

Abstract
In addition to polynucleotide polymerization, DNA polymerases and bacterial RNA polymerase can also remove nucleotides from the growing end of nucleic acid chains. For DNA polymerases this activity is an important factor in establishing fidelity in DNA synthesis. This report describes a novel in vitro activity of RNA polymerase II whereby it cleaves an RNA chain contained within an active elongation complex. These elongation complexes are arrested at a previously identified, naturally occurring transcriptional pause site in a human gene. The new 3′-end revealed by this cleavage remains associated with an active elongation complex and is capable of being extended by RNA polymerase II. Nascent RNA cleavage is evident after removal of free nucleotides and is dependent upon a divalent metal cation and transcription elongation factor SII. This function of SII could be important in its function as an activator of transcription elongation. It is also possible that the transcript cleavage activity of RNA polymerase II represents a proofreading function of the enzyme.
Transcription in prokaryotes and eukaryotes can be controlled at the level of transcript elongation (Spencer and Groudine, 1990; Kerppola and Kane, 1991). Thus, the ability of RNA polymerase to complete primary transcript synthesis can be regulated by cellular mechanisms. In eukaryotes the biochemical basis for this control is poorly understood, and the macromolecular composition of the target for this regulation, the RNA polymerase II elongation complex, has not been defined. Protein factors involved in transcription elongation have been identified (Sekimizu et al., 1976; Sawadogo et al., 1980; Price et al., 1987; Reinberg and Roeder, 1987; Rappaport et al., 1987; Sluder et al., 1989; Reines et al., 1989; Price et al., 1989; Flores et al., 1989; Bengal et al., 1990; Chafin et al., 1991), and intragenic sites which block transcript elongation by RNA polymerase II have been characterized (Maderious and Chen-Kiang, 1984; Bentley and Groudine, 1986, 1988; Kao et al., 1987; Skarnes et al., 1988; Rougvie and Lis, 1988, 1990; Resnekov et al., 1989; Resnekov and Aloni, 1989; Selby and Peterlin, 1990). One such factor is elongation factor SII (Sekimizu et al., 1976; Sawadogo et al., 1980; Reinberg and Roeder, 1987; Rappaport et al., 1987; Reines et al., 1989). This protein has been shown to bind to pure RNA polymerase II (Sawadogo et al., 1980; Horikoshi et al., 1984; Reinberg and Roeder, 1987; Rappaport et al., 1988). It enables the enzyme to read through discrete template signals that efficiently arrest transcription (Reinberg and Roeder, 1987; Rappaport et al., 1987; Reines et al., 1989; Sluder et al., 1989). Its mechanism of action is not completely understood, but its activity has been identified in a number of animal species and cell types.
DNA polymerases from eukaryotes and prokaryotes possess a 3′ to 5′ exonuclease activity that acts upon correctly and incorrectly base-paired DNA primers. Noncomplementary nucleotides are hydrolyzed from primers more efficiently than correct residues. Much evidence has established this as a proofreading activity which enhances the fidelity of DNA replication (Kornberg, 1980). Such a nuclease activity has not been identified in any DNA-dependent RNA polymerase.
By a process called pyrophosphorolysis both DNA and RNA polymerases can remove the 3′-most nucleotide from nucleic acid chains. Pyrophosphorolysis results in the removal of bases from RNA chains contained in active elongation complexes. It requires high levels of pyrophosphate and is a reversal of the polymerization reaction. Pyrophosphorolysis has proven useful in positioning template-engaged phage T4 RNA polymerase and Escherichia coli RNA polymerase at specific template locations (Kassevetis et al., 1986; Arndt and Chamberlin, 1990). It has been suggested that, in vivo, pyrophosphorolysis contributes to the fidelity of transcription by E. coli RNA polymerase (Kahn and Hearst, 1989).
Polymerase-associated nuclease activities have been reported for influenza virus RNA-dependent RNA polymerase and E. coli RNA polymerase. Influenza RNA polymerase can cleave RNA chains both endonucleolytically, to generate primers for viral transcription, and exonucleolytically, to remove mismatches from RNA primers (Ishihama et al., 1986). Recently, Chamberlin and co-workers (Surratt et al., 1991) identified a novel RNA-hydrolytic activity of E. coli RNA polymerase present in some ternary complexes. They showed that a small oligoribonucleotide (2, 3, 10, or 11 nucleotides long) could be excised from the 3′-terminus of a nascent transcript. It was suggested that endonucleolytic transcript cleavage in these particular complexes relieved phosphodiester bond strain resulting from specific contacts between RNA and RNA polymerase (Surratt et al., 1991). To date, no RNA-hydrolytic activity has been reported for eukaryotic DNA-dependent RNA polymerases. This report describes the identification and characterization of a new activity whereby RNA polymerase II in active elongation complexes efficiently cleaves nascent RNA chains.
