

Abstract
Previously, we showed that transient transfection with OCT4 not only produced high expression of Oct4 in skin keratinocytes, but also caused a generalized demethylation of keratinocyte DNA. We hypothesized that DNA demethylation alone might allow expression of endogenous OCT4. Here, we report that treatment with the cancer drug decitabine results in generalized DNA demethylation in skin keratinocytes, and by 48 hours after treatment, 96% of keratinocytes show expression of the endogenous Oct4 protein and the OCT4 repressor mir-145. This is true for keratinocytes only, as skin fibroblasts treated similarly show no OCT4 or mir-145 expression. Decitabine-treated keratinocytes also show increased mir-302c and proliferation similar to other Oct4+ cells. Treatment with doxorubicin, another cancer drug, induces expression of mir-145 only in cells that already express OCT4, suggesting that Oct4 regulates its own repressor. Co-treatment with decitabine and doxorubicin results first in increased OCT4 and mir-145, then a decrease in both, suggesting that OCT4 and mir-145 regulate each other. The novel strategy presented here provides a regulatable system to produce Oct4+ cells for transformation studies and provides a unique method to study the effects of endogenous Oct4 in cancer cells and the surrounding somatic cells.
Keywords: DNA demethylation, decitabine (also known as 5-aza-2′-deoxycytidine), doxorubicin, keratinocytes, Oct4, mir-145
INTRODUCTION
In the last several years, substantial importance has been placed on using cells from patients to make induced pluripotent stem (iPS) cells that could be used to repair their own damaged tissues. Several current studies report various technologies that differ from those used in the original iPS cell papers.1,2,3,4 However, these strategies remain problematic for human use because they are inefficient and expensive, or they use viral vectors to introduce exogenous DNA into the cells. Thus, development of a safe, efficient, and inexpensive strategy to reprogram cells for use in human therapy is of high importance. Previously, we developed a temporary, non-integrating reprogramming approach, which introduced OCT4 into human skin keratinocytes (HSKs) using a simple plasmid technology. It was temporary in that the transfected HSKs expressed Oct4 and reactivated other endogenous reprogramming factors for only three to four days. This was long enough to allow the HSKs to be transformed into neuronal or mesenchymal cells.5 It was relatively safe because the transfected HSKs only transformed if placed in neuronal or mesenchymal transformation medium. If they were kept in keratinocyte medium, they remained as normal HSKs. Thus, this method only temporarily reprogrammed the HSKs into cells that could be induced to change their differentiation lineage. Although this sounds ideal, it still involved insertion of exogenous DNA into human cells, thereby making the method problematic for human therapy.
During this previous work, we noted that transient expression of Oct4 in HSKs resulted in global demethylation in HSK DNA and expression of the endogenous Oct4, Sox2 and Nanog.5 This raised the question whether DNA demethylation alone might be sufficient to reactivate endogenous expression of reprogramming factors, such as Oct4, in HSKs, thereby avoiding the addition of exogenous DNA. Although such a possibility sounds unlikely, in 2004 Hattori reported that the demethylation drug decitabine (also know as 5-aza-2′-deoxycytidine) caused reactivation of the OCT4 gene in cultured trophoblast cells.6 A more recent report demonstrated that when the maintenance of DNA methylation was inhibited, progenitor cell fate could be manipulated.7 The drug of choice to produce DNA demethylation is decitabine, which works by incorporating into DNA CpG sites and sequestering the local DNA methyltransferase, resulting in loss of DNA methylation over time.8
Here, we report for the first time that the cancer treatment drug decitabine reactivates endogenous Oct4 and its regulator mir-145,9,10,11 in HSKs. We also show for the first time that another cancer treatment drug, doxorubicin, induces expression of endogenous mir-145 only in cells that already express OCT4. Moreover, expression of Oct4 and mir-145 did not require viral vectors, plasmids, reprogramming factors or extraneous DNA.
MATERIALS AND METHODS
Cell isolation and culture
Primary adult human skin keratinocytes (HSKs) and fibroblasts were isolated from normal skin, obtained from the Surgical Pathology Department at The University of Iowa Hospitals and Clinics with approval of The University of Iowa’s IRB. The skin specimens were not diseased or pathological; they were from amputations after accidents, mammary reductions, or from surgical reductions for obesity. To ensure sterility, strips of skin were soaked for 1 hour in medium containing 10% Antibiotic-antimycotic (Invitrogen, Carlsbad, CA). Epidermal sheets were mechanically separated from the dermal tissue after overnight incubation of the skin in Dispase II (24.0 U/ml, Roche, Indianapolis, IN) at 4°C. HSKs were isolated from the epidermal sheets by treatment in 0.25% trypsin (Invitrogen) for 30 mins. at 37°C. Cells were plated at a concentration of 8×105 cells/ml in keratinocyte serum free medium (KSFM, Invitrogen) + 1.5% Antibiotic-antimycotic on culture dishes coated with 0.25 μg/cm2 collagen type IV (BD Biosciences, Bedford, MA). Fibroblasts were isolated from the dermal sheets by treatment in 3.5% collagenase (Invitrogen). Cells were plated at a concentration of 2×105 cells/ml in DMEM + 10% FBS, 1.5% Antibiotic-antimycotic (all from Invitrogen) on culture dishes.
NTERA-2 cells (ATCC, Manassas, VA) were grown in DMEM + 10% FIBS + 1.5% Antibiotic-antimycotic. NTERA-2 is a pluripotent human testicular embryonal carcinoma cell line with the characteristics of human embryonic stem cells, including continual expression of OCT4.
Cell treatments
For transfection, HSKs were grown at 37°C in a 5% CO2 humidified incubator until 60–70% confluent, then transiently transfected with pMSCV-OCT4 using Turbofectin (Origene, Rockville, MD) at a concentration of 1 μg of plasmid to 2 μl of Turbofectin per cm2 for 48 hours.
For decitabine (Sigma, St. Louis, MO) treatment, Cells were grown at 37°C in a 5% CO2 humidified incubator for 8–24 hours until 80% confluent, then treated with 25 μM decitabine for the indicated amount of time.
