

Abstract
While novel retroviral vectors for use in gene-therapy products are reducing the potential for formation of replication-competent retrovirus (RCR), it remains crucial to screen products for RCR for both research and clinical purposes. For clinical grade gammaretrovirus-based vectors, RCR screening is achieved by an extended S+L− or marker rescue assay, while standard methods for replication-competent lentivirus detection are still in development. In this report, we describe a rapid and sensitive method for replication-competent gammaretrovirus detection. We used this assay to detect three members of the gammaretrovirus family and compared the sensitivity of our assay with well-established methods for retrovirus detection, including the extended S+L− assay. Results presented here demonstrate that this assay should be useful for gene-therapy product testing.
Keywords: replication-competent gammaretrovirus, gene therapy vectors, infectivity assay
INTRODUCTION
Gammaretroviral vectors were the first viral gene-therapy vectors to enter clinical trials and remain in use today.1 One potential hazard associated with the use of such vectors is the presence of replication-competent retroviruses (“RCR”) in the vector preparations, either as a result of recombination events between the plasmids used for vector production or between the plasmids and endogenous retroviral sequences in the packaging cell lines, or as a result of contamination in the laboratory.2–5 RCR are potentially pathogenic, and have in fact been shown to induce malignancy in mice and non-human primates.6,7 Therefore, it is critical that vector preparations be rigorously tested to exclude the presence of RCR, and the Food and Drug Administration (FDA) requires that all vector preparations be screened for RCR.8
At present, the FDA recommends inoculating an aliquot of each vector preparation into a permissive cell line and passaging the cells for at least three weeks (“amplification phase”). Cell-free media from the amplification phase can then be analyzed by a variety of methods, including S+L− and marker-rescue assays, to demonstrate the presence or absence of RCR (reviewed in Cornetta and Wilson9; for a description of the S+L− assay, see Bassin et. al.10).
We now describe a new method for the detection of RCR in gammaretrovirus-based gene-therapy products and the cell lines used to make them. The assay uses one of a series of DERSE (Detector of Exogenous Retroviral Sequence Elements) plasmids, stably maintained in a cell line permissive for the retrovirus of interest.11,12 Utilizing the unique reverse-transcription step of the retroviral infection cycle, a functional reporter gene is formed in the DERSE cell culture only after infection with an RCR. The DERSE assay can be constructed, potentially, to detect any RCR and can use any reporter gene.
In this report, we describe the inGluc-MLV-DERSE assay, using a 293T cell line that expresses the receptor for ecotropic MLVs (murine cationic amino acid transporter, mCAT1), and Gaussia Luciferase (Gluc) as the reporter gene.13–15 We use the inGluc-MLV-DERSE assay to detect three gammaretroviruses commonly used in construction of gene-therapy products: Moloney MLV (MoMLV); 4070A amphotropic MLV (4070A); and Gibbon Ape Leukemia Virus (GALV). Measurement of Gluc activity in the cell culture supernatant, or the Gluc gene in MLV-DERSE cell genomic DNA, indicates the presence of RCR in the test sample. The high enzymatic activity of Gluc and the requirement for RCR infection for Gluc expression result in an assay with a very high (104) signal:background ratio. The fact that Gluc is secreted from the cell means that a single infected culture can be periodically sampled over time without the need to harvest cells. Most appealingly, the inGluc-MLV-DERSE assay does not require specialized training to perform and compares well to current protocols used for RCR detection. In experiments not shown here, we have also obtained comparable results with other reporters, including GFP.
RESULTS
Generation of the inGluc-MLV-DERSE cell line
The DERSE plasmids use a strategy that was originally devised for detection of retrotransposition by retroelements.16–18 These plasmids consist of a retroviral genome with an intron-containing indicator sequence replacing the viral coding regions (Figure 1).12 The viral sequences retained in the plasmid provide all the functions necessary for viral replication and expression (except those functions that are provided by viral proteins). Importantly, the indicator sequence is oriented in a reverse direction relative to the viral sequences while the intron is oriented in a forward direction. In the absence of an RCR the cell transcribes the indicator sequence, using the viral sequences as a promoter, and removes the intron. The result is a minus-strand (non-coding) copy of the indicator RNA, flanked by viral cis-acting sequences that include the viral RNA packaging signal. If this cell is infected with an RCR, the minus-strand indicator sequence can be packaged by viral proteins encoded by the RCR. When this RCR goes on to infect a new cell, reverse transcription of the viral RNA (that now includes the minus-strand indicator sequence) results in a coding indicator gene that is subsequently integrated into, and expressed by, the newly infected cell. Here we describe the inGluc-MLV-DERSE cell line in which the indicator gene is the secreted Gaussia luciferase (Gluc) enzyme, the LTRs and other cis acting sequences are from MLV, and the host cell line is 293mCAT1. Gluc activity is measured by addition of coelenterazine to a small volume (10µL) of cell culture supernatant, followed by measurement of light output.
Figure 1. Schematic of inGluc-MLV-DERSE assay.