MATERIALS AND METHODS
Enzymes and Proteins
RNA polymerase II was purified from rat liver as described (Conaway et al., 1987) except the cytosol fraction (800 × g supernatant) was used as starting material for phosphocellulose (P-11, Whatman), DEAE-cellulose (DE52, Whatman), and DEAE-Sephadex A-25 (Sigma) chromatography. Rat liver transcription initiation factors B′ (partially purified βγ), α, and D (partially purified δ ε, and τ) were prepared as described (Conaway et al., 1987, 1990; Conaway and Conaway, 1989) except the DE52 step was omitted from the B′ purification.
Anti-RNA IgG (D44; Eilat et al., 1982) was purified by protein A-Sepharose chromatography from ascitic fluid from D44 tumor-bearing mice.
SII was purified from calf thymus as described (Rappaport et al., 1987). Partially purified bovine brain SII was prepared by chromatography of a cytosolic extract on phosphocellulose as described (Reines, 1991). The protein eluting from phosphocellulose at 0.6 M KCl was dialyzed to reduce the KCl concentration to 0.1 M and was frozen at −80 °C.
Protein concentration was determined with protein assay dye reagent (Bio-Rad) according to the manufacturer’s directions using bovine serum albumin (Sigma, Fraction V) as a standard. Unlabeled, fast protein liquid chromatography-purified NTPs were purchased from Pharmacia LKB Biotechnology Inc.
DNA Template
The plasmid pAdTerm-2 contains a TaqI fragment from the human histone H3.3 gene which bears a transcription arrest site for RNA polymerase II (site Ia; Reines et al., 1989). The fragment was inserted into the unique AccI site of pDNAdML, which contains the adenovirus major late promoter (−50 to +10) in the KpnI and XbaI sites of pUC18. When cleaved with NdeI this plasmid yields a runoff transcript of 530 nucleotides and a transcript of 205 nucleotides when polymerase stops at site Ia.
In Vitro Synthesis of RNA Polymerase II Elongation Complexes
Elongation complexes were formed on NdeI-linearized pAdTerm-2 plasmid DNA as described (Reines, 1991). Transcription was carried out using partially purified RNA polymerase II and general transcription factors isolated from rat liver (Reines et al., 1989, modification of Conaway et al., 1987). Briefly, 100–200 ng of DNA template was incubated with rat liver RNA polymerase II (0.5 μg; DEAE-Sephadex A-25 fraction) and fraction D (2 μg; carboxymethyl-Sephadex A-25 fraction) in 20 μl of 20 mM HEPES,1 pH 7.9, 20 mM Tris-HCl, pH 7.9, 2% (w/v) polyvinyl alcohol, 0.4 mg/ml acetylated-bovine serum albumin (Promega), 12 units of RNAguard (Pharmacia), 0.15 M KCl, 2 mM dithiothreitol, and 7% (v/v) glycerol for 30 min at 28 °C. Thirty-three μl of a solution containing fraction B′ (1 μg; hydroxyl-apatite fraction) and α (3 ng; TSK phenyl 5-PW fraction) in the same buffer without KCl, was added, and incubation continued for another 20 min. MgCl2, ATP, UTP, and [α-32P]CTP (Amersham Corp.; > 400 Ci/mmol) were added in 6 μl to final concentrations of 7 mM, 20 μM, 20 μM, and approximately 0.6 μM, respectively. In the absence of GTP, ternary complexes containing a 14-nucleotide transcript were synthesized since the first G residue appears in the transcript at position +15. Heparin (10 μg/ml) was added to ternary complexes followed by an 800 μM concentration of all four NTPs, and incubation continued for 15 min at 28 °C. This results in the synthesis of runoff transcript, transcript Ia, and a 325-nucleotide transcript. The latter two RNAs result from arrest of RNA polymerase II at two sites which have been described previously (Reines et al., 1987, 1989). They remain associated with transcriptionally active RNA polymerase II in elongation complexes (Reines, 1991).
Immunoprecipitation and Washing of Elongation Complexes
Elongation complexes were immunoprecipitated with a monoclonal antibody (D44) against RNA as described (Reines, 1991). Elongation complexes, formed as described above, were made 10 μg/ml in protein A-purified D44 IgG and incubated at 4 °C for 5 min. Ten μl of formalin-fixed Staphylococcus aureus (Bethesda Research Laboratories/Life Technologies) washed in reaction buffer (20 mM Tris, 3 mM HEPES, pH 7.9, 62 mM KCl, 2.2% polyvinyl alcohol, 3% (v/v) glycerol, 2 mM dithiothreitol, 0.5 mM EDTA, and 0.3 mg/ml acetylated bovine serum albumin) were added to each reaction equivalent and incubated at 4 °C for 5 min. Complexes were collected by centrifugation at 16,000 × g for 2 min in a microcentrifuge. Complexes were washed in 1.2 volumes of reaction buffer by gentle resuspension in reaction buffer and centrifugation. The final resuspension was in 55 μl of reaction buffer. Three cycles of resuspension and centrifugation were sufficient to reduce the nucleotide concentration to submicromolar levels.2 Reactions were stopped with an SDS-containing buffer (0.2 M Tris-HCl, pH 7.5, 25 mM EDTA, 0.3 M NaCl, 2% (w/v) SDS), and RNA was isolated as described (Reines, 1991).