For the doxorubicin treatments, NTERA-2 cells and HSKs were grown at 37°C in a 5% CO2 humidified incubator for 8 hours, then treated with doxorubicin for 16 hours. NTERA-2 cells were treated with 0.25 μg/ml doxorubicin. HSKs were treated with 15 μg/ml doxorubicin. Doses for each cell type were determined by a standard dosage curve. The highest dose of doxorubicin that did not kill the cells was used. The HSKs were highly resistant to the doxorubicin treatment as compared to the NTERA-2 cell line, and required a much higher dose to elicit a response.
For the double treatment, HSKs were grown at 37°C in a 5% CO2 humidified incubator overnight. The following morning they were treated with 25 μM decitabine for 31 hours. The next afternoon, the cultures were thoroughly rinsed to remove the decitabine, then treated with 15μg/ml doxorubicin for 16 hours. This resulted in a total treatment time of 47 hours.
Oct4 immunostaining
Cells were rinsed in PBS, then fixed with 4% paraformaldehyde for 20 min. After rinsing cells were permeabilized for 25 min. with 0.2% Triton-X in PBS, then blocked with 4% normal rabbit serum. Cells were then incubated for 2 hours at room temperature with the Oct4 antibody (Santa Cruz, CA, ms anti-OCT3/4 diluted 1:750 in 12% BSA). Cells were rinsed in high salt PBS, followed by rinse in normal PBS, then the secondary anti-mouse Alexa 594 antibody (Invitrogen) was added at a dilution 1:1000 in 12% BSA. After rinsing, the nuclei were stained in DAPI and cover slips mounted with vectashield (Invitrogen). Slides were stored at 4°C until photographed.
OCT4, mir-145, mir-302c expression
For analysis of microRNA expression, approximately 106 HSKs were plated on 100 mm culture dishes and allowed to grow overnight. The next morning cells were transfected or treated with decitabine and/or doxorubicin as described above. Control cells were untreated HSKs and in some cases also GFP-transfected HSKs. After 48 hrs, total RNA was isolated using TRIzol (Invitrogen) as per the manufacturer’s instructions. MicroRNA was collected following the manufacturer’s spin-column protocol (miRNeasy kit, Qiagen, Valencia, CA). cDNA was synthesized using the NCode VILO miRNA cDNA synthesis kit (#A11193-050 Invitrogen) as per manufacturers instructions.
miRNA was labeled and hybridized to the microarray using the GeneExplorer miRNA kit (#1101C-1199C, GenoSensor Corporation, Tempe, AZ) following the manufacturer’s instructions. After hybridization, the chip was dried and stained with 1:250 diluted SA-S dye. After washing and air drying the chip was scanned at the 635 nm Cy5 channel using the GenePix 4000B scanner from Axon. Expression data were analyzed using the GenePix Pro software. Three experiments using different HSKs and different microRNA isolates were performed.
Levels of expression of mir-145, mir-302c, and OCT4 were confirmed via qPCR. For OCT4, the forward and reverse primers, along with the probe, were from Applied BioSystems (Hs01895061_ul). Expression was quantified using the Express SYBR GreenER miRNA qRT-PCR kit (#A11193-051) according to manufacturer’s instructions. The forward primers for mir-145 and mir-302c were from Invitrogen (NCode miRNA Database, #3111 for has-mir-145, #3124 for has-mir-302c). They were used with the universal qPCR reverse primer and the Express SYBR GreenER universal qPCR super mix. 18S was the internal control. qPCR was performed in the BIO-RAD CFX96 Real-Time system C1000 thermal cycler.
Analysis of DNA methylation
To measure DNA methylation, cells were lysed and genomic DNA collected following the spin-column protocol (DNeasy Blood and Tissue kit, Qiagen). Global DNA methylation was measured with a modified enzyme-linked immunosorbent assay using the Methylamp Global DNA Methylation kit (Epigentek, Brooklyn, NY, USA) according to the manufacturer’s protocol. Triplicate samples of genomic DNA were bound in 96-well strip wells. Samples were blocked, and then labeled using an anti-methyl-cytosine primary antibody and an enzyme-conjugated secondary antibody. Following addition of the substrate solution for 2 to 5 min, the reaction was halted with 1N HCl and absorbance at 450 nm was measured. To quantify DNA methylation, samples were background subtracted and normalized to the 100% methylated DNA control samples.
Cell cycle analysis
HSKs were transiently transfected with pMSCV-OCT4-IRES-GFP or treated with decitabine alone. Controls were pMSCV-GFP-transfected HSKs for the OCT4-transfected cells or untreated HSKss for decitabine-treated cells. After 48 hours of treatment, cells were removed from the culture dishes using 0.05% Trypsin/EDTA (Invitrogen), centrifuged, and resuspended at a concentration of 2×106 cells per ml in PBS containing 0.1 mg/ml of propidium iodide. Analysis of DNA content was performed in The University of Iowa’s Flow Cytometry Core Facility using the Becton Dickenson LSR II. Since transfection efficiency was only around 10–20%, transfected cells were sorted for GFP to ensure that only OCT4+ cells or control GFP-transfected cells were analyzed. Decitabine-treated HSKs did not need to be sorted as 96% of these cells expressed Oct4. Experiments were performed three times. Percentage of cells in G1, S, and G2/M phases of the cell cycle was determined. Statistical significance was determined by paired t-test with controls.
