:2122–2124. [Google Scholar]
- (9).For a kilogram-scale preparation of (R)-2 from styrene oxide, see: Frizzle MJ, Caille S, Marshall TL, McRae K, Nadeau K, Guo G, Wu S, Martinelli MJ, Moniz GA. Org. Process Res. Dev. 2007;11:215–222..
- (10).We carried out a more comprehensive survey of the bases than that shown in Figure 1. Its detailed presentation is beyond the scope of this communication and will be described separately.
- (11).Yamashita Y, Odashima K, Koga K. Tetrahedron Lett. 1999;40:2803–2806. [Google Scholar]
- (12).Sommer S, Kuehn M, Waldmann H. Adv. Synth. Catal. 2008;350:1736–1750. [Google Scholar]
- (13).(a) Universite L, Attardo G, Tripathy S, Gagnon M. Methyl Sulfanyl Pyrimidines Useful as Antiinflammatories, Analgesics, and Antiepileptics. WIPO Pat. Appl. WO2010/132999 A1. 2010 [Google Scholar]; (b) Wagner S, Breyholz H-J, Law MP, Faust A, Höltke C, Schröer S, Haufe G, Levkau B, Schober O, Schäfers M, Kopka K. J. Med. Chem. 2007;50:5752–5764. doi: 10.1021/jm0708533. [DOI] [PubMed] [Google Scholar]
- (14).Martischonok V, Melikyan GG, Mineif A, Vostrovsky O, Bestmann H. J. Synthesis. 1991:560–564. [Google Scholar]
- (15).(a) Ilardi EA, Isaacman MJ, Qin Y.-c., Shelly SA, Zakarian A. Tetrahedron. 2009;65:3261–3269. [Google Scholar]; (b) Taber DF, Xu M, Hartnett JC. J. Am. Chem. Soc. 2002;124:13121–13126. doi: 10.1021/ja020816v. [DOI] [PubMed] [Google Scholar]
- (16).Partial decomposition of (4-bromophenyl)acetic acid was observed, presumably through a competing benzyne formation.
-
(17).The result with 2-pyridylacetic acid can be readily understood by considering the following resonance structures that reveal its enamide rather than enediolate reactivity:
 In addition, chelation between the carboxylate oxygen and pyridine nitrogen atoms is possible, which can disrupt the putative aggregate between the enediolate and the chiral lithium amide.
In addition, chelation between the carboxylate oxygen and pyridine nitrogen atoms is possible, which can disrupt the putative aggregate between the enediolate and the chiral lithium amide.
- (18).Shapiro G, Chesworth R. Tetrasubstituted Benzenes. WIPO Pat. Appl. WO2009/86277 A1. 2009 [Google Scholar]
- (19).We experimentally confirmed that the products are formed through enantioselective alkylation of the initially generated enediolate. Enolization of the product followed by a possible enantioselective protonation has been ruled out. See the Supporting Information (file Supporting Information 1, p S23) for details.
- (20).Preliminary studies revealed that under the standard conditions described herein, simple alkanoic acids (such as butyric acid) do not undergo highly enantioselective alkylation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.