

Abstract
Cell culture/xenograft and gene arrays of clinical material document that development of castration resistant prostate cancer (CRPC) cells involves acquisition of adaptive auto-regulation resulting in > 25 fold increase in Androgen Receptor (AR) protein expression in a low androgen environment. Such adaptive AR increase paradoxically is a liability in castrated hosts; however, when supraphysiologic androgen is acutely replaced. Cell synchronization/anti-androgen response document this is due to AR binding to replication complexes (RC) at origin of replication sites in early G1 associated with licensing/restricting DNA for single round of duplication during S-phase. When CRPC cells are acutely exposed to supraphysiologic androgen, adaptively increased nuclear AR is over-stabilizes preventing sufficient degradation in mitosis inhibiting DNA re-licensing and thus death in the subsequent cell cycle. These mechanistic results and the fact that AR/RC binding occurs in metastatic CRPCs directly from patients provides a paradigm shifting rationale for bipolar androgen therapy (BAT) in patient progressing on chronic androgen ablation. BAT involves giving sequential cycles alternating between periods of acute supraphysiologic androgen followed by acute ablation to take advantage of vulnerability produced by adaptive auto-regulation and binding of AR to RC in CRPC cells. BAT therapy is effective in xenografts and based upon positive results has entered clinical testing.
Keywords: Androgen Receptor, Origin of Replication, DNA Licensing, Orc, Cdc6, Cdt1, Mcm, Prostate Cancer, Bipolar Androgen Therapy
Introduction
During prostate carcinogenesis, molecular changes occur in prostate epithelial cells such that Androgen Receptor (AR) signaling is perverted from a growth suppressor to an oncogenic stimulator of malignant growth (1,2). Due to such an acquired addiction to AR signaling, androgen ablation therapy is the standard of care for metastatic prostate cancer since this not only inhibits proliferation, but also induces the apoptotic death of prostate cancer cells (1). While initially responsive to such “castration therapy”, metastatic prostate cancer cells adapt to a castration-resistant state for which there is no curative therapy (3). In the majority of cases, these castration resistant prostate cancer (CRPC) cells continue to express AR and their lethal growth is still stimulated by AR-dependent signaling despite greater than 90% suppression of serum androgen by castration therapy (4–5). Retention of AR dependence by such human CRPC cells is documented by their in vitro and in vivo growth inhibition following down regulation of their AR expression (7–10). These results validate that disrupting AR function is a rational therapeutic approach for CRPCs. This raises the question of how to disrupt AR function in patients progressing on hormonal therapy.
Historically, approaches have focused upon reducing the level of AR signaling in CRPCs by either lowering non-testicular sources of circulating and thus tumor androgen or by combining a competitive AR binding anti-androgen with castrating therapy. Unfortunately, these second-line approaches result in only incremental (i.e., < 5 months) increases in median survival (3). In addition, the demise of such second-line treated patients is nearly universally associated with a continuing rise in serum PSA, documenting that AR is still functioning (3). Experimental studies document that continued AR signaling by CRPCs can be the result of an increase in AR level within the cell (4). This increase can result in sufficient AR accumulation by mass action in the nuclei of CRPC cells to inhibit apoptotic death and stimulate cell proliferation despite a castrate level of androgen (4).
Such an increase in AR, however, creates a unique therapeutic vulnerability to selectively kill CRPCs. This is based upon the fact that AR is involved in DNA replication (11–13). DNA replication is dependent upon the process of DNA licensing to ensure that the genome is replicated only once per cell cycle. Restricted to early/mid-G1, DNA licensing consists of the coordinated assembly of pre-replication complex (pre-RC) machinery at specific origin of replication sites [ORS] (14) which in humans lacks canonically defined consensus sequences (15). Distributed at approximately 25 thousand sites throughout the human genome, ORS are first bound in early-G1 by the highly conserved origin recognition complex (ORC) composed of Orc1–6 subunits (16, 17). This is followed sequentially by binding of Cdc6 to ORC complex which is needed for subsequent loading of ring-shaped heptameric mini chromosome maintenance DNA helicase complex consisting of the Mcm2–7 plus Cdt1 onto DNA at ORS completing pre-RC formation required for G1-dependent DNA licensing (17, 18). Once DNA is licensed and the cell enters S-phase, the genome is replicated only once per cycle with re-replication prevented by S-phase activation of cyclin-dependent kinase (CDK)-induced inactivation, nuclear export, and eventual proteolytic degradation of the rate limiting RC proteins Orc1, Cdt1, and Cdc6 with the retention of Orc2–6 at ORS (18, 19). For re-licensing, RCs must be removed from ORS during G2/mitosis so that in early-G1 of the next cell cycle, ORS are fully accessible to binding of newly synthesized rate limiting RC proteins to initiate formation of new pre-RCs (19, 20).
If anti-androgen sensitive CRPC cells are exposed to AR antagonists (e.g., bicalutamide) before they complete pre-RC assembly, they do not undergo DNA replication and cell division (21). In contrast, if AR antagonism is initiated after pre-RC formation, anti-androgen sensitive CRPCs successfully complete S-phase and divide but do not re-enter the next cycle (21). These results raised the possibility that in CRPC cells, AR uniquely acquires oncogenic function as part of replication complexes (RCs). Since a common characteristic of the rate limiting RC proteins is that they are degraded during cell cycle progression and re-accumulate by early-G1 for formation of pre-RCs, we evaluated AR levels during cell cycle progression of CRPCs. These studies documented that AR is degraded via the proteasome during mitosis and rapidly re-synthesized in early-G1 (11). In contrast, AR is not degraded during mitosis in normal human prostate stromal cells whose growth is not AR stimulated (11). In a later study, we documented that if AR is not sufficiently degraded in mitosis by CRPCs, these cells do not completely re-license their DNA and thus they die in the next cell cycle (12). These results suggest that AR is a licensing factor needed for DNA replication in CRPCs
These replication results when combined with the observations that CRPC cells have increased AR (4) and that androgen binding stabilizes AR protein and increases its nuclear retention (22) provide a rationale for a paradigm shifting approach to CRPC. The rationale is that acute supraphysiologic androgen replacement to a patient progressing on chronic androgen ablated therapy increases nuclear AR to a point where insufficient removal of AR/RC from ORS in mitosis needed for full re-licensing occurs, thus inducing death in S-phase of the subsequent cell cycle. A subset of CRPC cells can survive such acute high androgen replacement if they adaptively down regulate their initially high AR expression. If they adapt by down regulating AR, however, they become vulnerable to apoptosis if subsequently re-exposed acutely to an androgen ablated environment. Thus, a paradigm shifting approach both to overcome and to take advantage of adaptive AR auto-regulation by CRPCs is to give castration resistant patients bipolar androgen therapy (i.e., BAT) consisting of multiple sequential cycles of a rapid supraphysiologic androgen followed by a rapid decline in serum androgen. In the present study, the mechanism for such BAT is presented and its therapeutic ability against prostate cancer xenografts validated.
Materials and Methods
Reagents
Testosterone was from Steraloids (Newport, RI) and the synthetic androgen, methytrienolone (i.e., R1881) from Perkin-Elmer(Boston, MA). Bicalutamide (i.e., casodex) was from LKT Laboratories (St Paul, MN).
Cell Culture
Development and characteristics of each of the human CRPC lines used has been reported (6,11, 23–25), except for the LNCaP/A- cells. All of these lines were grown in RPMI-1640 media (Invitrogen, Carlsbad, CA), except for LAPC-4 for which Iscove’s modified media (Invitrogen) was used. To these media was added 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) plus 100U/mL penicillin and 100 U/mL streptomycin sulfate except for the LNCaP/A- line. This latter line was established and maintained by Dr. Alan Meeker by long term growth in phenol red free RPMI-1640 media supplemented with 10% dextran coated charcoal (DCC) stripped FBS. Growth of these cells was determined with a MTT assay as described previously (26).
AR Protein Determination
Normal prostate tissue and localized prostate cancer were harvested from radical prostatectomy specimens from hormonally naïve patients under an approved IRB protocol as part of the SPORE Tissue Resource Core at Hopkins. These tissues were enzymatically dissociated into single cells and epithelial cells isolated by flow cytometry using an anti-EpCam antibody as described previously (23). AR protein levels were determined in these normal and malignant cells without culturing as well as a series of established prostate cancer xenograft and cell lines by densitometry of western blots as described previously (26). Protein lysates from 2.5 ×104 cells were loaded per lane for each tissue sample and the results normalized to AR level of EpCam flow sorted normal human prostate epithelial cells from hormonally naïve patient (i.e., denoted N-PrEC ).
AR m-RNA Determination
Normal prostate samples from organ donors (n=23) were obtained from Dr. Robert Getzenberg, Department of Urology, The Johns Hopkins School of Medicine as described previously (27). Radical prostatectomy specimens were used to isolate fresh frozen specimens from both areas of prostate cancer (N=30) or peripheral areas of normal prostate without cancer (N=6) collected at the Johns Hopkins Hospital as described previously (28). Castration resistant prostate cancer specimens (n=18) were autopsy specimens from 6 patients who died from prostate cancer, as previously reported (29). The use of surgical and autopsy specimens for molecular analysis was approved by the Johns Hopkins Medicine Institutional Review Boards. Standardized and established tissue processing procedures were followed prior to RNA extraction. Frozen tissue blocks were manually trimmed to enrich tissue lesions of interest (cancer or normal epithelium). Cryosections were cut from trimmed blocks. The first and last section from each block was reserved for pathological confirmation and estimation of the target tissue content (greater than 70%).