RESULTS
Global DNA demethylation by decitabine treatment is sufficient to induce endogenous Oct4 expression in HSKs
We have previously shown that transient expression of Oct4 in human skin keratinocytes (HSKs) induces the expression of endogenous Oct4 and decreases the global DNA methylation state in the treated cells to the level found in the pluripotent human testicular embryonal carcinoma cell line, NTERA-2.5 These findings along with the fact that the NTERA-2 cells show a continual expression of Oct4 suggested to us that Oct4 itself might directly affect the DNA methylation state of the HSKs. In the previous study, we transfected HSKs with a plasmid that expressed OCT4 under a CMV promoter. This resulted in extremely high levels of Oct4 expression in 10–20% of HSKs and required that we sort the cells to eliminate HSKs that did not express Oct4 before determining DNA methylation levels. For the experiments reported here, we wanted to avoid both of these problems and examine the effect of an endogenous level of Oct4 on DNA demethylation. Since it was reported that a short treatment with decitabine inhibits DNA methylation in various cell types,8 we tested this drug’s effect in the HSK cultures. We first treated cultured HSKs with various doses of decitabine and determined that a single dose of 25μM decitabine resulted in DNA demethylation with no cell death. To determine the best time frame, we treated HSKs for one to four days with 25 μM decitabine. Controls were untreated HSKs grown for the same number of days. The decitabine-treated HSKs showed less DNA methylation than their untreated controls, with the 24 hour treatment showing the least amount of DNA methylation (Fig. 1). No cell death was observed in the treated cultures. Next, we determined whether DNA demethylation was sufficient to induce expression of endogenous Oct4 in HSKs. Using immunocytochemical analysis, we examined HSKs treated with 25 μM decitabine for one to four days for expression of the Oct4 protein. Untreated HSKs did not express Oct4. Decitabine-treated HSKs showed nuclear expression of endogenous Oct4 at a much lower level than that seen in OCT4-transfected HSKs (Fig. 2 shows 48 hour samples). Counts of the numbers of Oct4+ cells revealed that at 24 hours of decitabine treatment, 62% of HSKs were Oct4+, and by 48 hours 96% of the HSKs were Oct4+. The percentages dropped to 4% and 2.5% at 3 and 4 days of decitabine treatment, respectively. Three independent experiments were performed with the same results. These combined results confirm that a single 48 hour treatment with 25 μM decitabine results in global demethylation of DNA and a concomitant temporary up-regulation of endogenous Oct4 protein in the HSKs. We also saw that the SOX2 and NANOG mRNAs were upregulated (data not shown). However, in these studies we do not know if this was a direct effect of the DNA demethylation or due to the Oct4 protein since we previously showed that the presence of Oct4 activated both SOX2 and NANOG.5
Figure 1. Treatment with decitabtine demethylates human skin keratinocyte (HSK) DNA.
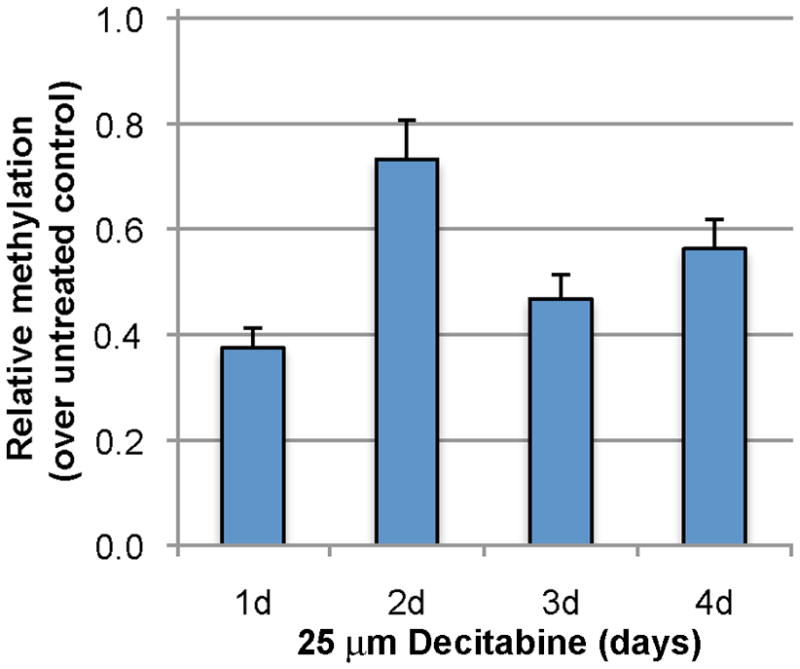
HSKs were cultured with 25 μM decitiabine for the noted number of days. Treated HSK cultures were normalized to untreated HSKs cultured for the same day. Note, HSKs treated for 24 hours show the least amount of methylation (the highest level of demethylation). n = 3 for each time point.
Figure 2. A single treatment with decitabine is sufficient to allow expression of the endogenous Oct4 protein in HSKs.
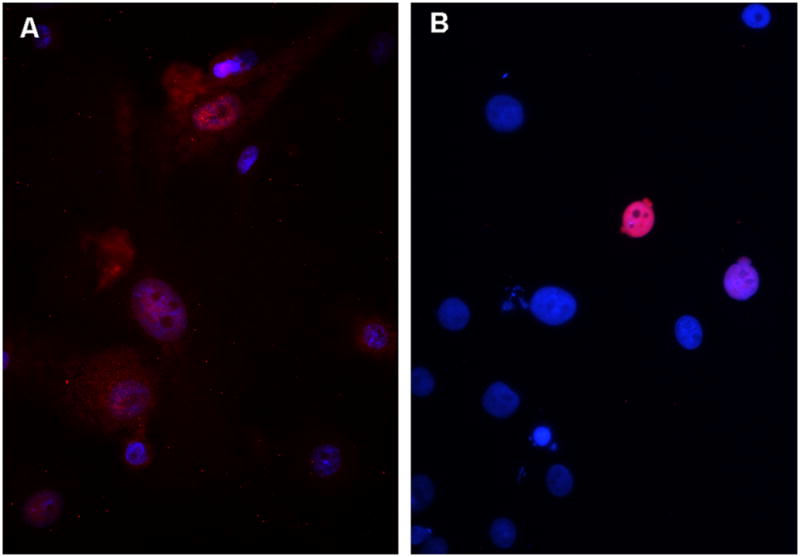
A) Immunofluorescent image of Oct4 expression in HSKs treated with one dose of 25 μM Decitabine for 48 hours. B) Immunofluorescent image of Oct4 expression in HSKs transfected with pMSCV-OCT4 for 48 hours. Note, transfection hits only ~10% of HSKs, and these cells have extremely high expression of nuclear Oct4. Decitabine treatment affects 96% of HSKs, and these cells show a low expression of Oct4. Blue = DAPI. Red/pink = antibody to Oct4.