The inGluc-MLV-DERSE plasmid consists of a Gaussia Luciferase (Gluc) sequence oriented in a reverse direction with respect to flanking MLV non-coding sequences. Within the non-coding Gluc sequence is an intron that is oriented in a forward direction relative to the viral non-coding regions (and can be spliced by the host cell). The plasmid is maintained in 293mCAT1 cells. In the absence of RCR, only minus-strand, spliced Gluc sequences are present in the cell. An RCR that infects the DERSE cell can package the RNA containing the minus-strand Gluc sequence. In the next round of infection reverse transcription of the encapsidated RNA produces a double-stranded DNA containing an uninterrupted Gluc gene. This gene is an intact, coding Gluc sequence that is subsequently integrated into the DNA of, and expressed by, the newly infected cell. Expression of Gluc is under the control of the cytomegalovirus (CMV) promoter.
Detection of virus with the inGluc cell line
We initially tested the inGluc-MLV-DERSE cell line in several ways. Firstly, we were interested in the ability of the cell line to detect RCR produced from a chronically infected cell line. To assess this we co-cultured the inGluc-MLV-DERSE cells with NIH-3T3 cells chronically infected with Moloney MLV (MoMLV). We observed a ~3-log increase in Gluc activity in the cell culture supernatant 3 days after the two cell lines were mixed, indicating that the MoMLV originating from the NIH-3T3 cells had infected and replicated in the inGluc-MLV-DERSE cells, resulting in expression of Gluc (Figure 2a).
Figure 2. MoMLV infection of inGluc-MLV-DERSE cells produces Gluc activity in the culture supernatant.

(a) inGluc-MLV-DERSE cells can detect RCR produced by a chronically infected cell-line. inGluc-MLV-DERSE cells were co-cultured with NIH-3T3 cells that were chronically infected with MoMLV. Three days after the co-culture was initiated, 10µL of culture supernatant was assayed for Gluc activity. Cultures and co-cultures were conducted in triplicate. Columns represent the mean of 3 cultures (standard error of mean error bars are present, but not visible at this scale). (b) Gluc activity in the cell culture supernatant accumulates over time and its level is directly related to the amount of virus used for infection. 96-well plates containing 1.25 × 103 inGluc-MLV-DERSE cells per well were infected with 0.03 ml of serial dilutions of MoMLV; Gluc activity in the supernatant was measured at 4 (circles), 5 (squares) and 6 (triangles) days post infection. The dashed line represents the average counts per second in supernatant from the mock-infected cultures. Points represent the mean of 3 infections (standard error of mean error bars are present, but not visible at this scale). The 10−1 dilution of the inoculum contained 1.5 × 103 focus-inducing units per 0.03 ml.10 (c) Gluc activity assay can be conducted directly in the cell-culture plate. inGluc-MLV-DERSE cells were infected with MoMLV and 3 days after infection Gluc substrate was added directly to the culture wells and Gluc activity measured. Columns represent the mean of 3 infections, error bars represent the standard error of mean.
We next investigated the relationship between the amount of virus present at the time of infection and the Gluc activity measured in the supernatant several days after infection. We infected the inGluc-MLV-DERSE cells with 10-fold serial dilutions of MoMLV and measured Gluc activity in the supernatant 4, 5 and 6 days after infection (Figure 2b). We found the Gluc activity level in the culture supernatant increased monotonically with the concentration of MoMLV used for infection. Further, we could follow the spread of MoMLV through the culture as periodic sampling of the culture supernatant demonstrated accumulation of Gluc activity over time. The dilution end-point for this assay was between 10−4 and 10−5 (Figure 2b); this result is in excellent agreement with the titer of this virus stock obtained by the direct S+L− focus assay10 (data not shown; see Figure legend for details.).
Finally, as Gluc is a secreted protein, we thought it interesting to see whether the Gluc assay could be conducted directly in the cell culture plate. We tested this by infecting the inGluc-MLV-DERSE cells with MoMLV in a black 96-well plate and then adding Gluc substrate directly to the cell culture well 3 days after infection (Figure 2c). The MoMLV-infected culture showed Gluc activity of ~107 counts per second (cps), the maximum activity measurable using our instrument, while a mock-infected culture had a background of ~103 cps. Thus it is possible to infect and detect RCR in the same plate. The ability to conduct the assay in this way may simplify automation of the assay and could have applications outside of gene-therapy product testing.
Real-time PCR for detection of RCR infection
We found measurement of Gluc activity in the supernatant to be a rapid and simple method for detecting infection of the inGluc-MLV-DERSE cell line. However, since infection of the cell line with a RCR results in the generation of a new gene (the spliced Gluc gene), it is also possible to use PCR as a read-out for the assay by targeting a primer/probe specifically to the coding Gluc sequences that flank the intron. We developed a duplex real-time PCR assay that simultaneously amplified the functional Gluc gene and, as a control for DNA quality and quantity, the single-copy CCR5 gene. Using this PCR, only the spliced, functional Gluc gene can be amplified (and thus amplification from the Gluc primer/probe set can only occur following RCR infection of the inGluc-MLV-DERSE cell line). Using a plasmid standard, we determined that the real-time PCR could detect 10 – 33 copies of Gluc in 100ng of inGluc-MLV-DERSE cell DNA (data not shown). To test the duplex real time PCR assay, we extracted genomic DNA from inGluc-MLV-DERSE cells 3 days after they were infected with serial dilutions of MoMLV. We performed duplex real-time PCR on the DNA and measured Gluc activity in the supernatant (Figure 3a, b, c and d). The Gluc probe Ct value was inversely related to the MoMLV concentration used for infection (Figure 3a and c). Further, the Gluc probe Ct values and the Gluc activity in the supernatant were in complete concordance - a culture deemed Gluc positive or negative in the duplex real-time PCR assay gave the same result in the Gluc activity assay (Figure 3a and d). As expected, CCR5 was amplified in all cases, confirming that the absence of Gluc probe fluorescence was not due to an absence, or poor quality, of DNA (Figure 3b).