Gene expression profiling was performed strictly according to the guidelines provided by the Agilent Whole Genome Expression Microarray system (Agilent Technologies, Santa Clara, CA).), using 2-color design as described previously (30). Briefly, each of the 77 RNA samples was linearly amplified and labeled with Cy3, and co-hybridized with a common reference sample derived from benign prostatic hyperplasia that was similarly amplified but labeled with Cy5. For each sample, expression ratios of Cy5/Cy3 for 44,000 genes/probes constituted the gene expression profile. Gene expression ratios for each sample were normalized independently, using the standard locally weighted least squares regression (LOWESS) procedure and the results presented as natural log of relative expression normalized to BPH tissue as described previously (30).
Immunocytochemical Staining
Cells were fixed with 10% buffered formalin and solubilized with 1% Triton X-100 before cytospun onto poly-lysine coated glass slides. Cell and tissue slides were processed for detection of AR, Ki-67 protein, or bromodeoxyuridine (BrdUrd) incorporation as described previously (25, 26).
Flow Cytometry studies
CWR22-Rv1 cells were transduced with an AR/GFP lenti- viral construct in which AR expression is constitutively driven by the promoter for the house-keeper gene, Elongation Factor-1 alpha, in order to induce unregulated over-expression of AR protein as described previously (31). GFP expressing cells were isolated by limited dilution cloning to isolate pure populations of CWR-22Rv1 transduced cells which were analyzed for their level of AR protein expression. To determine the percentage of cells in mitosis which express AR, methanol fixed cells were immunocytochemical stained for phospho-histone H3 (i.e., a mitotic marker) and positive cells in mitosis flow sorted and then stained for AR as described previously (11). To determine the cell death index (i.e., percentage of cancer cells dying) in both cell culture and xenograft tissue, single cells suspensions were enzymatically dissociated as described previously (23) and the cells were incubated with cell permeable DNA dye [i.e., Vybrant DyeCycle Ruby Stain (Invitrogen)] and analyzed for the percent of sub-G0/G1 cells on a BD FACSCalibur flow cytometer using BD CellQuest Pro software.
PSA Measurements
Levels of PSA in mouse plasma were determined by the Clinical Chemistry laboratory at Johns Hopkins using the Hybritech assays on the Beckman Access Immunoassay System (Beckman Coulter, Inc., Brea, CA).
AR binding to Replication Complexes (RC)
For the determination of whether AR binds to RCs, both cell lines and metastatic CRPC tissue were evaluated. Metastatic CRPC tissues from a variety of organ sites were tested and these tissues were generously provided by Dr. Ken Pienta. These tissues were obtained under an approved IRB protocol as part of the rapid autopsy program at the University of Michigan as described previously (5). Nuclear complex co-IP kit (cat#54001) from Active Motif (Carlsbad, CA) was used according to the manufacturer’s instructions to extract RC proteins from the nuclei of prostate cancer cells. This protocol starts with homogenizing 50 mgs of tissue (i.e., containing approximately 107 cells) or 107 cells harvested by trypsinization from cultures in 500ul of the hypotonic buffer plus detergent reagent provided in the kit. The resulting lysates are centrifuged at 14,000G to isolate cytosolic extract from the nuclear pellet. The nuclear pellet is re-suspended and digested with proprietary nuclear solubilization solution composed of a hypotonic digestion buffer containing 100mM PMSF, protease inhibitor cocktail, and a random cutting nuclease. After 90min at 4°C, the mixture is centrifugation at 14,000G for 10 min at 4°C. An aliquot of this soluble nuclear extract was treated with protease K and the DNA separated on 1.5% agarose gel followed by staining with ethidium bromide. An additional aliquot of unfractionated nuclear extract was collected and the remaining extract mixed with an equal volume of IP buffer which contained protease inhibitor to bring the final concentration to 150mM NaCl and 1% Triton X-100 detergent. The mixture was incubated overnight at 4°C with 4μg primary rabbit polyclonal IP antibody to the indicated specific protein and 40μl protein Agarose A/G beads (Santa Cruz). The antibodies used for IP were: AR (rabbit polyclonal N-20 sc-816 from Santa Cruz), Orc2 (rabbit polyclonal cat# 559266, BD Pharmingen), Cdc6 (rabbit polyclonal H-304 sc-8341 from Santa Cruz), Mcm2 (rabbit polyclonal cat#A300-191A, Bethyl Laboratories), and as a control, non-specific polyclonal rabbit IgG from Santa Cruz. The mixture was then centrifuged at 14,000G for 30 sec at 4°C and pelleted beads washed 3 times with BSA-supplemented wash buffer and 3 times in wash buffer. The washed beads were re-suspended with 1x loading buffer [i.e., 20mM Tris-HCl pH 7.8, 140mM NaCl, 1mM EDTA, 0.5% sodium deoxycholate, 0.5% Nonidet P-40 (NP-40) supplemented with PhosSTOP phosphatase inhibitor tablet and protease inhibitor tablet (Roche), and 1mM dithiothreitol]. An aliquot of the un-fractionated cytosolic and nuclear extracts were mixed with an equal volume of 2x loading buffer and these mixtures plus the re-suspended washed IP beads from the nuclear extract heated at 95°C for 5 min, centrifuged and the supernatants subjected to SDS PAGE/IB analysis as described previously (26). The antibodies used for IB detection of: AR (mouse monoclonal 441sc-7305 from Santa Crux); Orc1 (rat monoclonal 7A7 sc-23887, Santa Cruz); Orc2 (rat monoclonal 3G6 cat# 4736 from Cell Signaling); Cdc6 (mouse monoclonal 180.2 sc-9964 from Santa Cruz); Cdt1 (rabbit polyclonal H-300 sc-28262 from Santa Cruz), and Mcm2 (mouse monoclonal cat # 610700 from BD Pharmingen). See Figure 1 for details of the validation of the specificity of the antibodies used for IP and the efficiency of the IP protocol.
Figure 1. AR and RC protein expression in human prostate cancer cell lines and antibody validation for IP and IB analysis.
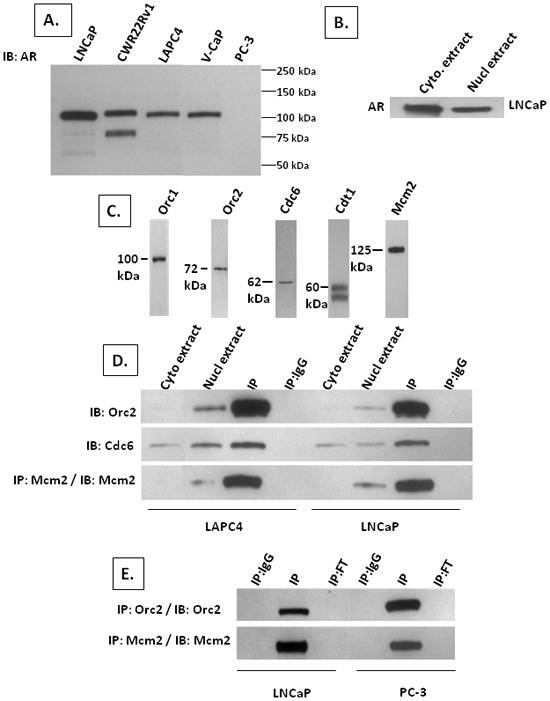
(A) IB of AR protein in AR-expressing LNCaP, CWR22Rv1, LAPC4, and VCaP cells compared to AR-non expressing PC-3 prostate cancer cells using an N-terminal specific anti-AR antibody. Nuclear protein extracts from 105 cells were loaded per well for each of the lines. (B) IB of AR in cytosolic (denoted cyto. extract) vs. nuclear (denoted nucl extract) extract from 105 LNCaP cells. (C) IB of Orc1, Orc2, Cdc6, Cdt1, and Mcm2 in nuclear extracts from 105 LNCaP cells. (D) IB for indicated protein in cytosolic (denoted cyto extract) vs. nuclear (denoted nucl extract) extracts and IB of IP using specific antibody against the indicated protein (denoted IP) or using non-specific IgG (denoted IP:IgG) from nuclear extracts of LAPC4 and LNCaP cells. (E) IB of IP using non-specific IgG (denoted IP:IgG) vs. specific antibody against the indicated protein (denoted IP) vs. flow through from the specific IP (denoted IP:FT) from nuclear extracts of LNCaP and PC-3 cells.
RC Protein Cross Linking
LNCaP cells growing in RPMI-1640 media plus 10% FBS were washed with phosphate buffer saline (PBS) and exposed for 30 min to 0.5mM of the cell-permeable cross-linker dithiobis[succinimdylpropionate] (DSP) in PBS to crosslink RC associated proteins as described previously (32). To terminate cross linking, cells were washed with 20mM Tris-HCl pH 7.4 and lysed in RIPA buffer [i.e., 25mM Tris-HCl pH 7.6, 150mM NaCl, 1% NP-40, 1% Na-deoxycholate, 0.1% SDS, plus protease inhibitor tablet (Roche)] to disrupt non-covalent protein-protein interactions. These RIPA buffered extracts were used for Orc2 co-IP followed by IB for Orc2, AR and Mcm2.