Treatment with decitabine increases mir-302c levels and induces cells to enter the proliferative phases of the cell cycle
To better understand the regulation of OCT4 in the decitabine-treated cells, we probed for microRNA expression in Oct4+ cells using the GeneExplorer miRNA array from GenoSensor. In three independent experiments, we found twenty-four microRNAs that were changed ten-fold or more in the Oct4-expressing HSKs. One microRNA that showed a change on the miRNA array was mir-302c. In human embryonic stem cells, members of the mir-302 cluster were shown to be regulated by direct binding of Oct4 and to regulate the cell cycle.12,13 Using qPCR, we found that a single 48-hour treatment with 25 μM decitabine increased mir-302c levels in HSKs only 1.9-fold over untreated controls (Fig. 3). Even so, FACS analysis for the amount of DNA content revealed that the HSKs were induced to proceed into the proliferative phases of the cell cycle. The percent of cells in the G1 phase of the cell cycle decreased significantly (p<0.01) from 82% in untreated control HSKs to 72% in the decitabine-treated cells, with a concomitant increase from 17% to 28% of cells in the S/G2/M phases of the cell cycle (Table 1). As a positive control, transfection with pMSCV-OCT4 showed a 6% increase in S/G2/M cells. However, some of this in was due to the transfection itself as the GFP transfection control showed an increase in S/G2/M cells of 3%. These data confirm our anecdotal observation that expression of Oct4 increases cell proliferation. Thus, in decitabine-treated HSKs Oct4 regulates mir-302c as reported for other cells.12
Figure 3. A single treatment with decitabine induces expression of endogenous OCT4, mir-145, and mir-302c in HSKs.
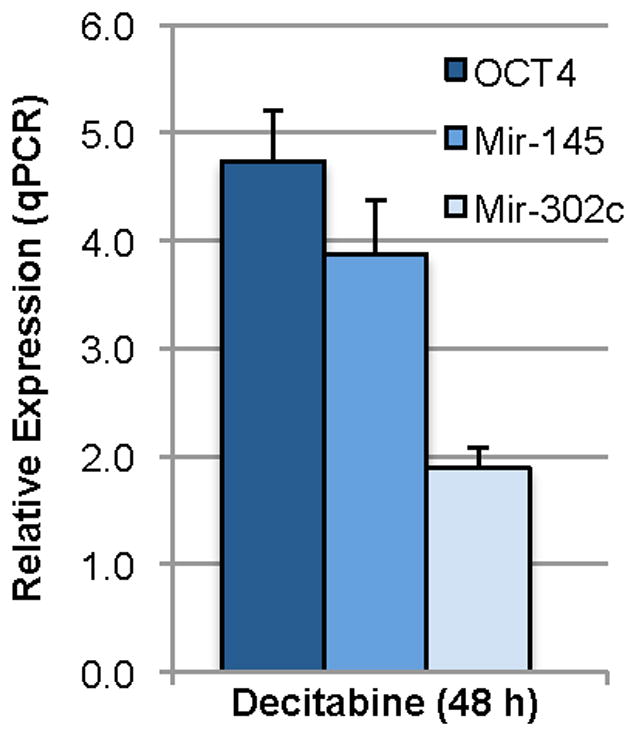
qPCR data from decitabine-treated HSKs were normalized to untreated HSK controls, which were arbitrarily set at 1.00. Decitabine treatment yields ~4.5-fold increase in OCT4, ~3-fold increase in mir-145, and ~2-fold increase in mir-302c expression over untreated control. n = 3 for each group.
Table 1.
Expression of Oct4 induces cells to enter S-phase of the cell cycle
| % G1-phase | % S-phase | % G2/M-phases | |
|---|---|---|---|
| Untreated HSKs | 82.0 ± 9.0 | 7.4 ± 0.9 | 9.9 ± 1.6 |
| GFP tf control | 79.0 ± 5.6 | 9.3 ± 1.7 | 11.1 ± 2.0 |
| OCT4 tf | 78.1 ± 0.8 | 9.0 ± 0.7 | 14.2 ± 3.1 |
| Decitabine | *72.0 ± 2.1 | *13.1 ± 1.3 | 15.0 ± 2.2 |
GFP tf = pMSCV-GFP transfection
OCT4 tf = pMSCV-OCT4 transfection
n = 3 for each group
p<0.01
A single treatment with decitabine increases expression of endogenous OCT4 and mir-145
Another microRNAs that changed in the GeneExplorer miRNA array was mir-145, which has been reported to be part of a double-negative feedback loop with OCT4.9 Moreover, in human embryonic stem (ES) cells not only was OCT4 shown to be a direct target of mir-145, but it was reported that the mir-145 promoter was directly repressed by OCT4.9 Using qPCR, we examined relative levels of OCT4 and mir-145 in the decitabine-treated HSKs. This treatment yielded a 4.7-fold increase in OCT4 expression. However, in contrast to what was reported for human ES cells, the decitabine-treated HSKs showed a 3.9-fold increase in mir-145 over untreated controls (Fig. 3). This appeared to be specific to the HSKs, as human skin fibroblasts treated similarly resulted in no increase in OCT4 and only a slight but non significant increase in mir-145 (data not shown).
mir-145 regulates OCT4 expression in decitabine-treated HSKs
It has been shown that increasing mir-145 expression in human ES cells increases differentiation and represses pluripotency genes, including OCT4.9 In order to determine whether OCT4 in the decitabine-treated HSKs is also regulated by mir-145, we induced expression of mir-145 using doxorubicin, a known cancer drug previously used to induce cells to express mir-145.14 To confirm that mir-145 would decrease OCT4 expression in our system, we first tested doxorubicin for its ability to induce mir-145 expression and to decrease OCT4 expression in the NTERA-2 testicular carcinoma cell line, which continually expresses OCT4. Cells were treated for 16 hours with varying doses of doxorubicin, then examined for expression of OCT4 and mir-145 by qPCR. Expression levels were normalized to untreated NTERA-2 cells. We found mir-145 expression increased in a dose dependent manner with a dose of 0.25 μg/ml showing the greatest increase in mir-145 and a concomitant decrease in OCT4 expression (Fig. 4). This dose was the highest dose that did not produce death in these cancer cells. Thus, in the NTERA-2 cancer cell line which already has OCT4 highly expressed, doxorubicin increases expression of mir-145, which then decreases OCT4 levels in the NTERA-2 cancer cells.