Electrophoresis and Autoradiography
RNA was dissolved in 80% (v/v) formamide, 0.025% (w/v) xylene cyanole, and 0.025% (w/v) bromphenol blue in TBE (89 mM Tris, 89 mM boric acid, pH 8.0, 1 mM EDTA) and denatured at 95 °C for 3 min. Samples were subjected to electrophoresis on 5% polyacrylamide gels in TBE. Gels were dried and exposed to X-Omat (Kodak) film at −80 °C with an intensifying screen. 32P-Labeled reference RNAs were synthesized from pKK34-121 (Thayer and Brosius, 1985) using E. coli RNA polymerase purifed as described (Gonzalez et al., 1977).
RESULTS
RNA polymerase II elongation complexes located at a well characterized site in a human histone gene were assembled on a linear DNA template (pAdTerm-2; Reines et al., 1989) from partially purified rat liver RNA polymerase II and general transcription initiation factors. The core adenovirus major late promoter (−50 to +10) was employed to drive transcription in these reactions. RNA was pulse labeled with [α-32P]CTP, and the complexes were immunoprecipitated using an anti-RNA monoclonal antibody (Reines, 1991). Three major RNAs were generated by transcription from this template: a runoff RNA of 530 nucleotides and RNAs resulting from the arrest of RNA polymerase II at histone site la (205 nucleotides) and a site further downstream in the plasmid (325-nucleotides) which has not been studied in detail (Reines et al., 1989).
Immunopurification of arrested elongation complexes efficiently separates them from unincorporated NTPs (Reines, 1991). It became evident that under some conditions the RNA chains in such complexes became shortened at their 3′-ends (asterisk, Fig. 1, lane 4). This activity required the presence of a protein fraction from bovine brain which contained elongation factor SII (Fig. 1, lanes 2 and 4). In the presence of this fraction and nucleotides, immunoprecipitated elongation complexes efficiently extended RNA chains to runoff length (Fig. 1, lane 3) as shown previously (Reines et al., 1989; Reines, 1991). A low but detectable level of transcript la persisted after either elongation or shortening (Fig. 1, lanes 3–5). These RNAs may represent transcripts released from the arrested complex. The shortening of a previously identified transcript of 325 nucleotides (Reines et al., 1989) was also apparent in these experiments. RNA polymerase II arrested at this location in the vector behaves identically to the enzyme at site Ia. Shortened RNA chains were associated with elongation complexes since, upon the addition of NTPs, these RNAs could be extended to runoff length (Fig. 1, lane 5). Cleavage must occur at the 3′-end of the nascent chain since the RNAs were labeled with [α-32P]CTP at positions 2, 4, 6, 9, 10, and 12 (transcription starts at +1). Hence, elongation complex-associated RNA chains undergo factor-dependent cleavage.
Fig. 1. Nascent RNA cleavage in the absence of nucleotides is factor dependent.

Elongation complexes arrested at a specific site (Ia) in a human histone gene were assembled on a linear DNA template from partially purified rat liver RNA polymerase II and general transcription factors and immunopurified as described under “Materials and Methods.” RNA was pulse labeled with [α-32P]CTP. Complexes were washed with buffer to remove solutes including unincorporated nucleotides and were made 7 mM in MgCl2 and 800 μM each in ATP, UTP, GTP, and CTP (lanes 1 and 3). Buffer (lanes 1 and 2) or 2 μg of partially purified SII from bovine brain (lanes 3–5) were added, and complexes were incubated at 28 °C for 30 min. Samples in lanes 1–4 were prepared for electrophoresis. The sample in lane 5 was treated as that in lane 4 except that it was chased with 800 μM each of ATP, UTP, GTP, and CTP for an additional 30 min at 28 °C before stoppage. RNA was analyzed by polyacrylamide gel electrophoresis and autoradiography. Three major RNAs were generated by transcription from pAdTerm-2 (Reines et al., 1989): a runoff RNA of 530 nucleotides (RO) and RNAs resulting from the arrest of RNA polymerase II at site Ia (205 nucleotides) and a site further downstream in the plasmid (325 nucleotides). The major cleavage product is indicated by an asterisk. Arrowheads indicate RNA size markers of 260, 380, 420, and 540 nucleotides synthesized from the plasmid pKK34-121 with E. coli RNA polymerase holoenzyme.
In the presence of pyrophosphate, E. coli RNA polymerase can extract as a NTP the 3′-terminal nucleotide of an RNA chain (Maitra and Hurwitz, 1967; Krakow and Fronk, 1969). Since pyrophosphate was not added to the reactions described here and immunopurified elongation complexes can be efficiently washed free of small molecules (Reines, 1991), cleavage by RNA polymerase II appeared hydrolytic and not pyrophosphorolytic. Furthermore, shortened transcripts remained elongation competent. Thus, the newly revealed 3′-ends must have contained a 3′-hydroxyl group although it was possible that the initial cleavage left a 3′-phosphoryl group which was subsequently removed by a distinct phosphatase activity.