Cell Synchronization
Cell synchronization was performed using two techniques. In the first technique, LNCaP cells were inoculated in phenol-red-free RPMI-1640 isoleucine-free media (Invitrogen) containing 6% dialyzed FBS (Hyclone) plus1nM testosterone as described previously (33). After 2 days, cells were harvested for determination of percentage of cells in cycle based upon immunocytochemistry for Ki-67expression and in S-phase based upon bromodeoxyuridine (BrdUrd) incorporation. The arrested cells were then released back into cycle by changing the media to isoleucine-containing RPMI-1640 media plus 10% FBS and 1nM testosterone and cells harvested at various time. In the second technique, LNCaP cells were inoculated on poly-lysine coated dishes in RPMI-1640 media containing 10% FBS and after one day, 50uM of the anti-androgen casodex added to the media. After 4 days, cells were harvested for IP/IB analysis and for Ki-67 expression and BrdUrd incorporation
Xenograft Studies
Using a Johns Hopkins Animal Care and Use Committee approved protocol, intact or castrated adult male triple immune-deficient NOG mice, as indicated, were inoculated subcutaneously (SC) in the flank with the indicated human prostate cancer cell lines in 200uL of Matrigel as described previously (24). For the BAT experiments, LNCaP/A- cells in Matrigel were inoculated into castrated male NOG mice with half the animals given no additional treatment and half implanted SC with 2 one cm long silastic implants filled with testosterone as described previously (25). These T-implants were removed after 2 weeks and then replaced 2 weeks later. Tumor volumes were determined using micro-caliper measurement as described previously (25). Serum testosterone was measured using a testosterone EIA test kit BioCheck Inc. (Foster City, CA). At harvest, tumor tissue was fixed in10% buffered formalin and processed for routine paraffin embedding and sectioning. Serial sections were stained with H&E and scored for mitotic index (i.e., percent of cells in mitosis) or used for immunocytochemical staining.
Statistics
All of the values are presented as means ± SE. Statisticalanalysis was performed by a one-way ANOVA with the Newman-Keulstest for multiple comparisons.
Results
Androgen Receptor is highly over-expressed by Castrate Resistant Prostate Cancer Cells
AR-expressing human prostate cancer xenografts derived from localized prostate cancers [i.e., PC82 (34) and CWR22 (35)] and cell lines from both localized prostate cancer [i.e., E006AA (36)] and lymph node metastasis [i.e., PacMetUT1 (37)] have been established derived from hormonally naïve patients. In addition, AR-expressing castrate resistant prostate cancer (CRPC) cell lines have been established from metastases harvested from patients progressing on androgen ablation therapy [i.e., LNCaP and LAPC4 from lymph node metastases and VCaP and MDA-PCA-2b from bone metastases (23)]. The level of AR protein expression by these xenograft/cell lines was compared to localized prostate cancer cells (L-PCA) and normal prostate epithelial cells (i.e., N-PrEC) analyzed directly from hormonally naïve patients without culturing. These comparisons documented that AR protein levels in localized as well as metastatic prostate cancer cells from hormonally naïve patients range from 1–6 fold that of normal prostate epithelial cells, Figure 2A. In contrast, AR is >25 higher in all of the CRPC lines (i.e., ranging from 27–90 fold higher compared to N-PrEC and from 9–30 fold higher compared to L-PCA cells harvested from hormonally naïve patients not adapted to a castration resistant state, Figure 2A.
Figure 2. AR is characteristically increased in human castrate resistant prostate cancer (CRPC) cell lines and CRPC clinical specimens.
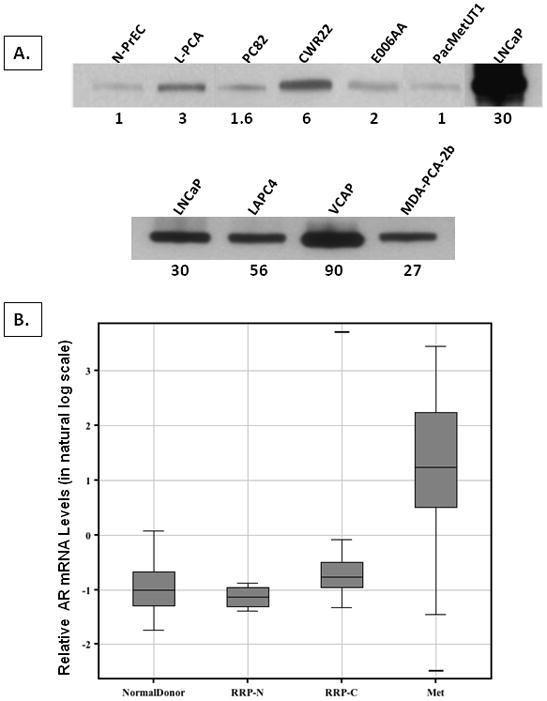
(A) Western blots of AR protein in EpCam flow sorted human normal human prostate epithelial cells without culturing (i.e., denoted N-PrEC), localized prostate cancer cells without culturing (i.e., denoted L-PCA) and a series of human prostate cancer xenografts (i.e., PC82 and CWR22) and prostate cancer cell lines (i.e., E006AA and PacMetUT1) from hormonally naïve patients and CRPC cell lines from androgen ablated patients (i.e., LNCaP, LAPC4, VCaP, MDA-PCA-2b). Number below each lane is the relative level of AR protein for the indicated prostate cancer xenograft/cell line normalized to AR expression in N-PrECs. Upper panel are the results of long film exposure time to detect AR in N-PrEC cells so that its densitometry level can be used to normalize AR expression in the indicated samples. Lower panel are the results of short film exposure time to determine the relative AR levels in 4 human CRPC cell lines (i.e., LNCaP and LAPC-4 derived from lymph node metastases and VCaP, and MDA-PCA-2b derived from bone metastases, all from different patients progressing on androgen ablation therapy). (B) Level of AR m-RNA expression in normal prostate tissues from 23 organ donors (denoted Normal Donor), normal prostate tissues from radical prostatectomy specimens from 6 hormone naïve patients (denoted RRP-N), localized prostate cancer from radical prostatectomy specimens from 30 hormone naïve patients (denoted RRP-C), and 18 metastases from 6 castration resistant patients obtained at rapid autopsy (denoted Met).
To evaluate whether this increase in AR expression by CRPC cell lines is an in vitro artifact of cell culture, mRNA expression of AR was compared in 23 normal prostates obtained from organ donors, 6 normal prostate tissues obtained from prostatectomy specimens, 38 localized prostate cancers from prostatectomy specimens of hormonally naïve patients, and 18 prostate cancer metastases obtained at warm autopsy from castrate resistant patients. AR RNA is over-expressed by 4-fold in metastatic CRPC compared to localized prostate cancer from hormonally naïve patients and more than 4-fold compared to normal prostate, Figure 2B. These results document that increased AR expression in CRPC cell lines is not an artifact of culture.
Adaptive Androgen Receptor auto-regulation is an acquired ability by CRPC cells
AR gene is amplified in prostate cancers and this occurs more frequently in cancers progressing on hormonal therapy than in hormonally naïve patients (38). AR amplification, however, only occurs in a minority of CRPCs (38). This suggests that the characteristic > 25 fold increase in AR protein expression usually is not due to initiating genetic events in prostate carcinogenesis but to molecular changes acquired during progression required for CRPC cells to selectively grow in a low androgen environment in androgen ablated patients. To evaluate this possibility, two independently derived androgen dependent human xenografts (i.e., PC82 and CWR22) derived from localized prostate cancer tissue from hormone naïve patients (34, 35) were used.
When xenografted into ten intact adult male immune-deficient mice whose normal serum testosterone (T) is 3.5 +/− 1.5 nM, which is the low end of the physiological range for an intact adult human male (39), both PC82 and CWR22 cancers grow in all animals, Figure 3A. While AR in these cancers is slightly elevated compared to normal prostates from hormonally naïve patients, (i.e., 1.6 +/− 0.4 in PC82 and 6.3+/− 1.5 fold in CWR22), this elevation is much lower than the >30 fold elevation characteristic of CRPCs, Figure 2A. Also unlike the CRPCs, none of ten PC82 cancers and only one of ten CWR22 cancers grew over a 1 year observation period when inoculated into castrated male mice whose serum T is < 300pM [i.e., high end of ablation range in castrated patients (39)]. These results document that not all androgen dependent human prostate cancer cells have the ability to adapt and become castrate resistant. Interestingly, the one CWR22 cancer which did eventually grow in a castrate was not palpable until 6 months, but after this lag-time, it grew at a rate comparable to that of CWR22 cancers growing in intact males, Figure 3A. These results are consistent with cancer cells in this one tumor stochastically acquiring molecular changes allowing its progression to a castration resistant state.