Figure 4. Doxorubicin treatment increases expression of mir-145 and decreases expression of OCT4 in NTERA-2 cells.
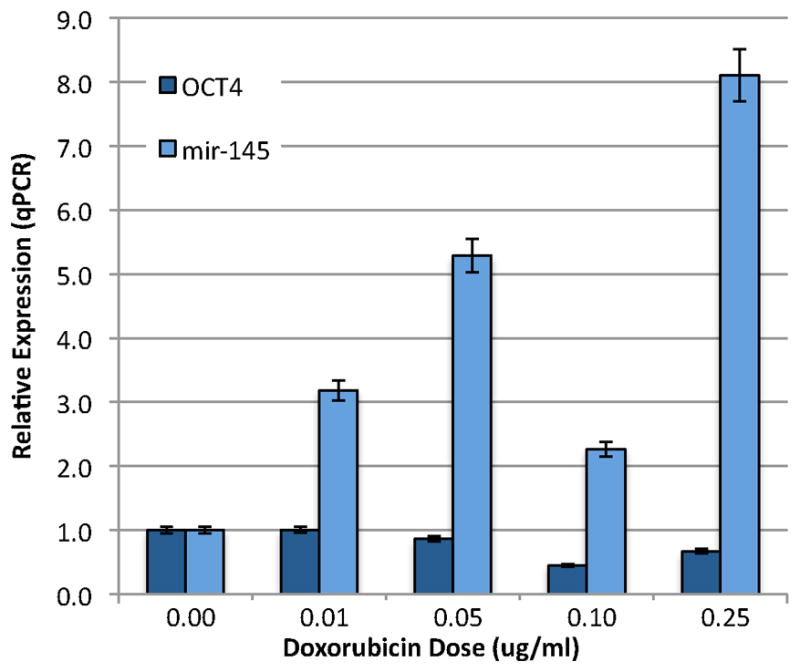
NTERA-2 cells, which continually express OCT4, were treated for 16 hours with the indicated doses of doxorubicin, then examined for expression of mir-145 and OCT4 mRNA by qPCR. Expression levels were normalized to untreated NTERA-2 cells, which were arbitrarily set at 1.0. Note, with increasing doses of doxorubicin, mir-145 expression levels increase with a concomitant decrease in OCT4 expression levels. n = 3 for each group.
Our next step was to test doxorubicin’s effect on HSKs. We performed a standard dosage curve and determined that the highest dose of doxorubicin that did not kill the HSKs was 15 μg/ml. Of note, the dose used for the NTERA-2 cells (0.25 μg/ml) had no effect on the HSKs. Thus, primary HSKs are much more resistant to the doxorubicin treatment as compared to the NTERA-2 cancer cell line. When we treated HSKs with 15 μg/ml doxorubicin for 16 hours, we found no change in expression levels of mir-145 or OCT4 (Fig. 5). This differed completely from the response we saw using the NTERA-2 cells. Since the NTERA-2 cells continually express OCT4, and the HSKs do not express OCT4, we hypothesized that OCT4 might need to be expressed in cells before mir-145 can be expressed, suggesting a regulatory loop. Since a single 48-hour treatment with decitabine increases expression of both OCT4 and mir-145 in the HSKs (Figs. 3 & 5), to test our hypothesis, we treated HSKs with decitabine for 31 hours to up-regulate both OCT4 and mir-145. We followed this with a 16-hour doxorubicin treatment to further increase mir-145 in OCT4+ HSKs, then examined cells by qPCR for expression levels of mir-145 and OCT4. This co-treatment regimen reduced the decitabine-induced increase in OCT4 by 13-fold and the mir-145 levels by 9-fold (Fig. 5), indicating that the repressive effect of mir-145 on OCT4 is more successful in cells that already express OCT4.
Figure 5. HSKs treated with decitabine followed by doxorubicin show reduced expression of mir-145 and OCT4.
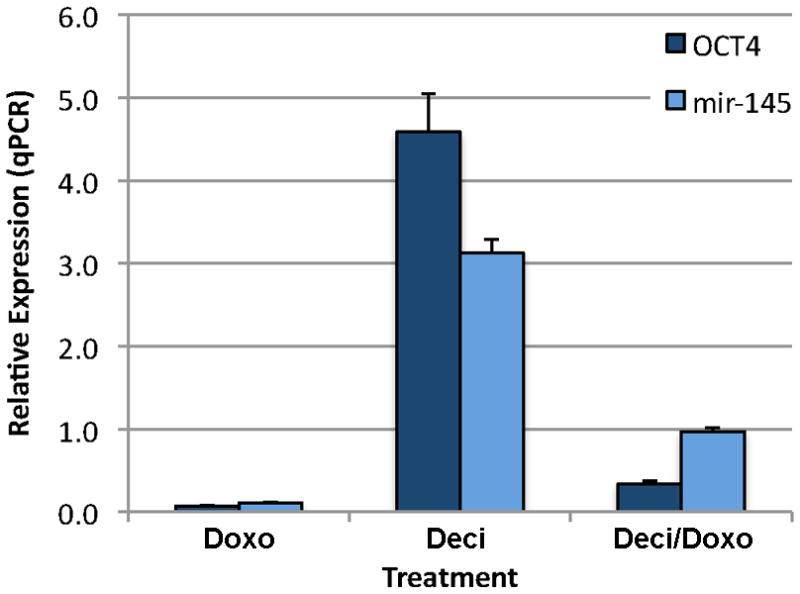
HSKs were treated with doxorubicin alone, decitabine alone, or decitabine for 31 hours followed by doxorubicin for 6 hours. Note, co-treatment reduces the decitabine-induced increase in OCT4 and mir-145 13-fold and 9-fold, respectively. n = 3 for each group.