To prove that RNA cleavage was mediated by elongation complex-associated RNA polymerase II and not free RNA polymerase II or a contaminating nuclease, the following experiments were performed. Arrested elongation complexes containing [32P]RNA were disrupted with SDS. RNA was isolated and added to washed, intact elongation complexes. The mixture was incubated under conditions that allow transcript shortening. Only the fraction of RNA chains corresponding to the amount of RNA in an intact elongation complex was shortened (Fig. 2, lanes 2 and 3). The remaining RNA in the mixture remained intact (Fig. 2, lanes 1 and 2). These results rule out the possibility that RNA cleavage was autocatalytic or carried out by a diffusible factor such as a contaminating ribonuclease. Furthermore, in the presence of α-amanitin at a concentration that specifically inhibits transcription by mammalian RNA polymerase II (1 μg/ml), washed elongation complexes could not shorten any transcripts (Fig. 2, lane 4). These data are most consistent with the model that elongation complex-associated RNA polymerase II can hydrolyze nascent RNA.
Fig. 2. RNA polymerase II must be in an elongation complex to cleave nascent transcripts.

Elongation complexes containing 32p-labeled runoff RNA (RO), transcript Ia, and the 325-nucleotide RNA were synthesized as described in the legend of Fig. 1. The RNAs were isolated and run on a polyacrylamide gel (lane 1) or mixed with intact, washed elongation complexes (lane 2). This mixture, and washed elongation complexes alone (lanes 3 and 4), were incubated with 7 mM MgCl2 and 2 μg of partially purified bovine brain SII at 28 °C for 15 min. One reaction (lane 4) also received α-amanitin (1 μg/ml) before incubation. RNA was isolated and analyzed by electrophoresis as described under “Materials and Methods.” Migration positions of RNAs are indicated as described in the legend of Fig. 1.
To prove that the factor required for RNA shortening was in fact elongation factor SII, a preparation of calf thymus SII purified to apparent homogeneity (Rappaport et al., 1988) was tested. This protein has been shown to activate transcription elongation through a regulated pause site in the adenovirus genome (Rappaport et al., 1988) and site Ia in the human histone H3.3 gene (Reines et al., 1989; SivaRaman et al., 1990). The addition of calf thymus SII to nucleotide-free elongation complexes was sufficient to stimulate transcript shortening (Fig. 3, lanes 2 and 5). This was also true for a highly purified preparation of rat liver SII and recombinant mouse SII synthesized in vitro.3 Also, the detergent sarkosyl, which inhibits SII-dependent, but not SII-independent, transcript elongation by RNA polymerase II (Reines et al., 1989), inhibited RNA cleavage (Fig. 4A, lane 1). This further supports the contention that SII was responsible for activating nascent transcript shortening by RNA polymerase II. These data also suggest that SII can interact with an elongation complex in the absence of NTPs.
Fig. 3. Pure calf thymus SII and a divalent metal activate RNA cleavage.
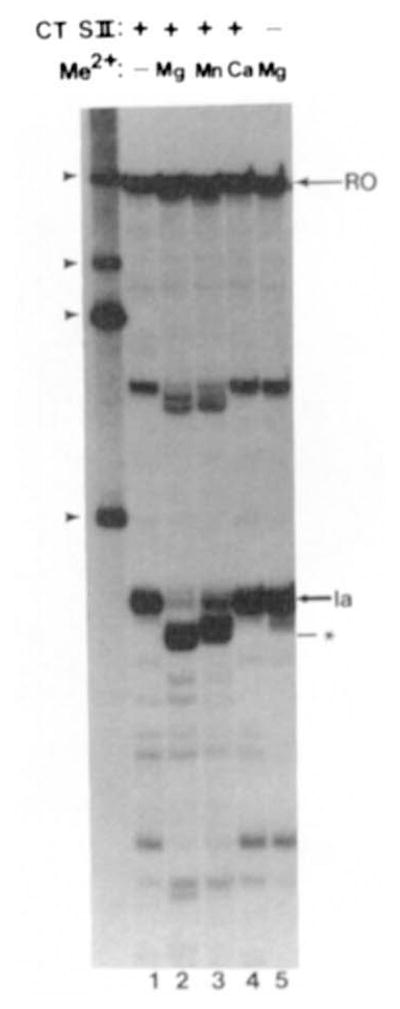
Washed elongation complexes were incubated with 36 ng of pure calf thymus SII (+) or buffer (−). Reactions were adjusted to 7 mM in MgCl2, MnCl2, or CaCl2 as indicated and incubated at 28 °C for 30 min. RNA was isolated and separated on a 5% polyacrylamide gel as described under “Materials and Methods.” Migration positions of RNAs are indicated as described for Fig. 1.