Figure 3. In Vivo adaptive AR auto-regulatory response of castrate resistant CWR22RH cancer to varying androgen environments.
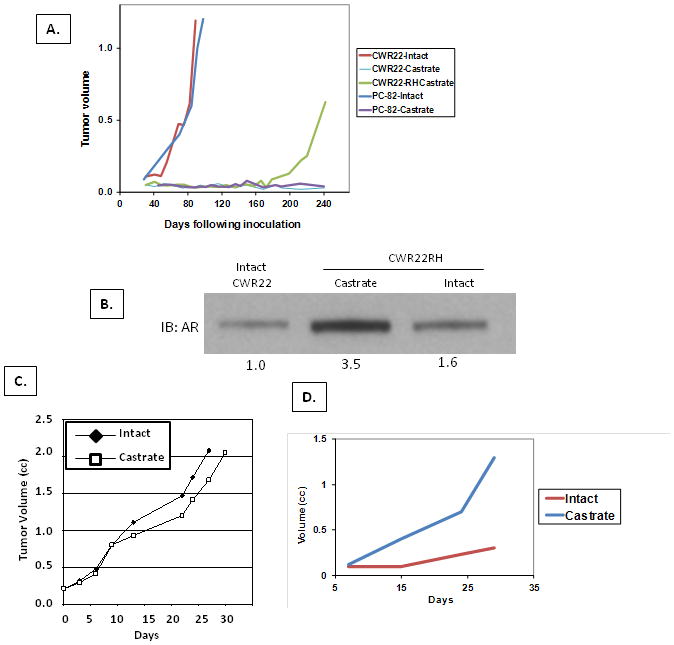
(A) In vivo growth response of PC82 and CWR22 tissue xenografted into intact (n=10) vs. castrated (N=10) mice. For both cancers, all intact animals developed growing tumors, while only a single castrate resistant cancer (i.e. termed CWR22RH) grew in one of the castrates inoculated with CWR22. Mean tumor volumes for the intact PC82 and CWR22 groups vs. the growth of the single CWR22RH tumor which developed in a castrated host are presented. (B) AR level in parental CWR22 growing in intact host vs. CWR22RH grown in intact vs. castrated host. Number below blot is the relative AR expression vs. CWR22 in intact host. (C) Growth of CWR22RH in intact (n=8) vs. castrate (n=8) hosts. (D) Growth of CWR22-Rv1(4X/AR) cells transduced to express 4fold higher AR than parental cells in intact (n=5) vs. castrated (N=5) host.
In this single castrate resistant cancer (i.e., termed CWR22RH), AR level is enhanced by 3.5 fold compared to the parental CWR22 tumors growing in intact mice, Figure 3B. Since CWR22 cancers in intact hosts already express a 6 fold higher level of AR than normal prostate epithelial cells, Figure 2A, this means that CWR22RH cancer cells growing in a castrate express AR at a > 20-fold higher level than normal prostate epithelial cells in an intact host. This raises the issue of whether such increased AR expression is constitutive in such CRPC cells or whether these cells acquire an adaptive ability to auto-regulate AR expression to optimize growth under varying androgen conditions. To address this question, the original castrate resistant CWR22RH cancer was serially passaged five times in castrated hosts. Such serial passage in castrated hosts results in CWR22RH cancers which eventually grow equally well in intact and castrated animals, Figure 3C. While their growth is similar in either low or high serum androgen, their level of AR is inversely related to the level of serum androgen (i.e., low androgen-high AR in castrate vs. high androgen-low AR in intact host), Figure 3B. These results document that the development of castration resistance can involve acquiring adaptive auto-regulation of AR to optimize growth under changing host levels of androgen.
Paradoxical death of castrate resistant prostate cancer cells constitutively expressing high AR in intact vs. castrated hosts
The previous results raise the issue of the consequence of disrupting this adaptive AR auto-regulation upon the growth of CRPC cells under different levels of systemic androgen. To test this, CWR22-Rv1 cells were transduced with an AR/GFP lenti-viral construct in which AR expression is driven by a strong constitutive promoter and GFP expressing clones isolated and screened for AR expression by Western blot analysis. A clone [i.e., termed CWR22-Rv1(4X/AR)] which constitutively expressed a 4-fold enhanced level of AR regardless of androgen level in the media was expanded and the cells inoculated into intact and castrated mice. Paradoxically, growth of CWR22-Rv1(4X/AR) cells is much better in castrates than in intact hosts, Figure 3D. This difference in growth is not due to a difference in the percentage of cancer cells in cycle (i.e.,> 40% of the cells express the cell cycle marker Ki-67 in both intact and castrated animals), but instead is due to a 3-fold (p<0.05) increase in the percentage of cancer cells dying in intact vs. castrated animals (i.e., 44+/− 7% dying in intact treated vs. 15+/− 2 % in castrated hosts) determined by flow cytometric analysis.
These results document that if AR expression is too high, exposure to a physiologic level of androgen paradoxically stimulates death of CWR22-Rv1 cells in vivo. These results raise 2 issues. First, is this response unique to CWR22-Rv1 cells and second, if not, is such androgen-dependent stimulation of death in vivo due to a direct effect on the cancer cells themselves or via indirect effects requiring other host cells? To answer these 2 questions, LNCaP cells was utilized since this additional CRPC line grows continuously in culture, Figure 4A, and secretes the AR-dependent marker, PSA (i.e.,> 200 ng PSA /106cell/day) in the presence of the castrate level of androgen provided by 10% FBS-containing media (39). In this low androgen environment, these CRPC cells express AR protein at >10-fold higher level than hormonally responsive primary cancers from untreated patients and >30-fold higher than normal prostate epithelial cells, Figure 2A. The level of AR in these cells is not a constitutive characteristic, however, but can be dynamically regulated. For example, when these cultured cells are rapidly exposed to the anti-androgen, bicalutamide, AR is down regulated by >3 fold, Figure 4B. Associated with AR downregulation is a profound (p<0.05) inhibition of growth, Figure 4A, a 10-fold inhibition in PSA secretion (i.e., reduced to < 20 ng PSA/106cell/day), and decreased expression of the cell cycle marker Ki-67 from 79+/− 11 % to 11+/− 4% following exposure to bicalutamide. In contrast to AR downregulation by bicalutamide, when LNCaP cells are acutely exposed to a supraphysiological level of the non-metabolizable synthetic androgen, methyltrienolone [aka R1881] (i.e., 10nM R1881 which is biologically equivalent to >25nM testosterone), AR is stabilized and its nuclear level is rapidly increased by another 2-fold, Figure 4B, resulting in AR being 60-fold higher than in normal prostate epithelial cells. This results in enhanced AR signaling as documented by a significant (p<0.05) 4-fold increase in PSA secretion (i.e., to a level >800ng/ PSA /106cell/day) detectable within 1 day of androgen exposure. Paradoxically, however, such androgen-induced 2-fold increase in nuclear AR results in profound (p<0.05) growth inhibition, Figure 4A. Importantly, unlike the growth inhibition produced by bicalutamide, growth inhibition induced by acute exposure to supraphysiologic androgen is not associated with a decrease in the percentage of cells in cycle (i.e., 72+/− 14% Ki-67 positive). Instead, flow cytometric analysis documented that this growth inhibition induced by acute exposure to high androgen is due to a 3-fold increase (p<0.05) in cell death ( i.e., going from 11+/−3 % dying without the high androgen to 34+/−6% of cells dying with 10nM R1881).
Figure 4. Growth and AR response of parental LNCaP cells vs. low androgen adapted LNCaP/A- cells to acute exposure to either anti-androgen or androgen supplementation.
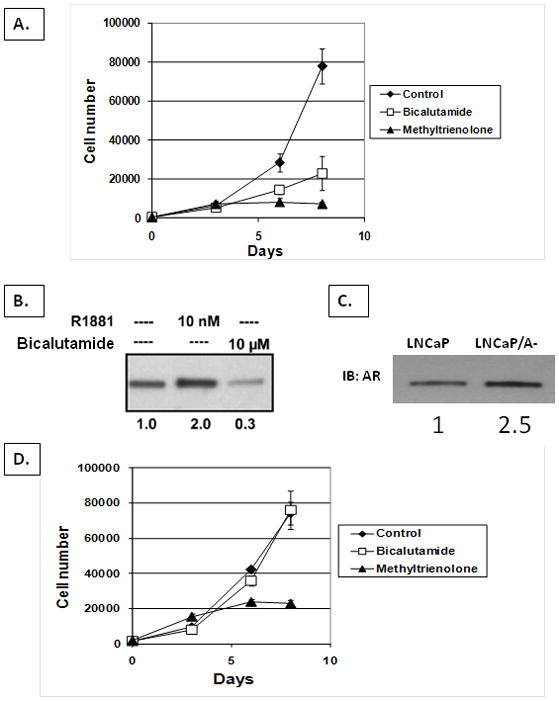
(A) In vitro growth response of LNCaP cells to acute exposure to 10uM of the anti-androgen, bicalutamide or 10nM of the synthetic androgen, methyltrienolone. (B) Western blot of AR in LNCaP cells after indicated treatment. Numbers below each lane are the relative level of AR in untreated vs. bicalutamide or methyltrienolone treated LNCaP cells. (C) Western blot of AR in LNCaP vs. LNCaP/A- cells. Numbers below each lane are the relative level of AR in LNCaP vs. LNCaP/A- cells. (D) In vitro growth response of LNCaP/A- cells to acute exposure to 10uM of bicalutamide or 10nM of methyltrienolone.