DISCUSSION
In the studies presented here, we show that a single dose of decitabine not only demethylates HSK DNA, but also induces expression of Oct4 in these cells. Furthermore, the induced OCT4 mRNA is down-regulated by microRNA mir-145. This agrees with published findings that mir-145 suppresses growth of cancer cells by targeting OCT4.10,11 There is still some controversy about how the decitabine treatment works to inhibit cancer cells. One possibility is that the decitabine works through a combination of several mechanisms, including DNA methyltransferase (Dnmt) inhibition, mutations, and toxic effects in the cells.15 In this scenario, the decitabine treatment up-regulates both double-stranded DNA repair genes and p53 via a DNA-enzyme complex, which is formed when decitabine covalently binds Dnmt. Any degradation compounds that are produced during this action could be incorporated into DNA by polymerases, and could be toxic to the cells.15 Another possibility is that decitabine works by directly incorporating into DNA CpG sites opposite methylated CpG sites and binds the DNA methyltransferase (Dnmt) enzyme, thereby sequestering its activity. This causes loss of DNA methylation in one daughter DNA strand because Dnmt is not available to remethylate the hemi-methylated sites created during the first round of DNA replication.8 However, the demethylation effect is reversible as removal of the decitabine releases the Dnmt and cells become remethylated. Thus, the cells are not permanently altered by decitabine treatment. This latter mechanism is the likely explanation for the decitabine effect that we see in the HSKs as the cells survive when the decitabine treatment is discontinued. Moreover, since the methyltransferase inhibitory effect of decitabine results in a loosening of the chromatin and transcription of genes that are silenced by cytosine methylation,16 the fact that we see transcription of the normally silenced endogenous OCT4 gene further supports the latter mechanism.
We hypothesized that HSKs would be susceptible to a decitabine pre-treatment because we had already discovered that transient expression of Oct4 alone partially and temporarily reprogrammed HSKs.5 Moreover, this was unique for the primary HSKs, as we were unsuccessful in reprogramming primary skin fibroblasts using transient expression of Oct4 alone. In the studies presented here, we find that the HSKs greatly increase both OCT4 and mir-145 in response to the decitabine treatment. The HSKs may be unique in this response because when we treat primary skin fibroblasts with decitabine, they show no changes in OCT4 or mir-145 expression. In our previous studies, transfected HSKs expressed Oct4 for only three to four days, just long enough to allow them to be transformed into alternative cell lineages.5 Once the HSKs began their transformation, they turned off Oct4. Notably, when Oct4 was transiently expressed in HSKs via a plasmid for forty-eight hours, a temporary global demethylation occurred in the HSK DNA,5 suggesting that DNA demethylation and reactivation of endogenous Oct4 expression in HSKs might be linked. This hypothesis was supported by published reports that decitabine could reactivate Oct4 in cultured trophoblast cells.6 Furthermore, several reports demonstrated that inhibiting Dnmt1 resulted in changes in cell fate: adult pancreatic beta cells were converted into alpha cells;7 human umbilical cord mesenchymal stem cells were differentiated into cardiac cells;17 and conversion of human embryonic stem cells into cardiomyocytes was enhanced by decitabine;18
At this time, we do not know what other genes are directly activated by the decitabine demetheylation treatment. We do know that both the Oct4 target genes, SOX2 and NANOG are activated; however, we do not know if they were directly affected by the demethylation or by the presence of the Oct4 protein, which we have previously shown could activate these two genes.5 We also know that several miRNA are expressed. Despite these unknowns, we have found that the effects of a non-lethal dose of decitabine are reversible. Moreover, we saw no adverse effects on the keratinocytes after 48 hours of treatment with a non-lethal dose. Additionally, the normal keratinocytes are highly resistant to the doxorubicin treatment as compared to the NTERA-2 cancer cells, which suggests that choosing the appropriate dose may have only slight and reversible effects on the surrounding normal cells. Both of these cancer treatment drugs are used for several types of cancers. Since they are given systemically, their effect will be in all tissues, both cancerous and normal. Our data suggest that if the drugs are given in too high a dose, they could activate Oct4 in normal keratinocytes, which could induce the keratinocytes to aberrantly proliferate. However, such effects on keratinocytes are reversible, and thus may result in no permanent damage to the skin.
Although it is likely that the decitabine demethylation treatment induces expression of several genes, some of which may have interactions with Oct4, the fact that we found changes in two microRNAs (mir-302c and mir-145) that are specifically regulated by Oct4 lends credence to our hypothesis that endogenous OCT4 is reactivated when DNA is demethylated. Most microRNAs regulate their target mRNAs via post-transcriptional binding to a region in the mRNA’s 3′ UTR (untranslated region). The binding destabilizes the single-stranded mRNA and prevents its translation into protein. In embryonic stem (ES) cells, which continuously proliferate, several microRNAs have been identified. Of these, eight (mir-302 a*-a-b*-b-c*-c-d and 367) specifically regulate the cell cycle in human ES cells,13 and three (mir-291-3p, mir-294, and mir-295) directly inhibit known cell cycle inhibitors.19,20 It is generally known that ES cells continually proliferate because they have a shortened G1-phase of the cell cycle because they are able to bypass the normal G1/S checkpoint, and that this is due to expression of several microRNAs.12,21 In human ES cells, mir-302a regulates the cell’s transition from G1-phase to S-phase by directly repressing the cyclin D1 mRNA, resulting in low levels of cyclin D1 protein and loss of the G1/S checkpoint.12,22 When the mir-302 cluster was inhibited, the number of ES cells in G1 increased.12 Anecdotally, we observe that the Oct4-expressing HSKs appear to proliferate much faster than untreated HSKs, suggesting that Oct4 is somehow inducing the cells to proceed through the cell cycle. Since the mir-302 cluster is regulated by direct binding of Oct4,12 our data suggest that Oct4 expression in the HSKs induce the cells to proliferate, while also up-regulating the cyclin D1 regulator mir-302c. In support of this, we find significantly fewer decitabine HSKs in the G1-phase of the cell cycle and more cells in S/G2/M-phases. This correlates with other published data in which exogenous expression of mir-302 in HeLa cells, fibroblasts, or skin cancer cells induce rapid proliferation and increase the number of cells in S-phase, while decreasing the number of cells in G1-phase.12,23
Our data suggest that a balance exists between OCT4 and mir-145 similar to what is found in ES cells. In human ES cells OCT4 is highly expressed, while mir-145 is low. It was reported that when mir-145 is increased in human ES cells, it represses untranslated regions of the pluripotency reprogramming factors OCT4, SOX2, and KLF4.9 Furthermore, the Oct4 binds and represses the mir-145 promoter. 9 Thus, OCT4 and mir-145 regulate each other. We show that DNA demethylation can induce expression of both OCT4 and mir-145 in HSKs. Moreover, once OCT4 is expressed in HSKs, increasing mir-145 levels cause a decrease in the levels of both OCT4 and mir-145. This is suggestive of a double negative/positive regulatory loop, with the Oct4 protein increasing the level of mir-145, which in turn represses the OCT4 mRNA. The loss of OCT4 mRNA, results in less Oct4 protein to bind and induce mir-145, resulting in a decrease of mir-145.