Figure 3. inGluc-MLV-DERSE cell infection can be detected using PCR directed to the Gluc gene.
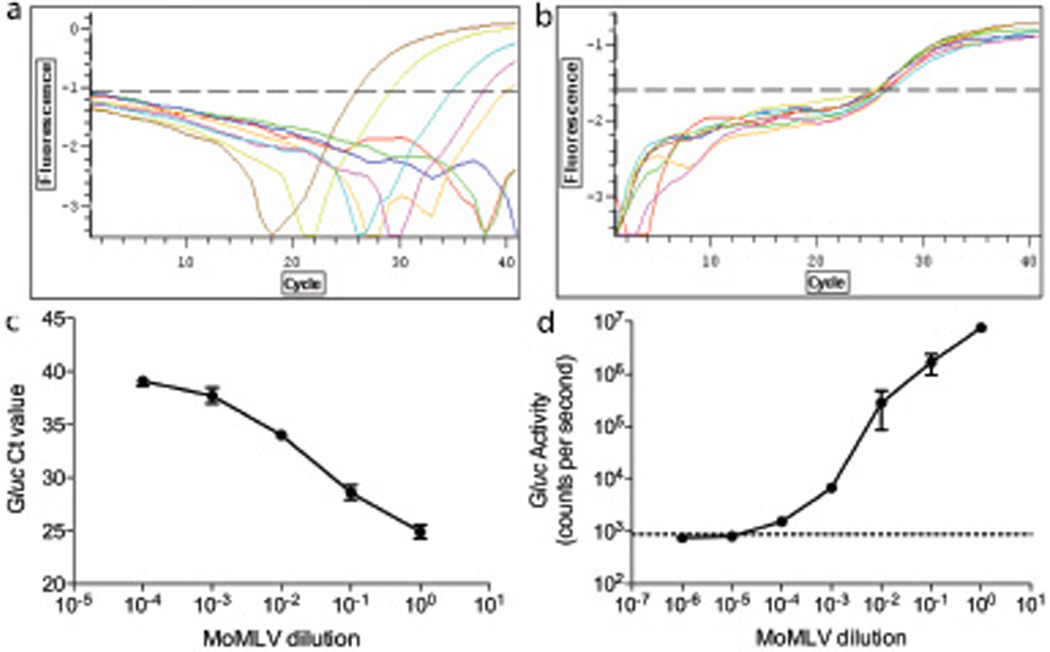
2.5 × 105 inGluc-MLV-DERSE cells in 20 cm2 dishes were infected, in triplicate, with 1 ml of serial dilutions of an MoMLV preparation that had a titer of 1.5 × 106 focus-inducing units per ml in the direct S+L− focus assay. Three days after infection, cell culture supernatant and cellular genomic DNA were collected. Duplex real-time PCR, directed to the Gluc and CCR5 genes, was conducted on 100ng of genomic DNA from inGluc-MLV-DERSE cells infected with undiluted (brown); 10−1 (mustard); 10−2 (aqua); 10−3 (pink); 10−4 (orange); 10−5 (green) or 10−6 (blue) dilutions of MoMLV; or on 100ng of genomic DNA from mock infected inGluc-MLV-DERSE cells (red). (a) Fluorescence from Gluc probe. (b) Fluorescence from CCR5 probe. (c) Gluc probe Ct values from the duplex real time PCR, plotted against the dilution of MoMLV used for infection of the inGluc-MLV-DERSE cells. Gluc copy numbers were calculated as 9749, 1923, 179, 35 and 19 for the undiluted through 10−4 infections, respectively. Error bars represent standard error of mean for 5 PCRs. (d) Gluc activity in 10µL of cell culture supernatant from MoMLV-infected inGluc-MLV-DERSE cells. The dashed line represents average counts per second in supernatant from the mock-infected cultures. Error bars represent standard error of mean for 3 infections.
In a separate experiment, we infected the inGluc-MLV-DERSE cells with MoMLV and the day after infection performed real-time PCR on the cellular DNA and a Gluc activity assay on the supernatant (results not shown). In this case, we found that the Gluc gene was amplified by real-time PCR but Gluc activity was not detected in the supernatant, presumably because Gluc protein levels had not accumulated sufficiently. This suggests that the real-time PCR can detect infection of the inGluc-MLV-DERSE cell line marginally earlier than measurement of Gluc activity in the supernatant. However, the extra time and manipulations (which could increase the risk of contamination) required to extract DNA and perform the real-time PCR, as well as the fact that the experiment is terminated once DNA is collected, may make the Gluc activity more suitable for many purposes.
Comparison of the inGluc-MLV-DERSE assay with other methods for gammaretrovirus detection
To support the results from the inGluc-MLV-DERSE assay, we compared the assay with two established methods for detecting retrovirus – the reverse transcriptase (RT) assay and immunoblotting against viral proteins. We infected the inGluc-MLV-DERSE cells with serial dilutions of 4070A amphotropic MLV, grew the cells, and collected the supernatant for Gluc activity assay, RT assay or immunoblot (Figure 4a, b and c). Within 16 days of the initial infection, we observed that all cultures infected with the 10−5 dilution of 4070A and none of the cultures infected with the 10−6 dilution of 4070A had Gluc activity in the supernatant, making 10−5 the dilution end-point for this particular virus stock using this assay (Figure 4a). This result is in reasonable agreement with the results of the standard, extended S+L−-based assay19, which gave a titer of 106 infectious units/ml for the inoculum (data not shown). Gluc activity was first detected in the cultures infected with the 10−5 dilution 6 days after infection (Figure 4a).