These results document that acute exposure to a high level of androgen directly induces death of CRPC cells initially expressing a high level of AR. To further evaluate whether this association between high AR expression and androgen induced death is causally linked, LNCaP cells were chronically maintained in phenol red free RPMI-1640 media with 10% DCC stripped FBS which has an even lower level of androgen than in androgen ablated patients (39). These cells, termed LNCaP/A- cells adapt to growth in the lower androgen media by additionally increasing their expression of AR protein by 2.5-fold compared to the parental LNCaP cells, Figure 3C. This results in LNCaP/A- cells expressing an AR level that is 75 times higher than normal prostate epithelial cells and 25 times higher than hormone naïve prostate cancers. Associated with this 75-fold increase in AR is complete resistance to growth inhibition by bicalutamide, Figure 4D, even though anti-androgen treatment does lower PSA secretion 2.6-fold (p<0.05) from 54+/−7ng PSA/106cell/day to 21+/−5ng PSA/106cell/day. While resistant to bicalutamide, the growth of these LNCaP/A- cells is still profoundly (p<0.05) inhibited by acute exposure to a supraphysiologic level of androgen (i.e., 10nM R1881), Figure 4D. This paradoxical androgen induced growth inhibition is not associated with a decrease in the percent of cells in cycle based upon expression of the cell cycle marker Ki-67 on day 8 (i.e., 85+/− 9% in R1881 treated vs. 91+/− 5% in untreated cells), but is associated with a greater than 3-fold (p<0.05) increase in cell death index (i.e., 8+/− 2 % in untreated cells vs. 27+/− 6% dying in R1881 media). Associated with this paradoxical androgen-induced increase in death is a dramatic increase (p<0.05) in the percent of flow sorted mitotic cells which retain detectable AR expression (i.e., 85+/− 11% vs. > 0.5% in untreated cells). These growth inhibition changes, however, are not associated with decrease secretion of the AR-dependent PSA gene into the media (i.e., 54+/−7ng PSA/106 cells/day in untreated cells vs. 84+/− 35 ng PSA/106cells/day in R1881 treated cells) documenting that AR-dependent transcription is not inhibited by added androgen. These combined results document that CRPC cells have the ability to adaptively up-regulate their AR level for optimal growth under decreasing androgen environment, but if the androgen level is acutely increased, paradoxically, such adaptive auto-regulation is a liability though over-stabilization of AR in mitosis.
Paradoxical death of castrate resistant prostate cancer cells induced by acute exposure to supraphysiologic androgen involves AR binding to replication complexes
Previous studies documented that in CRPC cells whose proliferation is stimulated by AR signaling, AR must be degraded sufficiently in mitosis or these cells do not completely re-license their DNA for replication completely and thus die in the next cell cycle (12). These results suggest that AR is a licensing factor needed for DNA replication in CRPC cells. This would explain why these malignant cells adaptively auto-regulate to maintain an optimal level of nuclear AR for DNA replication under varying environmental androgen levels. This would also provide a mechanism for the paradoxical death induced by acute switching of CRPCs from castrate to a supraphysiologic level of androgen via over stabilizing AR/RC complexes preventing full re-licensing in the next cell cycle.
The possibility that AR becomes a licensing factor in CRPC cells is supported by co-IP/IB analysis which have documented that AR binds RCs in proliferating human LNCaP cells (13). To evaluate whether this is a general observation or unique to LNCaP cells, similar co-IP/IB analysis of AR binding to RC was performed on additional well-characterized human CRPC in vitro lines were evaluated, all of which are routinely maintained in vitro in media supplemented with 10 % FBs containing a castrate level of androgen (39). To evaluate whether AR binds with the RC in CRPCs, a proprietary protocol was used to extract RC proteins from the nuclei of 3 additional CRPC lines (i.e., LAPC4, VCaP, and CWR22Rv1), beside LNCaP, whose growth is also stimulated by AR even when grown in a low androgen environment (i.e., 10% FBS containing media). This protocol fragments nuclear DNA into sizes of less than 100 base pairs (bp), Figure 5A, allowing efficient extraction of DNA-bound multi-protein complexes. The fact that the DNA is less than 100bp is significant since this limits non-specific co-IP of unrelated multi-protein complexes bound to different, but closely located DNA binding locations. Nuclear extracts were initially analyzed by IB for AR protein. These results (i.e., first lane in Figure 5B) confirm that AR is present in the nuclei of these growing CRPCs even in media containing only castrate level of testosterone. Additional aliquots of these chromatin extracts were then incubated with the validated anti-Orc2, Mcm2, and Cdc6 antibodies to co-IP RC associated proteins. In all 4 human CRPC lines tested, AR is associated with RC licensing proteins Orc2, Mcm2, and Cdc-6, Figure 5B. The association of AR with RC proteins is similarly detected using AR specific antibody for co-IP followed by IB for the RC licensing proteins, Figure 5C.
Figure 5. AR binds RC proteins in AR-dependent but not AR-independent human castrate resistant prostate cancer cells.
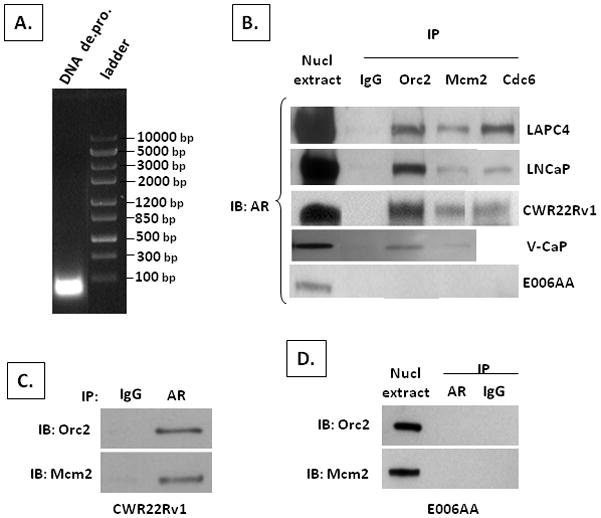
(A) Average base pair (bp) size of DNA in nuclear extract. (B) IB of AR in nuclear extracts (denoted nucl extract) and from co-IP using a non-specific IgG antibody (denoted IgG) vs. antibody specific for either Orc2, Mcm2, or Cdc6 on nuclear extracts from indicated human prostate cancer line (C) IB for Orc2 and Mcm2 from co-IP using an AR specific antibody on nuclear extracts of CWR22Rv1 cells. (D) IB for Orc2 and Mcm2 in nuclear extracts (denoted nucl extract) and from co-IP using an AR specific antibody (denoted AR) vs. a non-specific IgG (denoted IgG) on nuclear extracts from E006AA cells.
To determine the biological specificity of association of AR with RC proteins in CRPC lines, similar co-IP studies were preformed with E006AA cells. This line was established from a localized prostate cancer from a hormonally naïve patient (36). Like the other lines derived from hormonally naïve patients, E006AA cells expresses AR at a level comparable to normal PrECs, Figure 2A, even though these cells have amplified their AR gene and are AR-independent due to a mutation in the AR dimerization domain rendering AR non-functional for growth stimulation or PSA expression (24). Despite this mutation, however, AR is present within the nuclei of E006AA cells even when grown in castrate level testosterone, Figure 5B. There is no binding between AR and RC licensing proteins in these AR-independent E006AA cells regardless of whether antibodies for the RC proteins, Figure 5B, or AR, Figure 5D, are used for co-IP.
To further document that binding of AR with RC proteins is not an artifact of cellular fractionation, LNCaP cells were exposed to the cell-permeable cross-linker dithiobis[succinimdylpropionate] (DSP) before isolating nuclei. DSP was used because previous studies documented that exposure of cells to this agent results in crosslinking RC proteins without crosslinking protein to DNA (32). DSP treated cells were lysed in RIPA buffer which contains 0.1 % SDS and a series of detergents to disrupt non-specific protein-protein interactions. These RIPA buffered lysates were then evaluated by IP/IB for the co-incident presence of RC proteins and AR. DSP treatment cross links Orc2 to RC proteins, like Mcm2, Figure 6A in AR dependent LNCaP confirming previous reports with other cell types (32). Orc2 is also cross linked to AR in the AR dependent LNCaP cells, Figure 6A, documenting that AR is bound to RC even before the cells are fractionated.
Figure 6. AR binding to Orc2 is chemically cross-linkable and present in castration resistant metastatic prostate cancer tissue directly from patients.