We have not used extraneous gene transfection to increase OCT4 and mir-145 levels, rather we produce our results using approved cancer treatment drugs. Decitabine and doxorubicin are often used together to treat a variety of cancers in humans. It is thought that demethylation via decitabine helps the doxorubicin and other cancer drugs be more effective at killing the cells.24,25 In our studies, doxorubicin alone had little effect on the HSKs, but it did up-regulate mir-145 in the NTERA-2 cancer cell line, which resulted in a decrease in OCT4 in these cells. The decitabine treatment alone resulted in an increase in both OCT4 and mir-145 in HSKs. Notably, the combination of decitabine, followed by doxorubicin decreased expression of both mir-145 and OCT4 in the HSKs. It is not known whether malignant skin cancers, such as squamous cell carcinomas, express the microRNAs, mir-145 or mir-302. As for OCT4, there is still some controversy about its expression in cutaneous carcinoma cells. An analysis of a variety of 115 benign and cancerous specimens found no expression of OCT4 mRNA in any cancerous or normal specimen.26 This differed from an earlier study, which found Oct4 mRNA expression in immortalized cells, tumor cells, and cell lines, but not in differentiated normal cells.27 Oct4 and Nanog expression has also been found in oral squamous cell carcinomas from patients who became resistant to the chemotherapeutic agent cisplatin.28 It is not known whether DNA demethylation directly activates Oct4 expression in skin melanocytes or in melanoma skin cancer cells. However, when such cells were treated with decitabine, they seemed to activate melanocyte differentiation genes, such as SOX9 or genes that encode the antigen MAGE-1 on a melanoma cell line that is recognized by cytolytic T-cells,29,30 suggesting that decitabine affects non-keratinocyte cells differently than keratinocytes. It was reported that treatment with decitabine changed expression of only 0.67% of genes in melanoma skin cancer cells.31 Moreover, the genes that did change expression were conserved in tumor and normal cells. They concluded that decitabine-mediated gene activation requires either DNA promoter demethylation or activators that induce transcription of hypomethylated promoters, and that this accounted for the low number and specificity of genes that were activated. Our data support this conclusion and suggest that even if human cancers express OCT4, the current decitabine/doxorubicin treatment regimen may cause little permanent damage to the surrounding normal cells.
In this paper we present two novel findings. First, the cancer drug decitabine reactivates endogenous Oct4 and the OCT4 repressor mir-145 in human skin keratinocytes. Second, contrary to the mechanism reported for ES cells, Oct4 induces expression of mir-145 in the keratinocytes. Moreover, it is the loss of Oct4 that cause down-regulation of mir-145 in the keratinocytes. Additionally, we show that the cancer drug doxorubicin induces expression of mir-145 only in cells that already express OCT4. This latter result may demonstrate a benefit for normal tissues in cancer patients who are treated with the combination of decitabine and doxorubicin. Thus, the novel strategy that we present here provides a regulatable system to produce cells expressing Oct4 for transformation studies and provides a unique method to study the effects of endogenous Oct4 in cancer cells and the surrounding somatic cells.
Acknowledgments
We thank the members of the Dana Levasseur lab (Iowa) for helpful discussions, and members of the UI Flow Cytometry Core and the DNA Core for technical assistance. This research was funded in part by a grant from the National Institute of Arthritis Musculoskeletal and Skin at the National Institutes of Health to JRB (R01AR053619). The funding source had no involvement in study design, data collection, analysis, or interpretation.
Abbreviations
- HSKs
human skin keratinocytes
- ES cells
embryonic stem cells
- Deci
decitabine
- Doxo
doxorubicin
- Oct4 or OCT4
octamer-binding transcription factor 4, also known as POU5F1 (POU domain, class 5, transcription factor 1)
- qPCR
quantitative polymerase chain reaction
Footnotes
AUTHOR CONTRIBUTIONS
The authors make the following declarations about their contributions: Conceived and designed the experiments: JRB, MCW, SC. Performed the experiments: SC, SW, ATC. Analyzed the data: JRB, SC. Wrote the paper: JRB.