Figure 4. Comparison of inGluc-MLV-DERSE assay with RT assay and immunoblot.

1.25 × 105 inGluc-MLV-DERSE cells cells in a 6-well (9.6 cm2) plate were infected with 1 ml of dilutions of a 4070A MLV preparation that had a titer of 106 infectious units/ml in an S +L–-based assay.19 The supernatant was collected and cells split at 3, 6, 9, 13 and 16 days post infection (dpi). (a) Using the Gluc activity assay, the virus dilution end-point (10−5) is first detected between 3 (circles) and 6 (squares) days post infection. The dashed line represents average counts per second in supernatant from mock-infected cultures. Error bars represent standard error of mean for 3 infections. (b) RT assays of the culture supernatants give the same dilution end-point as the Gluc activity assay. 1µL of supernatant from 9 (circles), 13 (squares) and 16 (triangles) dpi were assayed for RT activity. The dashed line represents average counts per minute (cpm) in supernatant from mock-infected cultures. Error bars represent standard error of mean for 3 RT assays. (c) Gluc activity and p30CA levels, as estimated by immunoblot with MLV30 antiserum, gave the same dilution end-point. Virus pellets from 1mL of supernatant from cultures infected with 10−3, 10−4, 10−5 (9 and 13dpi) and 10−6 dilutions of 4070A (13 dpi) were used in the immunoblot.
We next measured RT activity in the supernatant collected during growth of the 4070A-infected inGluc-MLV-DERSE cells. Using this particular RT activity assay, we first detected RT activity in supernatant collected 13 days after infection of the inGluc-MLV-DERSE cells with a 10−5 dilution of 4070A (Figure 4b). RT activity was not detected in supernatant from cells infected with a 10−6 dilution of 4070A, even at 16 days after infection. While the Gluc activity assay proved to be more rapid than the RT assay under these experimental conditions, both assays gave the same dilution endpoint for this 4070A stock in this experiment (Figure 4a and b).
Additionally, we used a broadly reactive antiserum to MLV p30CA in an immunoblot of virus pellets prepared from the supernatants of the 4070A-infected cultures (Figure 4c). Supernatant p30CA levels, as estimated by immunoblot, were proportional to Gluc activity (Figure 4a and c). Under the conditions used, the Gluc activity assay was more sensitive than the immunoblot for RCR detection. Thus, while supernatant from the culture infected with a 10−5 dilution of 4070A demonstrated Gluc activity 6 days after infection, p30CA was not observed in the supernatant until 12 days after infection (Figure 4a and c). Again, both assays gave the same endpoint for this 4070A stock, as p30CA was not observed in supernatants collected from cells infected with a 10−6 dilution of 4070A.
Comparison of DERSE assay with FDA-compliant protocols for RCR detection
To determine the usefulness of the inGluc-MLV-DERSE assay for testing gene-therapy products, we used a single GALV stock and compared Gluc activity assay results from infection of the inGluc-MLV-DERSE line (conducted at NCI) with an extended S+L− assay conducted at the Indiana University Vector Production Facility.20 Using the S+L− assay, the GALV stock was found to contain 105 tissue culture-infectious doses (TCID50) per ml (not shown). We infected the inGluc-MLV-DERSE cells with 10-fold dilutions of the virus, in triplicate, and then passaged the cells and monitored Gluc activity in the cell culture supernatant. Twelve days (and 3 cell passages) after infection, 3/3 of the cultures infected with a 10−5 dilution of GALV and 1/3 of the cultures infected with a 10−6 dilution of GALV showed maximum Gluc activity in the supernatant (Figure 5). Using identical infection conditions, we repeated this experiment twice (that is, with 2 different GALV dilution series): in the second and third experiments 3/3 of the cultures infected with a 10−5 of GALV were positive and 0/3 of the 10−6 infected cultures (results not shown). Thus, the results from the inGluc-MLV-DERSE assay are in excellent agreement with those from the S+L− assay.
Figure 5.

Comparison of GALV titer using the inGluc-MLV-DERSE assay with that from the S+L− assay. 1.25 × 105 inGluc-MLV-DERSE cells in 9.6 cm2 wells were infected in triplicate with 1 ml of serial dilutions of a GALV stock that had an S+L− determined end-point of between 10−5 and 10−6 infectious units/ml.20 Cells were passaged and Gluc activity in the supernatant was monitored at 3 (circles), 6 (squares), 10 (triangles) and 12 (inverted triangles) days post infection (dpi). A representative experiment of three is shown. The dashed line represents average counts per second in supernatant from mock-infected cultures. Error bars represent standard error of mean for the triplicate infections of one experiment. For clarity, Gluc activity is only shown for the positive culture infected with the 10−6 dilution of GALV, and not for the negative cultures.