Fig. 4. Kinetics of SII-activated transcript cleavage by RNA polymerase II.

Washed elongation complexes were incubated with 2 μg of partially purified bovine brain SII and 7 mM MgCl2. Incubation proceeded at 28 °C for 0, 2, 5, or 60 min (panel A) or 0, 20, 50, 80, 110, and 300 s (panel B). One sample (panel A, lane 1) was made 0.25% (w/v) in sarkosyl before incubation at 28 °C for 30 min. RNA was isolated and analyzed by electrophoresis as described under “Materials and Methods.” Migration positions of RNAs are indicated as described for Fig. 1.
RNA chain elongation, pyrophosphorolysis, and an endonucleolytic RNA cleavage reaction identified for E. coli RNA polymerase in isolated ternary complexes all require magnesium (Krakow and Fronk, 1969; Surratt et al., 1991). RNA polymerase II-mediated transcript cleavage observed here also required a divalent metal ion (Fig. 3). The chloride salt of the metals, magnesium, manganese, and calcium, were tested for co-factor activity in RNA polymerase II-mediated transcript shortening. Magnesium chloride at 7 mM efficiently supported RNA cleavage (Fig. 3, lane 2). This concentration of manganese chloride resulted in a less efficient rate of cleavage (Fig. 3, lane 3, and see below). Transcript cleavage was undetectable with 7 mM calcium (Fig. 3, lane 4). Thus, in its requirement for a divalent metal ion, nascent RNA cleavage by RNA polymerase II resembled the RNA synthetic, hydrolytic, and pyrophosphorolytic reactions of E. coli RNA polymerase.
The kinetics of RNA cleavage were examined. Partially purified SII and MgCl2 were mixed with washed elongation complexes. RNA was isolated from the reaction after various lengths of time at 28 °C. By 2 min, all of transcript Ia was truncated (Fig. 4A, lane 3). The pattern of products generated suggested that there was a preferred site to which truncation was limited (Fig. 4, lanes 3–5). After 1 h, however, or in the presence of higher concentrations of SII, smaller transcripts were detected, demonstrating the potential for more extensive cleavage of RNA. Samples taken at very early time points showed that approximately half of transcript Ia was cleaved after 20 s (Fig. 4B). Complete cutting resulted after 50 s. Transcript Ia was cleaved into a discrete intermediate that had a half-life of approximately 5 min (Fig. 4). This intermediate appeared to be a precursor to the major cleavage product (asterisk, Fig. 4). The major cleavage product persisted for at least 1 h under these conditions (Fig. 4A, lane 5). This kinetic analysis suggested that a single RNA chain underwent a stepwise nucleolytic process which resulted in progressive RNA shortening. Cutting was, however, discontinuous, in that different rates of formation and disappearance of various intermediates were observed. Cleavage also followed a preferred order, advancing from the 3′-end to the 5′-end of the nascent RNA substrate.
The 3′ to 5′ exonuclease of E. coli DNA polymerase I excises the 3′-most nucleotide from a template-paired DNA primer. This activity is readily detected in the absence of deoxynucleoside triphosphates. Excision can, however, be inhibited by the cognate deoxynucleoside triphosphate (kornberg, 1980). For example, thymidine triphosphate inhibits the removal of a thymidine residue from the end of a primer annealed to poly(dA). To test the effect of the presence of one nucleotide upon transcript shortening, the cleavage reaction was carried out in the presence of each individual NTP. Cleavage takes place in the presence of each one of the four NTPs (Fig. 5). Without a kinetic study it cannot be concluded that the rate of cleavage was unaltered under conditions of single nucleotide replacement. It is clear, however, that a high level of any single NTP does not prevent cleavage. Judging from the different lengths of the major cleavage products shown in Fig. 5 (GTP > ATP > UTP ≈ CTP), it appears that 3′-ends are generated which can be differentially reextended by RNA polymerase II depending upon the template sequence at the RNA 3′-terminus and the presence of the cognate NTP.
Fig. 5. Ribonucleoside triphosphates do not inhibit nascent transcript cleavage by RNA polymerase II.

Washed elongation complexes were incubated with 7 mM MgCl2 and either buffer (lane 1) or 2 μg of partially purified bovine brain SII (lanes 2–6) for 30 min at 28 °C. Samples also contained 800 μM ATP (lane 3). UTP (lane 4), GTP (lane 5), or CTP (lane 6) or no nucleotide (lanes 1 and 2) during this incubation. Migration positions of RNAs are indicated as described for Fig. 1.
DISCUSSION
This report describes a new enzymatic activity for a eukaryotic DNA-dependent RNA polymerase. RNA polymerase II from rat liver can remove a large number of residues from the growing end of nascent transcripts contained within functional elongation complexes. Detection of cleavage activity is dependent upon a divalent cation, elongation factor SII, and the removal of free nucleotides. It has not yet been determined whether cleavage releases mononucleotides (an exonuclease) or oligonucleotides (an endonuclease). The 3′-most product of the cleavage reaction, unlabeled in these experiments, is currently being sought.