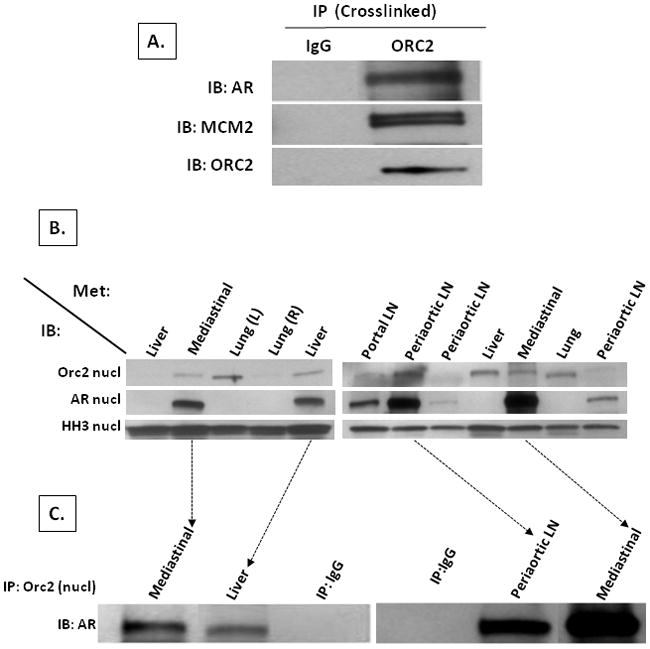
(A) LNCaP cells were treated for 30 minutes with 0.5mM of the cell-permeable cross-linker dithiobis[succinimdylpropionate] and whole cell extracts made with RIPA buffer. IB for AR and Mcm2 from co-IP using a non-specific IgG (denoted IgG) vs. Orc2 specific antibody (denoted ORC2) on these RIPA buffer whole cell extracts. (B) IB for Orc2 and AR in nuclear extracts from human metastatic prostate cancer tissues obtained from indicated sites at rapid autopsy. Histone H3 (denoted HH3) expression was evaluated as a marker of nuclear extract efficiency. (C) IB of AR from co-IP using non-specific IgG (denoted IP:IgG) vs. Orc2 specific antibodies on nuclear extracts of the metastatic prostate cancer tissue with the highest nuclear AR expression.
To investigate whether AR binds with RC licensing factors is an artifact of cell culture, metastatic prostate cancers harvested at rapid autopsy from CRPC patients with no in vitro culturing was initially evaluated for expression of AR and Orc2 proteins, Figure 6B. Seven out of 12 castration-resistant metastatic tissues had a sufficient growth fraction to allow detection of nuclear Orc2 expressed by the small subset of malignant cells in cycle. This remarkably low growth fraction (i.e., median % Ki-67 positive cancer cells is 4.6) and variable (i.e., range % Ki-67 positive cancer cells is 0.25–26) is characteristic of metastatic CRPC (5, 40). Interestingly, the 7 tissues which had detectable nuclear Orc2 were the tissues with the highest AR expression, Figure 6B. In the 4 metastatic tissues with the highest nuclear AR, co-IPs of nuclear extracts with anti-Orc2 antibody demonstrated specific binding between AR and Orc2, Figure 6C, confirming that the previous cell line based results are not an artifact of cell culturing.
To determine when during the cell cycle androgen occupied AR binds with RCs, LNCaP cells were synchronized via culturing in isoleucine-free media containing 6% dialyzed FBS plus 1nM testosterone for 40 hrs (33). This protocol arrests cells in G0/early-G1 as documented by the decline (p<0.05) in the percent of cells expressing Ki-67 [i.e., a marker present in all parts of the cell cycle except G0 (41)] from 86 +/− 11% to 5+/− 1% within 40 hrs of synchronization. This induced G0-arrest is also confirmed by the determination that < 5% of the cells incorporate BrdUrd as opposed to 38+/− 7% of unsynchronized cells incorporating BrdUrd. Upon addition of regular growth media containing isoleucine, 10% FBS and 1nM testosterone, the G0/early-G1 arrested cells are released and progress through the cell cycle undergoing peak DNA replication as determined by BrdUrd incorporation between 24–36 hrs. Thus cells were harvested at various times during the first 48-hour post G0/early-G1 release and lysates analyzed by IB, Figure 7A. These results document that Orc2, Cdt1, and Mcm2 are expressed in G0/early-G1 and their expression remains constant throughout G1-progression, as reported previously (42). In contrast, AR, Orc1, Cyclin D1, Cdk4, Cdc6, and Cdk2 expression are low in early-G1 and increases during progression through G1, as reported previously (11, 19, 20, 42–44). AR and Orc1 reach maximum expression within two hours of G1-progression while Cyclin D1 and Cdk4 peak at eight hr, Figure 7A. Induction of maximal expression of Cdc6 and Cdk2, which are markers of mid-G1 and entrance into S-phase respectively (19), occurs by twenty-four hours of release, Figure 7A.
Figure 7. AR binds with RCs in early-G1 in AR-dependent castration-resistant prostate cancer cells.
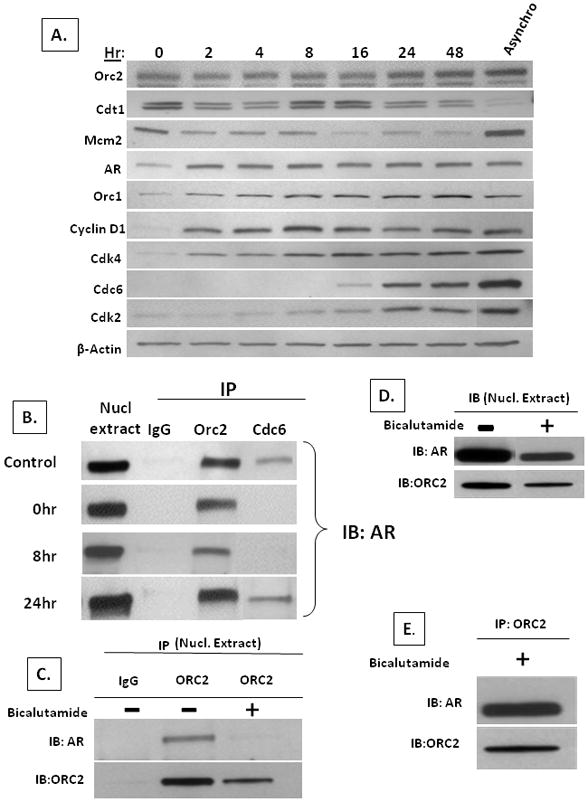
(A) Temporal change in expression detected by IB of Orc2, Cdt1, Mcm2, AR, Orc1, Cyclin D1, Cdk4, Cdc6, and Cdk2 in LNCaP cells progressing through cycle after release from early-G1 arrest. Protein lysates from 105 cells were loaded per lane. Expression in asynchronously growing LNCaP cells (denoted Asynchro). (B) IB of AR from nuclear co-IP using non-specific IgG (denoted IgG) vs. specific antibody to either Orc2 or Cdc6 in asynchronously growing LNCaP cells (denoted control) vs. cells arrested in early-G1 (denoted 0hr) or 8, or 24 hr post-release. (C) IB of AR and ORC2 from nuclear co-IP using non-specific IgG (denoted IgG) vs. specific antibody to Orc2 in asynchronously growing LNCaP cells (denoted bicalutamide -) vs. cells treated for 4 days with 10uM bicalutamide (denoted bicalutamide+). (D) IB of AR and ORC2 from nuclear extracts of bicalutamide untreated (denoted-) vs. treated (denoted+) from (C). (E) IB of AR and ORC2 from nuclear co-IP using specific antibody to Orc2 on LNCaP cells treated for 4 days with 10uM bicalutamide.
Based upon these temporal expression studies, LNCaP cells were arrested in G0/early-G1 and harvested at 0, 8 and 24 hours post release into cycle. Using anti-Orc2 and anti-Cdc6 antibodies, co-IPs were performed on nuclear lysates and analyzed for the presence of AR at these specified time points. These studies documented that nuclear AR is bound with Orc2 at all of the time points; however, nuclear AR is only associated with Cdc6 as cells enter mid-G1 phase (i.e., at 24-hours post release), Figure 7B. These results demonstrate that nuclear AR initially binds with RCs in early-G1 as the pre-RC licensing complexes are forming.
To determine whether such AR binding to RCs is required for AR-dependent CRPC’s proliferation, LNCaP cells were exposure for 4 days to 10uM of the anti-androgen antagonist, bicalutamide which inhibited AR stimulated growth, Figure 7A. Using anti-Orc2 antibody, co-IPs were performed on nuclear lysates from these bicalutamide treated vs. untreated cells and analyzed for the presence of AR. This treatment results in the inhibition of AR binding to Orc2, Figure 7C, even though Orc2 and AR are still present within the cell nuclei, Figure 7D. Coincident with this bicalutamide-induced inhibition of AR binding to Orc2, 90% of the cells arrest in G0/early-G1as documented by only 10+/−3% of the bicalutamide treated cells express Ki-67. Such bicalutamide-induced G0/early-G1arrest is also confirmed by the determination that < 5% of the cells incorporate BrdUrd. These results support that AR binding to RC is required for proliferation of LNCaP cells.