References
- 1.Okita K, Hong H, Takahashi K, Yamanaka S. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat Protoc. 2010;5:418–428. doi: 10.1038/nprot.2009.231. nprot.2009.231 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. stemcells.2008-1075 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yusa K, Rad R, Takeda J, Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods. 2009;6:363–369. doi: 10.1038/nmeth.1323. nmeth.1323 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amabile G, Meissner A. Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med. 2009;15:59–68. doi: 10.1016/j.molmed.2008.12.003. S1471-4914(09)00016-1 [pii] [DOI] [PubMed] [Google Scholar]
- 5.Racila D, Winter M, Said M, Tomanek-Chalkley A, Wiechert S, Eckert RL, et al. Transient expression of OCT4 is sufficient to allow human keratinocytes to change their differentiation pathway. Gene Ther. 2011;18:294–303. doi: 10.1038/gt.2010.148. gt2010148 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hattori N, Nishino K, Ko YG, Ohgane J, Tanaka S, Shiota K. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J Biol Chem. 2004;279:17063–17069. doi: 10.1074/jbc.M309002200. M309002200 [pii] [DOI] [PubMed] [Google Scholar]
- 7.Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell. 2011;20:419–429. doi: 10.1016/j.devcel.2011.03.012. S1534-5807(11)00118-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 9.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. S0092-8674(09)00252-9 [pii] [DOI] [PubMed] [Google Scholar]
- 10.Wu Y, Liu S, Xin H, Jiang J, Younglai E, Sun S, et al. Up-regulation of microRNA-145 promotes differentiation by repressing OCT4 in human endometrial adenocarcinoma cells. Cancer. 2011;117:3989–3998. doi: 10.1002/cncr.25944. [DOI] [PubMed] [Google Scholar]
- 11.Yin R, Zhang S, Wu Y, Fan X, Jiang F, Zhang Z, et al. microRNA-145 suppresses lung adenocarcinoma-initiating cell proliferation by targeting OCT4. Oncol Rep. 2011;25:1747–1754. doi: 10.3892/or.2011.1252. [DOI] [PubMed] [Google Scholar]
- 12.Card DA, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, et al. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008;28:6426–6438. doi: 10.1128/MCB.00359-08. MCB.00359-08 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, et al. Human embryonic stem cells express a unique set of microRNAs. Dev Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. S0012160604001381 [pii] [DOI] [PubMed] [Google Scholar]
- 14.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, et al. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A. 2009;106:3207–3212. doi: 10.1073/pnas.0808042106. 0808042106 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogstad DK, Herring JL, Theruvathu JA, Burdzy A, Perry CC, Neidigh JW, et al. Chemical decomposition of 5-aza-2′-deoxycytidine (Decitabine): kinetic analyses and identification of products by NMR, HPLC, and mass spectrometry. Chem Res Toxicol. 2009;22:1194–1204. doi: 10.1021/tx900131u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95–101. [PubMed] [Google Scholar]
- 17.Qian Q, Qian H, Zhang X, Zhu W, Yan Y, Ye S, et al. 5-Azacytidine Induces Cardiac Differentiation of Human Umbilical Cord-Derived Mesenchymal Stem Cells by Activating Extracellular Regulated Kinase. Stem Cells Dev. 2011 Jun 1; doi: 10.1089/scd.2010.0519. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501–508. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. ng.250 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Blelloch R. Cell cycle regulation by MicroRNAs in embryonic stem cells. Cancer Res. 2009;69:4093–4096. doi: 10.1158/0008-5472.CAN-09-0309. 0008-5472.CAN-09-0309 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H. Stem cell division is regulated by the microRNA pathway. Nature. 2005;435:974–978. doi: 10.1038/nature03816. nature03816 [pii] [DOI] [PubMed] [Google Scholar]
- 22.Fluckiger AC, Marcy G, Marchand M, Negre D, Cosset FL, Mitalipov S, et al. Cell cycle features of primate embryonic stem cells. Stem Cells. 2006;24:547–556. doi: 10.1634/stemcells.2005-0194. 2005-0194 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin SL, Chang DC, Chang-Lin S, Lin CH, Wu DT, Chen DT, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA. 2008;14:2115–2124. doi: 10.1261/rna.1162708. rna.1162708 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.George RE, Lahti JM, Adamson PC, Zhu K, Finkelstein D, Ingle AM, et al. Phase I study of decitabine with doxorubicin and cyclophosphamide in children with neuroblastoma and other solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer. 2010;55:629–638. doi: 10.1002/pbc.22607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan HH, Porter AG. DNA methyltransferase I is a mediator of doxorubicin-induced genotoxicity in human cancer cells. Biochem Biophys Res Commun. 2009;382:462–467. doi: 10.1016/j.bbrc.2009.03.065. S0006-291X(09)00523-3 [pii] [DOI] [PubMed] [Google Scholar]
- 26.Katona TM, Billings SD, Montironi R, Lopez-Beltran A, Cheng L. Expression of OCT4 transcription factor in cutaneous neoplasia. Appl Immunohistochem Mol Morphol. 2007;15:359–362. doi: 10.1097/PAI.0b013e31803006eb00129039-200712000-00001. [pii] [DOI] [PubMed] [Google Scholar]
- 27.Tai MH, Chang CC, Kiupel M, Webster JD, Olson LK, Trosko JE. Oct4 expression in adult human stem cells: evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis. 2005;26:495–502. doi: 10.1093/carcin/bgh321. bgh321 [pii] [DOI] [PubMed] [Google Scholar]
- 28.Tsai LL, Yu CC, Chang YC, Yu CH, Chou MY. Markedly increased Oct4 and Nanog expression correlates with cisplatin resistance in oral squamous cell carcinoma. J Oral Pathol Med. 2011;40:621–628. doi: 10.1111/j.1600-0714.2011.01015.x. [DOI] [PubMed] [Google Scholar]
- 29.Alcazar O, Achberger S, Aldrich W, Hu Z, Negrotto S, Saunthararajah Y, et al. Epigenetic regulation by decitabine of melanoma differentiation in vitro and in vivo. Int J Cancer. 2011 doi: 10.1002/ijc.26320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber J, Salgaller M, Samid D, Johnson B, Herlyn M, Lassam N, et al. Expression of the MAGE-1 tumor antigen is up-regulated by the demethylating agent 5-aza-2′-deoxycytidine. Cancer Res. 1994;54:1766–1771. [PubMed] [Google Scholar]
- 31.Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine. Mol Pharmacol. 2004;65:18–27. doi: 10.1124/mol.65.1.1865/1/18. [pii] [DOI] [PubMed] [Google Scholar]