It is clear that SII is a factor that enables transcribing RNA polymerase II both to lengthen and to shorten RNA chains. Since SII has been shown to bind to RNA polymerase II (Sawadogo et al., 1980; Horikoshi et al., 1984; Reinberg and Roeder, 1987; Rappaport et al., 1988), it can be considered an accessory factor of polymerase which is not required for transcription initiation in vitro but which activates otherwise latent activities of the enzyme. It should be noted that an integral subunit of vaccinia virus DNA-dependent RNA polymerase shares significant amino acid homology to mouse SII (Ahn et al., 1990). It will be interesting to learn if vaccinia virus RNA polymerase has the capability for nascent transcript hydrolysis and if so what the importance of that function is for RNA synthesis by the virus.
SII-dependent elongation could be a means by which the transcription machinery is coupled to the RNA-processing machinery. RNA processing, i.e. splicing and cleavage of the primary transcript for the formation of mRNA 3′-ends, requires specific signals in the RNA sequence. Pausing within a gene at a site such as Ia, which is within an intron, could serve to regulate the rate of emergence or retraction of these signals from the elongation complex. After an appropriate nucleoprotein complex has formed on the RNA, the progress of polymerase could be stimulated by factor-dependent control over RNA chain elongation or RNA chain shortening, both of which have now been demonstrated in vitro. Others have offered similar models to describe the apparent coupling of primary transcript polyadenylation to the termination of transcription by RNA polymerase II at the end of a gene (Logan et al., 1987; Connelly and Manley, 1988). It remains possible, however, that nascent transcript cleavage may only take place in vitro in isolated nucleotide-starved RNA polymerase II elongation complexes.
What is the function of the RNA cleavage activity carried out by RNA polymerase II? Since nuclease activities of DNA polymerases are involved in proofreading the accuracy of strand synthesis, it is possible that this is the function of the SII-stimulated nuclease activity of RNA polymerase II. This is consistent with the observation that the reaction is not sequence specific; there is no extensive sequence similarity between site Ia and the site at +325, both of which stop RNA polymerase II. Transcript shortening has also been detected in numerous other elongation complexes distributed along this template.2 Thus, RNA cleavage appears to be a general activity of the enzyme. In addition, a single RNA chain can be cleaved repeatedly. In fact, after extensive incubation with SII, transcript Ia can be reduced in size by approximately 100 nucleotides.2
In this regard it is interesting to point out that the accessory subunits of DNA polymerase III and bacteriophage T4 DNA polymerase influence both the processivity of chain elongation and the proofreading exonuclease activity of these enzymes (Alberts et al., 1975; Huang et al., 1981; Bedinger and Alberts, 1983; Reems et al., 1991). DNA polymerase III holoenzyme, with its associated auxiliary factors, can read through DNA regions that impede the progress of the polymerase alone (LaDuca et al., 1983). Thus, SII resembles auxiliary factors for DNA polymerase III since they both influence the enzyme’s nuclease activity and the efficiency of elongation of nascent nucleic acid chains. The exonuclease function of E. coli DNA polymerase III, a 10-subunit enzyme, resides in a single subunit, ε (Scheuermann and Echols, 1984). RNA polymerase II contains 8–12 subunits (Sawadogo and Sentenac, 1990). Many of these subunits have not been ascribed a function. It will be of interest to determine which subunit(s) of RNA polymerase II contains the nuclease function and if this activity is involved in transcriptional proofreading.
Given that the nuclease activity requires SII, an elongation factor, I suggest an alternative, although not mutually exclusive, hypothesis for the function of the SII-stimulated RNA cleavage by RNA polymerase II. RNA cleavage could be integral to, or even required for, SII’s function in stimulating transcript elongation. Transcript truncation could allow polymerase to make repeated attempts at chain elongation through a site at which elongation is blocked. A model (Fig. 6) that assimilates a current understanding of transcription elongation through site Ia illustrates how RNA cleavage could be important for the stimulation of transcription elongation by SII.
Fig. 6. Model for SII-dependent transcription elongation through histone site Ia possible role of RNA cleavage and DNA unbending.

Solid circle, RNA polymerase II; bold parallel lines, DNA duplex; thin solid line, RNA transcript; Ia, transcription arrest site Ia. See “Discussion” for explanation.