If this conclusion is correct, then in LNCaP cells acquiring resistance to bicalutamide inhibition of their growth, AR should still bind to RCs even in the presence of bicalutamide. To test this prediction, the bicalutamide-resistant LNCaP/A- variant described earlier was used. These cells adapted to growth in extremely low androgen by up-regulating their AR expression by 2.5 fold, Figure 3C, allowing these cells to grow at an identical rate in media with or without the addition of bicalutamide, Figure 3D. These bicalutamide-resistant LNCaP/A- cells were exposed for 4 days to bicalutamide during which time neither their growth nor AR binding to Orc2 is inhibited, Figure 7E, documenting that such AR binding to RC is not a non-specific effect but is tightly coupled with proliferation of these CRPCs. In addition, these results document that adaptive AR elevation allows AR in LNCaP/A- cells to continue to bind to RCs even in an extremely low androgen environment.
Bipolar Androgen Therapy (BAT) Increase Death of CRPCs in Xenografts
The previous results document that the characteristic AR auto-regulation by CRPCs paradoxically becomes a liability, when a supraphysiological level of androgen is acutely replaced since it prevents timely and complete degradation of AR bound at the ORS, stalling DNA re-licensing in the next cycle. These results provide a rationale for testing whether sequential cycling between periods of acute supraphysiologic followed by acute ablated androgen can be used to take advantage of the unique vulnerability produced by adaptive auto-regulation of AR and it’s binding to RC in CRPC cells. To determine whether such bipolar androgen therapy (BAT) is therapeutic against CRPC in vivo, the previously described LNCaP/A- cells were xenografted into castrated NOG mice and half of the animals were implanted with two testosterone (T) filled-silastic capsules. These T-implants rapidly elevate (p<0.05) serum testosterone by more than 60-fold going from< 300 pM in the untreated castrates (n=10) vs.18.3+/−1.7nM in the T-implanted mice (n=10) within 1 day. This elevated androgen levels is supraphysiologic for NOG mice since serum T in intact adult NOG mice (n=50) is only 3–4nM, but this elevated level would be in the high normal range (i.e., 10–35nM) for humans (39). After two weeks of high androgen replacement, these implants were removed causing the high serum T to acutely decrease to <0.3nM within 1day. After two weeks of low androgen, the group initially given T-implants were re-implanted to cyclically re-elevate the serum T to >18nM and a week later, serum and tumor tissue was harvested. This cyclic BAT results in greater than 70% inhibition (p>0.05) of LNCaP/A- tumor growth, Figure 8A. However, in the setting of this androgen induced growth inhibition, serum PSA level normalized per gram of tumor tissue increases nearly 4-fold when the supraphysiological level of androgen is present (i.e., from 24.5+/− 3.3 ng/ml per gram of tumor in un-supplemented castrates to 95.5 +/− 25.2 ng/ml per gram of tumor in androgenized animals).
Figure 8. Morphologic, Ki-67, and AR Response of casodex resistant LNCaP/A-to Bipolar Androgen Therapy.
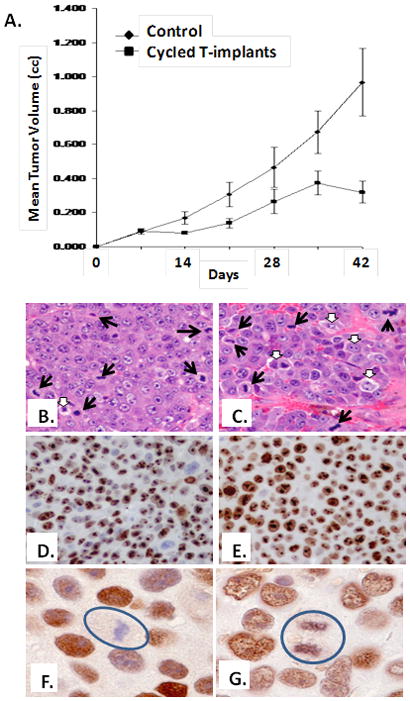
(A) In vivo growth response of LNCaP/A- cells to bipolar androgen therapy. Castrated male NOG mice were inoculated with LNCaP/A- cells and half (n=10) of the mice received nothing further and the other half of the castrated animals (n=10) received bipolar androgen therapy consisting of implantation with testosterone filled capsules (n=10) which rapidly produce a super-physiologic level of androgen. After 2 weeks of a high level, the serum androgen was then rapidly reduced to a castrate level by removing the T-implants and after 2 additional weeks of low androgen, the animals were re-implanted with T-capsules. H&E histology (B) in untreated castrates vs. (C) castrates treated with bipolar androgen therapy as described in (A). Black arrows indicate cancer mitoses and white arrows indicate dying cancer cells. Cancer cell Ki-67 expression is high in both in untreated castrates (D) vs. castrates treated with bipolar androgen therapy (E). AR is undetectable in cancer cell mitosis, denoted by circle, in untreated castrates (F) vs. AR expression in mitosis, denoted by circle, in castrates treated with bipolar androgen therapy (G).
The growth inhibited cancers in the animals receiving BAT had a slightly lower mitotic index which was 0.32+/− 0.04 % vs. 0.51+/− 0.06% in the non-cycled castrate mice, Figure 8B&C, but a similar percentage of cancer cells in cell cycle (i.e., both had >75% of cells expressing Ki-67), Figure 8D&E. These results are consistent with growth inhibition in the animals treated with BAT being due not to a decrease in proliferation but instead to an increase in tumor cell death as the cells progress through cell cycle. This was confirmed based upon an increase (p<0.5) in the cell death index which was 15+/− 4.3% in un-supplemented vs. 39+/−8.2% in cycled mice. In addition, this enhanced death response is associated with an increase (p<0.5) in the percentage of cells in S-phase which was 15+/− 7 % in castrated animals vs. 30+/−6% in androgen-cycled animals; coupled with a decrease (p<0.05) in cells in G0/G1 which was 49+/−9% vs. 25+/− 8 % and G2/M which was 17+/−2% vs. 5 +/−3% in castrated vs. androgen cycled host, respectively. Immunocytochemistry documented that AR is present within the nuclei of LNCaP/A- cells even when they are growing in un-supplemented castrated hosts, Figure 8F. While bipolar androgen cycling did not increase the percentage of cells with nuclear AR (i.e., >90% for both situations), it did induced the aberrant present of AR in mitotic cells, Figure 8G. This is unlike the situation in un-supplemented castrates where AR is never present in any mitotic cells, Figure 8F.
Discussion
The present studies document that human prostate cancer cells stochastically adapt to castration by a >25-fold increase in AR protein level and that this increase is coupled with AR acquiring an oncogenic role in DNA replication via binding to replication complexes (RC) during early G1 licensing. It is unclear as to why AR binding to RC is apparently selected for as an oncogenic event in CRPC cells. This question is presently being studied. Regardless of the answer, the present studies have also discovered that such an adaptive AR increase can become a liability in a castrated host if supraphysiologic androgen is acutely replaced since this induces death of these cells. Mechanistically, this paradoxical therapeutic response involves androgen’s ability to further increase and stabilize the enhanced level of nuclear AR in CRPC cells to a point where insufficient removal of AR/RC from ORS occurs in mitosis. Since this is needed for full re-licensing, cell death is induced in S-phase of the subsequent cell cycle.
Translating these pre-clinical in vitro and in vivo results has lead to the development of a paradigm shifting approach away from chronic androgen ablation to a protocol in which patients progressing on androgen ablation are acutely cycled between sequential periods of exposure to supraphysiologic androgen followed by androgen ablation. In addition, while these pre-clinical studies were in process, Haffner et al published results demonstrating that repletion of androgen to androgen-depleted cells resulted in DNA double strand breaks (DSBs) that were mediated through the recruitment of AR and topoisomerase II beta (TOP2B) to AREs (45). These DNA double strand breaks can be stabilized with the use of etoposide, a potent TOP2B inhibitor. Therefore, on the basis of the combined results demonstrating effects of supraphysiologic androgen on re-licensing and potential for production of stabilized DSBs in conjunction with etoposide, we have designed a clinical trial in which castrated men are given intramuscular injections of 400 mg testosterone cypionate to rapidly produce supraphysiologic levels of serum T that are maintained for at least 2 weeks post-injection These men also received two weeks dosing with oral etoposide beginning with each new injection.
Overall this regimen has been well tolerated with the major side effect being nausea induced by the etoposide. The high dose T is well-tolerated in these patients with relatively low metastatic burden with none of the first 6 patients experiencing worsening of pain or other prostate cancer- related morbidity. The dose of 400 mg testosterone cypionate produces supra-phyisologic levels. However, while serum T approached near-castrate levels at the end of each cycle, no men achieved true castrate levels while on this cycling therapy. Two of the first four patients who completed 3 cycles of treatment experienced a >50% overall decline in their PSA levels. While these initial results demonstrate the safety of the regimen and suggest a potential for therapeutic benefit, more patients must be treated to better determine the benefits and toxicity associated with this BAT approach and, therefore, accrual to this trial is ongoing. In addition, in future trials, consideration will be given to design a regimen that produces the maximal BAT through the cyclical production of acute increases in serum T to supraphysiologic levels followed by a rapid return to a more complete androgen-depleted condition.
Acknowledgments
Grant support: NIH Grant R01DK52645, NIH Prostate SPORE Grant 2P50-CA58236, the Maryland Stem Cell Research Fund MSCRF-II-0428 and the One-in-Six Foundation, Akron, OH generously supported this research. Jason D’Antonio was supported by a Urology Training Grant (NIH T32DK07552-21A1).