Finally, using the same GALV stock, we investigated the sensitivity of the inGluc-MLV-DERSE assay when infecting with an RCR in the presence of excess replication-incompetent retroviruses, as might be the situation when testing gene-therapy products. High concentrations of vector at the time of infection have been observed to interfere with detection of RCR, likely due to competition for cellular entry receptors.21,22 To determine the effect of excess vector on our assay, we infected 1.25×105 inGluc-MLV-DERSE cells with serial dilutions of GALV. The dilutions were made either in media or in 7.6 × 106 infectious units of a GALV-based gene-therapy vector shown to be RCR-negative using an FDA-compliant extended S+L− assay. As expected, in multiple experiments, no RCR were detected in the vector preparation by the inGluc-MLV-DERSE assay. However, we did observe somewhat lower Gluc activity in cell cultures infected with GALV in the presence of vector, compared to cultures infected with GALV alone. This observation was most notable in cultures infected with the 10−5 dilution of GALV (Figure 6a and b). Importantly though, the rate at which GALV infection proceeded through the culture was the same regardless of the presence or absence of gene-therapy vector (Figure 6a). As there was no effect of excess gene-therapy vector on the kinetics of RCR spread through the culture, the reduction in Gluc activity in samples assayed soon after infection was a result of interference by the vector at the time of virus attachment to the cell, and not interference of the vector at a later stage in the retroviral cycle. Most importantly, however, within the resolution of our assay, the dilution end-point for the GALV stock was the same (i.e. 10−5) both in the presence and absence of gene-therapy vector (Figure 6b); as in Figure 5, this result also agreed with the titer of the GALV preparation previously determined by passaging on S+L− cells (data not shown).
Figure 6. Effect of clinical-grade GALV vector on the infectivity of inGluc-MLV-DERSE cells.

inGluc-MLV-DERSE were infected as in Fig. 5 with dilutions of GALV prepared either in media (circles) or in the presence of 7.6 × 106 infectious units of clinical-grade GALV vector (squares), in triplicate. Cells were passaged and Gluc activity in the supernatant was monitored at 3, 7, 10 and 13 days post infection. (a) Kinetics of virus spread in cultures infected with the 10−5 dilution of GALV. (b) The dilution endpoint of the GALV RCR is determined to be 10−5, regardless of the presence or absence of GALV vector. Gluc activity was measured in the supernatant of cultures from 10 days after infection. The dashed line represents average counts per second in supernatant from the mock-infected cultures. Error bars represent standard error of mean for 3 infections.
DISCUSSION
While advances in gene-therapy vector preparation have reduced the risks of RCR formation during gene-therapy vector production, screening of these products remains essential. Screening for RCR in both gammaretrovirus- and lentivirus-based vectors is a time-consuming and meticulous process. In particular, the S+L− assay used for screening gammaretrovirus based vectors requires counting of RCR-induced foci in a cell monolayer. This is a tedious and somewhat subjective measurement. Furthermore, the FDA-compliant biologic assays are generally a two-stage process, with vector product (or co-culture when testing cell lines and patient cells) being first passaged on a permissive cell line for 3 weeks (to amplify any RCR) prior to conducting a 4–6 day S+L− assay or a marker rescue assay (RCR detection).
The inGluc-MLV-DERSE assay described here does not require the identification of foci, a substantial improvement on the S+L− assay. Further, the DERSE assay combines the amplification and detection stages for relatively rapid, real-time monitoring of RCR infection of the inGluc-MLV-DERSE cell culture. We show here the assay sensitively detects RCR in supernatant and produces results that are comparable to other RCR detection techniques (Figure 4 and 5). Moreover, the inGluc-MLV-DERSE assay is capable of directly detecting RCR by simple cocultivation with a producer cell line (Figure 2a). Thus it should be useful for screening both gene-therapy products and the packaging cell lines used for vector production (though the latter were not investigated here).
What makes the DERSE assay unique, however, is that the indicator gene (or protein) is only formed if the inGluc-MLV-DERSE cells are infected with RCR. This should result in a high signal: background ratio, with resulting enhancement of detection sensitivity, regardless of the indicator gene used. Furthermore, as far as we know, any replication-competent gammaretrovirus would register in this assay. Thus, in addition to applications in gene-therapy product testing, the generic DERSE assay will undoubtedly have multiple applications for basic RCR research.
In this work, we chose Gaussia luciferase as our indicator gene as it has two advantages over other reporter proteins. Firstly, Gluc is a predominantly secreted protein and thus there is no need for cell lysis, as is required for assays of firefly luciferase. In fact, since Gluc is secreted from the cell, the Gluc activity assay can be conducted directly in the cell culture plate (Figure 2c). Secondly, this highly active enzyme can produce luminescent signal intensities of at least 107 counts per second, a signal ~104 times greater than the background of our assay. The combination of these two properties of the Gluc enzyme means that, potentially, this particular DERSE assay could become fully automated rather simply (again, with possible applications outside of gene-therapy product testing).
For gamma-RCR detection, we chose to maintain the inGluc-MLV-DERSE vector in mCAT1-expressing 293T cells (293mCAT1 cells). We show here that these cells are sufficient for infection and rapid amplification of gamma-RCR that are commonly used with vectors for gene delivery, namely GALV, MoMLV and 4070A. Additionally, we have used the inGluc-MLV-DERSE 293mCAT1 cell line to detect several other gammaretroviruses including members of the xenotropic MLV family. 5 While we did not compare the infectability of these cells against other cells used with GALV, MoMLV or 4070A, previous work has demonstrated that 293 cells are most suitable for GALV amplification.20 We would expect the 293mCAT1 cells used here to be equally suitable for GALV amplification when compared to 293 cells. While Mus Dunni cells are currently recommended for amplification of MLV-based vectors, we did not investigate an inGluc-MLV-DERSE assay in these cells.22 It may well be that Mus Dunni cells offer enhanced virus amplification when assessing MLV-based vectors; it is important to note that the indicator plasmid can be maintained in any cell line deemed optimal for growth and detection of a particular retrovirus.