It has been shown that site Ia, and other transcription arrest signals, are coincident with bent regions of DNA (as measured by the anomalous migration upon gel electrophoresis of site Ia-containing DNA; Kerppola and Kane, 1990). Point mutations that change the bent character of this region decrease the sequence’s ability to block transcription by pure RNA polymerase II. Furthermore, anomalous migration of this fragment is temperature dependent. This may mean that the extent of bending is variable, i.e. that bent DNA is in equilibrium with unbent or “less” bent DNA (Koo et al., 1986). This may in turn explain why approximately half of the polymerase molecules can pass by site Ia unimpeded (Reines et al., 1989; Reines, 1991). Assume for this argument that, as suggested by Kerppola and Kane (1990), steric hindrance by the bend prevents further elongation and that the presence of polymerase at Ia prevents bent DNA from assuming a less bent, readthrough-permissive configuration. Perhaps, by inducing RNA cleavage (Fig. 6, step 1), SII facilitates the movement of polymerase relative to the DNA. Note that whether the enzyme is an endonuclease or exonuclease is insignificant to this model as the function of RNA cleavage is to move the protein away from the DNA bend. Movement of polymerase serves to allow bent DNA to isomerize into an unbent form (Fig. 6, step 2). In the presence of NTPs, formerly arrested polymerases are now able to elongate RNA chains (Fig. 6, step 3). The ability to elongate will depend upon the equilibrium between bent and unbent DNA. Some polymerases may need multiple rounds of RNA cleavage and movement before encountering an unbent region through which they can elongate. In general then, SII’s function could be to increase the probability that an arrested elongation complex will find a region of the template passable and may be one of many mechanisms for stimulating transcription elongation by RNA polymerase II.
Acknowledgments
I acknowledge the following individuals for a critical reading of the manuscript and stimulating discussions: Drs. J. Boss, M. Chamberlin, L. Coluccio, J. Conaway, R. Conaway, C. Kane, and K. Wilkinson.
Footnotes
The abbreviations used are: HEPES, 4-(2-hydroxyethyl)-1-piper-azineethanesulfonic acid; SDS; sodium dodecyl sulfate.
D. Reines, unpublished results.
P. Ghanouni and D. Reines, unpublished results.
This work was supported by American Cancer Society Grant IRG 182 and by National Institutes of Health Grant GM-46331.
References
- Ahn BY, Gershon PD, Jones EV, Moss B. Mol Cell Biol. 1990;10:5433. doi: 10.1128/mcb.10.10.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B, Morris CF, Mace D, Sinha N, Bittner M, Moran L. ICN-UCLA Symp Mol Biol. 1975;3:241. [Google Scholar]
- Arndt KM, Chamberlin MJ. J Mol Biol. 1990;213:79. doi: 10.1016/S0022-2836(05)80123-8. [DOI] [PubMed] [Google Scholar]
- Bedinger P, Alberts BM. J Biol Chem. 1983;258:9649. [PubMed] [Google Scholar]
- Bengal E, Flores O, Reinberg D, Aloni Y. Mol Cell Biol. 1990;11:1195. doi: 10.1128/mcb.11.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL, Groudine M. Nature. 1986;321:702. doi: 10.1038/321702a0. [DOI] [PubMed] [Google Scholar]
- Bentley DL, Groudine M. Cell. 1988;53:245. doi: 10.1016/0092-8674(88)90386-8. [DOI] [PubMed] [Google Scholar]
- Chafin DR, Clausen TJ, Price DH. J Biol Chem. 1991;266:9256. [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 1989;86:7356. doi: 10.1073/pnas.86.19.7356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway JW, Bond M, Conaway RC. J Biol Chem. 1987;262:8293. [PubMed] [Google Scholar]
- Conaway JW, Reines D, Conaway R. J Biol Chem. 1990;265:7552. [PMC free article] [PubMed] [Google Scholar]
- Connelly S, Manley J. Genes & Dev. 1988;2:440. doi: 10.1101/gad.2.4.440. [DOI] [PubMed] [Google Scholar]
- Eilat D, Hochberg M, Fischel R, Laskov R. Proc Natl Acad Sci U S A. 1982;79:3818. doi: 10.1073/pnas.79.12.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores O, Maldonado E, Reinberg D. J Biol Chem. 1989;264:8913. [PubMed] [Google Scholar]
- Gonzalez N, Wiggs J, Chamberlin MJ. Arch Biochem Biophys. 1977;182:404. doi: 10.1016/0003-9861(77)90521-5. [DOI] [PubMed] [Google Scholar]
- Horikoshi M, Sekimizu K, Natori S. J Biol Chem. 1984;259:608. [PubMed] [Google Scholar]