We wish to thank Lee Blosser and Ada Tam of the Johns Hopkins Flow Cytometry Core for the FACS sorting, and Dr. Angelo DeMarzo and Jessica Hicks of the Johns Hopkins Cancer Center Histology Core for cell and tissue processing and immunocytochemical staining. We also wish to thank Dr. Angelo DeMarzo and Helen Fador of Department of Pathology, the Johns Hopkins School of Medicine for generously providing normal and malignant prostate tissues from surgical specimens as part of the SPORE Tissue Resource Core at Hopkins; Robert Getzenberg of the Brady Urological Institute, the Johns Hopkins School of Medicine for generously providing the organ donor normal prostate tissues; Dr. Ken Pienta of the University of Michigan for generously providing the VCaP cell line and metastatic prostate cancer tissue samples; Dr. Alan Meeker of The Johns Hopkins School of Medicine for generously providing the LNCaP/A- cell line; Dr. Fritz Schroeder of Erasmus University, Rotterdam for providing the PC82 xenograft; Dr Thomas Pretlow of Case-Western Reserve School of Medicine for generously providing the CWR22 xenograft; Dr. Shahriar Koochepour of the LSU Health Science Center, School of Medicine for generously providing the E006AA cell line; Dr D. A. Troyer of U Texas Health Science Center for generously providing the PacMetUT1 cell line; and Drs. William Isaacs, William Nelson, Angelo DeMarzo, Robert Getzenberg and Alan Meeker from The Johns Hopkins School of Medicine for the critical review and suggestions of this manuscript.
Abbreviations
- AR
androgen receptor
- ARE
androgen response element
- BAT
Bipolar Androgen Therapy
- Cdc6
cell division cycle 6 homolog
- CDK
cyclin-dependent kinase
- Cdt1
chromatin licensing and DNA replication factor
- DBD
DNA binding domain
- LBD
Ligand binding domain
- Mcm
mini-chromosome maintenance
- ORC
origin recognition complex
- ORS
origin of replication sites
- Pre-RC
pre-replication complex
- RC
replication complex
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972–82. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 2.Vander Griend DJ, Litvinov I, Isaacs JT. Androgen Receptor (AR) suppresses human prostate epithelial cell proliferation via AR/β-Catenin/TCF-4 complex inhibition of c-Myc transcription. PLos-one. 2012 doi: 10.1002/pros.22828. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 6.Vander Griend DJ, D’Antonio J, Gurel B, et al. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells. Prostate. 2010;70:90–9. doi: 10.1002/pros.21043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–13. [PubMed] [Google Scholar]
- 8.Li TH, Zhao H, Peng Y, Beliakoff J, et al. A promoting role of androgen receptor in androgen-sensitive and -insensitive prostate cancer cells. Nucleic Acids Res. 2007;35:2767–76. doi: 10.1093/nar/gkm198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snoek R, Cheng H, Margiotti K, et al. In vivo knockdown of the androgen receptor results in growth inhibition and regression of well-established, castration-resistant prostate tumors. Clin Cancer Res. 2009;15:39–47. doi: 10.1158/1078-0432.CCR-08-1726. [DOI] [PubMed] [Google Scholar]
- 11.Litvinov IV, Vander Griend DJ, Antony L, et al. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc Natl Acad Sci U S A. 2006;103:15085–90. doi: 10.1073/pnas.0603057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vander Griend DJ, Litvinov IV, Isaacs JT. Stabilizing androgen receptor in mitosis inhibits prostate cancer proliferation. Cell Cycle. 2007;6:647–51. doi: 10.4161/cc.6.6.4028. [DOI] [PubMed] [Google Scholar]
- 13.D’Antonio J, Vander Griend D, Isaacs J. DNA licensing as a novel androgen receptor mediated therapeutic target for prostate cancer. Endocr Relat Cancer. 2009;16:325–32. doi: 10.1677/ERC-08-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annu Rev Biochem. 2002;71:333–74. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 15.Tabancay AP, Forsburg SL. Eukaryotic DNA replication in a chromatin context. Curr Top Dev Biol. 2006;76:129–84. doi: 10.1016/S0070-2153(06)76005-7. [DOI] [PubMed] [Google Scholar]
- 16.Siddiqui K, Stillman B. ATP-dependent assembly of human origin recognition complex. J Biol Chem. 2007;282:32370–32383. doi: 10.1074/jbc.M705905200. [DOI] [PubMed] [Google Scholar]
- 17.Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol. 2005;6:476–86. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arias EE, Walter JC. Strength in numbers: preventing re-replication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 19.Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122:915–926. doi: 10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 20.Fujita M. Cdt1revisited: complex and tight regulation during the cell cycle and consequences of deregulation in mammalian cells. Cell Div. 2006;1:22. doi: 10.1186/1747-1028-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai VU, Cifuentes E, Menon M, et al. Androgen receptor regulates Cdc6 in synchronized LNCaP cells progressing from G1 to S phase. J Cell Physiol. 2005;204:381–7. doi: 10.1002/jcp.20422. [DOI] [PubMed] [Google Scholar]
- 22.Kemppainen JA, Lane MV, Sar M, Wilson EM. Androgen receptor phosphorylation, turnover, nuclear transport, and transcriptional activation. Specificity for steroids and antihormones. J Biol Chem. 1992;267:968–74. [PubMed] [Google Scholar]
- 23.Vander Griend DJ, Karthaus WL, Dalrymple S, et al. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008;68:9703–11. doi: 10.1158/0008-5472.CAN-08-3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Antonio JM, Vander Griend DJ, Antony L, et al. Loss of Androgen Receptor-Dependent Growth Suppression by Prostate Cancer Cells Can Occur Independently from Acquiring Oncogenic Addiction to Androgen Receptor Signaling. PLoS ONE. 2010;5:e11475. doi: 10.1371/journal.pone.0011475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001;61:5038–44. [PubMed] [Google Scholar]
- 26.Uzgare AR, Isaacs JT. Enhanced redundancy in Akt and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004;64:6190–9. doi: 10.1158/0008-5472.CAN-04-0968. [DOI] [PubMed] [Google Scholar]
- 27.Prakash K, Pirozzi G, Elashoff M, et al. Symptomatic and asymptomatic benign prostatic hyperplasia: molecular differentiation by using microarrays. Proc Natl Acad Sci U S A. 2002;99:7598–603. doi: 10.1073/pnas.112191399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bova GS, Fox WM, Epstein JI. Methods of radical prostatectomy specimen processing: a novel technique for harvesting fresh prostate cancer tissue and review of processing techniques. Mod Pathol 1993. 1993;6:201–7. [PubMed] [Google Scholar]
- 29.Liu W, Laitinen S, Vihinen M, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med. 2009;15:559–65. doi: 10.1038/nm.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn TA, Chen S, Faith DA, et al. A novel role of myosin VI in human prostate cancer. Am J Pathol. 2006;169:1843–54. doi: 10.2353/ajpath.2006.060316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Litvinov I, Antony L, Dalrymple SL, et al. PC-3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006;66:1329–38. doi: 10.1002/pros.20483. [DOI] [PubMed] [Google Scholar]
- 32.Fujita M, Ishimi Y, Nakamura H, et al. Nuclear organization of DNA replication initiation proteins in mammalian cells. J Biol Chem. 2002;277:10354–361. doi: 10.1074/jbc.M111398200. [DOI] [PubMed] [Google Scholar]
- 33.Cifuentes E, Croxen R, Menon M, et al. Synchronized prostate cancer cells for studying androgen regulated events in cell cycle progression from G1 into S phase. J Cell Physiol. 2003;195:337–45. doi: 10.1002/jcp.10317. [DOI] [PubMed] [Google Scholar]
- 34.Hoehn W, Schroeder FH, Reimann JF, et al. Human prostatic adenocarcinoma: some characteristics of a serially transplantable line in nude mice (PC82) Prostate. 1980;1:95–104. doi: 10.1002/pros.2990010113. [DOI] [PubMed] [Google Scholar]
- 35.Wainstein MA, He F, Robinson D, et al. CWR22: androgen-dependent xenograft model derived from a primary human prostatic carcinoma. Cancer Res. 1994;54:6049–52. [PubMed] [Google Scholar]
- 36.Koochekpour S, Maresh GA, Katner A, et al. Establishment and characterization of a primary androgen-responsive African-American prostate cancer cell line, E006AA. Prostate. 2004;60:141–52. doi: 10.1002/pros.20053. [DOI] [PubMed] [Google Scholar]
- 37.Troyer DA, Tang Y, Bedolla R, et al. Characterization of PacMetUT1, a recently isolated human prostate cancer cell line. Prostate. 2008;68:883–92. doi: 10.1002/pros.20758. [DOI] [PubMed] [Google Scholar]
- 38.Koivisto P, Kononen J, Palmberg C, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–9. [PubMed] [Google Scholar]
- 39.Sedelaar JP, Isaacs JT. Tissue culture media supplemented with 10% fetal calf bovine serum contains a castrate level of testosterone. Prostate. 2009;69:1724–9. doi: 10.1002/pros.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]