We used several cell densities and flask/plate sizes while developing this assay, eventually settling on the use of 6-well plates as these provided us with ample cells for DNA extraction and supernatant for RT assay or immunoblot, without using large amounts of space or reagents. While such a small plate size is likely not compatible with the requirement to test 5% of the final gene-therapy product volume, it should be sufficient for research purposes. In setting these parameters, it is crucial to remember that infection by gammaretroviruses requires that the host cell be dividing, while relatively high cell densities are optimal for virus spread.20,22 In general, we found that passaging the cells such that they were confluent in 3–4 days provided a good environment for rapid RCR amplification. Using the standardized 6-well plate assay, we observed that inGluc-MLV-DERSE cells initially infected with a few virus particles had detectable Gluc activity in the culture supernatant 6 – 7 days after infection and became completely infected 10 – 14 days after infection (Figure 4 and 5). While vigilant RCR detection may still benefit from a 3-week amplification stage (to ensure detection of low numbers or slowly replicating RCR), the inGluc-MLV-DERSE assay remains a more rapid method for detecting small quantities of virus than the currently used two-stage protocols as it combines the amplification and detection steps.
Similar to previous reports, we found that the presence of excess gene-therapy vector interferes somewhat with the initial amplification of RCR (Figure 6).21,22 As expected, this was most clearly observed at the limiting dilution (10−5) of GALV where the ratio of RCR: vector is lowest. However, at least for this combination of RCR and vector, the vector did not detectably affect the titer of GALV obtained in the inGluc assay. Thus, the data indicate that the effect of vector is minimal, and is presumably similar to the decrease in sensitivity observed in the extended S+L− assay.
It can easily be imagined that an RCR would be partially replication-defective, and would replicate too slowly to be detected by the new assay. However, the results indicate that the inGluc signal appears very rapidly (see Fig. 4 and 6A), so that efficiently replicating viruses are detected well before the 3 weeks required by the FDA. It seems likely that inefficiently replicating viruses will be detected sooner by the new assay than by standard assays. In fact, we have shown that the inGLuc assay could detect MLVs produced by the VCaP and EKVX tumor cell lines5, although these MLVs had very poor infectivity.5,23 It is also conceivable that the efficient packaging of the inGluc genome might competitively inhibit the spread of an RCR in the indicator cells; in this case, modulation of its expression level and/or its packaging signal might further enhance the sensitivity of the assay.
Yet another advantage of the DERSE assay is that a generic PCR, targeted to the indicator gene rather than the RCR itself, can be used to detect RCR (Fig. 3). Thus, unlike traditional PCR-based assays for RCR detection, PCR directed towards the Gluc gene is a functional assay, as it will only be positive in the presence of an RCR. Further, the PCR overcomes the requirement for virus-specific primer and probe design: any RCR capable of rescuing the inGluc vector, regardless of its viral origin or the recombination event(s) that generated it, will result in the production of a functional Gluc gene.
Importantly, the assay concept presented here is highly versatile and can be extended for detection of, potentially, any RCR simply by replacing the MLV non-coding regions with those of the retrovirus of interest. We have previously reported on DERSE plasmids comprising inGluc or inluc in the context of HTLV non-coding sequence.11,12 Of particular interest to the gene-therapy community, HIV-DERSE plasmids containing HIV genomic sequences in place of the MLV sequences described here, should allow detection of RCR (sometimes called “RCL”) in lentivirus-based gene-therapy vectors.12
Furthermore, the indicator gene can be replaced with any gene of interest allowing a researcher to customize the assay read-out to suit available equipment. We have successfully detected gamma-RCR infection of an inGFP-MLV-DERSE/LnCaP cell culture by using either FACS analysis or fluorescence microscopy to measure the appearance of GFP-positive cells following RCR infection (not shown). Here again, the fact that GFP is expressed only after RCR infection of the inGFP-MLV-DERSE cell line offers the possibility of a large signal: background ratio, particularly in comparison to other fluorescence-based RCR detection techniques that rely on enhancement, rather than initiation, of GFP expression.24 Observation of GFP-positive cells using fluorescence microscopy may represent a very simple method for RCR detection.
The inGluc-MLV-DERSE assay is a rapid and sensitive method for detection of RCR. End-point dilution experiments using the inGluc-MLV-DERSE assay gave results that were consistent with established retroviral detection methods (RT assay, immunoblot, extended S+L− assay). The presence of a great excess of gene-therapy vector did not change the limit of RCR detection at the resolution used (10-fold RCR dilutions). Further, the assay gave reproducible results over multiple experiments. While further assay development would be required to implement this method into the FDA guidelines, the work presented here demonstrates the potential of this assay for RCR detection in gene-therapy products. Future work may involve development of similar assays for lentiviral vectors. In conclusion, the DERSE assay is a versatile method for detecting RCR which should be useful not only for the gene-therapy community but for any researcher studying or working with RCR.