- Huang C-C, Hearst JE, Alberts B. J Biol Chem. 1981;256:4087. [PubMed] [Google Scholar]
- Ishihama A, Mizumoto K, Kawakami K, Kato A, Honda A. J Biol Chem. 1986;261:10417. [PubMed] [Google Scholar]
- Kahn JD, Hearst JE. J Mol Biol. 1989;205:291. doi: 10.1016/0022-2836(89)90342-2. [DOI] [PubMed] [Google Scholar]
- Kao SY, Caiman AF, Luciw PA, Peterlin BM. Nature. 1987;330:489. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- Kassevetis GA, Zentner PG, Geiduschek EP. J Biol Chem. 1986;261:14256. [PubMed] [Google Scholar]
- Kerppola T, Kane CM. Biochemistry. 1990;29:269. doi: 10.1021/bi00453a037. [DOI] [PubMed] [Google Scholar]
- Kerppola T, Kane CM. FASEB J. 1991;5:2833. doi: 10.1096/fasebj.5.13.1916107. [DOI] [PubMed] [Google Scholar]
- Koo HS, Wu HM, Crothers DM. Nature. 1986;320:501. doi: 10.1038/320501a0. [DOI] [PubMed] [Google Scholar]
- Kornberg A. DNA Replication. W. H. Freemanand Co; San Francisco: 1980. pp. 127–130. [Google Scholar]
- Krakow JS, Fronk E. J Biol Chem. 1969;244:5988. [PubMed] [Google Scholar]
- LaDuca DJ, Fay P, Chuang C, McHenry C, Bambara R. Biochemistry. 1983;22:5177. doi: 10.1021/bi00291a018. [DOI] [PubMed] [Google Scholar]
- Logan J, Falck-Pederson E, Darnell JE, Shenk T. Proc Natl Acad Sci U S A. 1987;84:8306. doi: 10.1073/pnas.84.23.8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderious A, Chen-Kiang S. Proc Natl Acad Sci U S A. 1984;81:5931. doi: 10.1073/pnas.81.19.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra U, Hurwitz J. J Biol Chem. 1967;242:4897. [PubMed] [Google Scholar]
- Price DH, Sluder AE, Greenleaf AL. J Biol Chem. 1987;262:3244. [PubMed] [Google Scholar]
- Price DH, Sluder AE, Greenleaf AL. Mol Cell Biol. 1989;9:1465. doi: 10.1128/mcb.9.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappaport J, Reinberg D, Zandomeni R, Weinmann R. J Biol Chem. 1987;262:5227. [PubMed] [Google Scholar]
- Rappaport J, Cho K, Saltzman A, Prenger J, Golomb M, Weinmann R. Mol Cell Biol. 1988;8:3136. doi: 10.1128/mcb.8.8.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reems JA, Griep MA, McHenry CS. J Biol Chem. 1991;266:4878. [PubMed] [Google Scholar]
- Reinberg D, Roeder RG. J Biol Chem. 1987;262:3331. [PubMed] [Google Scholar]
- Reines D. J Biol Chem. 1991;266:10510. [PMC free article] [PubMed] [Google Scholar]
- Reines D, Wells D, Chamberlin MJ, Kane CM. J Mol Biol. 1987;196:299. doi: 10.1016/0022-2836(87)90691-7. [DOI] [PubMed] [Google Scholar]
- Reines D, Chamberlin MJ, Kane CM. J Biol Chem. 1989;264:10799. [PubMed] [Google Scholar]
- Resnekov O, Aloni Y. Proc Natl Acad Sci U S A. 1989;86:12. doi: 10.1073/pnas.86.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnekov O, Kessler M, Aloni Y. J Biol Chem. 1989;264:9953. [PubMed] [Google Scholar]
- Rougvie AE, Lis JT. Cell. 1988;54:795. doi: 10.1016/s0092-8674(88)91087-2. [DOI] [PubMed] [Google Scholar]
- Rougvie AE, Lis JT. Mol Cell Biol. 1990;10:6041. doi: 10.1128/mcb.10.11.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawadogo M, Sentenac A. Annu Rev Biochem. 1990;59:711. doi: 10.1146/annurev.bi.59.070190.003431. [DOI] [PubMed] [Google Scholar]
- Sawadogo M, Sentenac A, Fromageot P. J Biol Chem. 1980;255:12. [PubMed] [Google Scholar]
- Scheuermann RH, Echols H. Proc Natl Acad Sci U S A. 1984;81:7747. doi: 10.1073/pnas.81.24.7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimizu K, Kobayashi N, Mizuno D, Natori S. Biochemistry. 1976;15:5064. doi: 10.1021/bi00668a018. [DOI] [PubMed] [Google Scholar]
- Selby MJ, Peterlin BM. Cell. 1990;62:769. doi: 10.1016/0092-8674(90)90121-t. [DOI] [PubMed] [Google Scholar]
- SivaRaman L, Reines D, Kane CM. J Biol Chem. 1990;265:14554. [PubMed] [Google Scholar]
- Skarnes WC, Tessier DC, Acheson NH. J Mol Biol. 1988;203:153. doi: 10.1016/0022-2836(88)90099-x. [DOI] [PubMed] [Google Scholar]
- Sluder AE, Greenleaf AL, Price DH. J Biol Chem. 1989;264:8963. [PubMed] [Google Scholar]
- Spencer C, Groudine M. Oncogene. 1990;5:777. [PubMed] [Google Scholar]
- Surratt CK, Milan SC, Chamberlin MJ. Proc Natl Acad Sci U S A. 1991;88:7983. doi: 10.1073/pnas.88.18.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer GC, Brosius J. Mol Gen Genet. 1985;199:55. doi: 10.1007/BF00327509. [DOI] [PubMed] [Google Scholar]