MATERIALS AND METHODS
Preparation of the inGluc-MLV-DERSE plasmid
The inGIuc cassette was inserted as a SmaI to Acc65 fragment into the pBabe-puro retroviral vector between the BsrGI and the blunt-ended SalI sites.25 The inGluc cassette consists of the CMV immediately early promoter, the human codon-optimized Gaussia luciferase coding sequences interrupted by the second intron of the rabbit β-globin gene at position 144, and the SV40 polyA signal.14
Preparation of inGluc-MLV-DERSE cells
293mCAT1 cells were seeded at 1×106 cells/10cm plate in DMEM containing 10% fetal calf serum, 100 Units/mL penicillin, 100µg/mL streptomycin and 0.292 mg/mL glutamine (DM). The following day cells were transfected with 10µg of the inGluc-MLV-DERSE plasmid using Transit 293 from Mirus Bio LLC (Madison, WI), according to the manufacturer’s instructions. The inGluc-MLV-DERSE plasmid contains the pac gene conferring puromycin resistance. Stable transfectants were selected in 5µg/mL puromycin and were subsequently maintained in 1µg/mL puromycin (DM-Puro).
Gaussia Luciferase Activity Assay
The BioLux™ Gaussia Luciferase Flex Assay Kit from New England Biolabs (Ipswich, MA) was used to assay for Gluc activity. Coelenterazine was prepared according to the manufacturer’s instructions. The stabilizer reagent was not used. As Gaussia luciferase is a secreted protein, 10µL of cell culture supernatant was assayed (with 50µL of coelenterazine reagent). Light output was measured in a microbeta Trilux luminescence and liquid scintillation counter for 1 second per sample.
Viruses
MoMLV was obtained from 0.45µm filtered culture supernatant from a chronically infected NIH 3T3 cell line,26 and the titer of this virus preparation was determined by direct S+L− focus assay.10 4070A MLV was purchased from the American Type Culture Collection (#VR-1445) and was titered as described.19 GALV SEATO virus was a kind gift from Mary Beth Eiden, NIH, and its titer was measured by the S+L− assay as described;20 this assay was initiated by addition of 1 ml of the virus preparation to 1 × 106 293 cells. Clinical grade retroviral vector expressing the Fanconi A gene and pseudotyped with the GALV Env was produced by the Indiana University Vector Production Facility. The vector stock had previously screened negative for RCR using the extended S+L− assay according to current FDA requirements for clinical products. The vector titer was determined by quantitative PCR using the methods previously described27 with the following probe and primer set: Forward: 5'-CGC AAC CCT GGG AGA CGT CCC-3', Reverse: 5'-CGT CTC CTA CCA GAA CCA CAT ATC C-3'; Probe: 5'-FAM-CCG TTT TTG TGG CCC GAC CTG AGG-TAMRA-3'.
Infection and co-culture of inGluc-MLV-DERSE cells
inGluc-MLV-DERSE cells were seeded one day prior to infection. On the day of infection the cells were treated with 20µg/mL DEAE-Dextran for 30 minutes. DEAE-Dextran was removed and cells were rinsed twice with growth medium. Virus and POLYBRENE® (8µg/mL final concentration) were added to cells and the cultures incubated for 4 hours. Virus was replaced with DM-Puro. Experiment-specific cell quantities and volumes are given below.
In-well Gluc activity assay
inGluc-MLV-DERSE cells were seeded at 1250 cells/well/100µL in DM-Puro, in a black µclear, Advanced TC, 96-well plate from Greiner Bio-one (Frickenhausen, Germany). 30µL of DEAE-Dextran and MoMLV were used and virus was replaced with 50µL DM-Puro. 3 days after infection, 100µL of coelenterazine reagent was added to each well.
Co-culture assay
inGluc-MLV-DERSE cells and NIH-3T3 cells chronically infected with MoMLV were seeded at 1250 cells (each) per well of a 96-well plate. No DEAE-dextran treatment, POLYBRENE®, or puromycin were used.
4070A and GALV end-point experiments
inGluc-MLV-DERSE cells were seeded at 1.25×105 cells/well/2mL in a 6-well plate. 1mL of DEAE-Dextran and virus were used. Cells were passaged such that they were confluent every 3 – 4 days.
Immunoblot
Supernatants from infected inGluc-MLV-DERSE cells were first filtered through a 0.45µm filter. Supernatants were then centrifuged through a 20% sucrose cushion at 110,000×g for 1 hour. The virus pellet was resuspended in 2xNuPAGE sample buffer (Invitrogen, Carlsbad, CA). Antiserum prepared against purified MLV p30CA (MLV30) was used.28 Detection used Western Lightning Plus-ECL from Perkin Elmer (Waltham, MA).
Reverse Transcriptase (RT) Assay
5µL of inGluc-MLV-DERSE cell culture supernatant (0.45µm filtered) was mixed with 25µL of RT assay buffer (62.5mM Tris pH 8.0, 25mM KCl, 0.6mM MnCl2 (added immediately before use), 10mM DTT (added immediately before use), 5µg/mL oligo(dT), 50µg/mL poly(rA), 0.25% v/v Nonidet P40 and 3.75 Ci/mmol [α-32P]-TTP (added immediately before use)). Oligo(dT) 12-18mer was purchased from Gene Link (Hawthorne, NY), poly(rA) from GE Healthcare (Piscataway, NJ) and [α-32P]-TTP from Perkin Elmer (Waltham, MA). Assays were incubated for 4 hours at 32°C and then 5µL of the mixture was spotted onto a DEAE filter mat (Perkin Elmer, Waltham, MA). The filter mat was dried (85°C for 10 minutes) and then washed 4 times for 10 minutes each in 2xSSC then twice for 5 minutes each in ethanol. The filter mat was dried again (85°C for 10 minutes) and then placed in a 32P cassette (Perkin Elmer, Waltham, MA). 32P activity was measured in a microbeta Trilux luminescence and liquid scintillation counter for 1 minute per sample.
Duplex Real Time PCR
Genomic DNA was extracted using the QIAmp DNA Mini kit (Qiagen, Hilden, Germany). Amplification of the Gluc gene was achieved using 3’Gluc-F (5’-CAG GAA TCT CAG GAA TGT C-3’); InGluc (5’-FAM-TGG GAC AGG CAG ATC AGA CAG-BHQ1-3’) and SpGluc-R (5’-GGG AAG TTG CCC GGG AA-3’). The 5 nucleotides on the 5’end of the SpGluc-R primer target Gluc sequence 5’ of the intron, while the 12 nucleotides on the 3’end of the SpGluc-R primer target Gluc sequence 3’ of the intron. In this way, SpGluc-R targets only the functional Gluc gene. CCR5 was amplified using CCR5-F (5-CCAGAAGAGCTGAGACATCCG-3); CCR5-PR (5’-HEXTCCCCTACAAGAAACTCTCCCCGG-BHQ1-3’); and CCR5-R (5’-GCCAAGCAGCTGAGAGGTTACT-3’).28 AmpliTaq Gold polymerase and associated buffer were purchased from Applied Biosystems/Life Technologies (Foster City, CA). A master mix of PCR reagents was prepared such that the final concentrations in the PCRs were: 1xPCR buffer; 300nM dNTPs; 2.5mM MgCl2; 50nM CCR5-F; 50nM CCR-R; 50nM CCR-PR; 500nM Gluc-F; 500nM SpGluc-R; 100nM InGluc and 0.06 Units/µL polymerase. Reactions were heated to 95°C for 15 minutes followed by 40 cycles of 95°C for 30 seconds and 60°C for 90 seconds. Reactions were performed using a DNA engine Opticon 2 instrument (MJ Instruments, now BioRad, Hercules, CA). Fluorescence profiles are presented on a log scale and are global minimum background subtracted as described by the Opticon Monitor 2 software.
ACKNOWLEDGEMENTS
We thank Jane Mirro, Gerry Princler, Patricia Lloyd and Shawn Hill for superb technical assistance, and Vineet KewalRamani for many helpful discussions. This work was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Work at Indiana University is supported by the NIH, National Center for Research Resource (National Gene Vector Biorepository P40 RR024928).
Footnotes
CONFLICT OF INTEREST
KC is founder of Rimedion, Inc. but there is no overlap with the work reported here. The authors declare no conflict of interest.
REFERENCES
- 1.Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, et al. Gene transfer into humans--immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 2.Chong H, Starkey W, Vile RG. A replication-competent retrovirus arising from asplit-function packaging cell line was generated by recombination events between the vector, one of the packaging constructs, and endogenous retroviral sequences. J Virol. 1998;72:2663–2670. doi: 10.1128/jvi.72.4.2663-2670.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrett E, Miller AR, Goldman JM, Apperley JF, Melo JV. Characterization of recombination events leading to the production of an ecotropic replication- competent retrovirus in a GP+envAM12-derived producer cell line. Virology. 2000;266:170–179. doi: 10.1006/viro.1999.0052. [DOI] [PubMed] [Google Scholar]
- 4.Patience C, Takeuch Y, Cosset FL, Weiss RA. MuLV packaging systems as models for estimating/measuring retrovirus recombination frequency. Dev Biol (Basel) 2001;106:169–179. discussion 253-163. [PubMed] [Google Scholar]
- 5.Sfanos KS, Aloia AL, Hicks JL, Esopi DM, Steranka JP, Shao W, et al. Identification of replication competent murine gammaretroviruses in commonly used prostate cancer cell lines. PLoS One. 2011;6:e20874. doi: 10.1371/journal.pone.0020874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornetta K, Nguyen N, Morgan RA, Muenchau DD, Hartley JW, Blaese RM, et al. Infection of human cells with murine amphotropic replication-competent retroviruses. Hum Gene Ther. 1993;4:579–588. doi: 10.1089/hum.1993.4.5-579. [DOI] [PubMed] [Google Scholar]
- 7.Donahue RE, Kessler SW, Bodine D, McDonagh K, Dunbar C, Goodman S, et al. Helper virus induced T cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J Exp Med. 1992;176:1125–1135. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.U.S. Food and Drug Administration. Guidance for Industry - Supplemental Guidance for Testing for Replication Competent Retrovirus in Retroviral Vector Based Gene Therapy Products and During Follow-up of Patients in Clinical Trials Using Retroviral Vectors. 2006 doi: 10.1089/10430340150218440. [DOI] [PubMed] [Google Scholar]
- 9.Cornetta K, Wilson CA. Safety of retroviral vectors: Regulatory and technical considerations. In: Dropulic B, Carter B, editors. Concepts in genetic medicine. Hoboken N.J.: John-Liss; 2008. pp. 277–288. [Google Scholar]
- 10.Bassin RH, Tuttle N, Fischinger PJ. Rapid cell culture assay technique for murine leukaemia viruses. Nature. 1971;229:564–566. doi: 10.1038/229564b0. [DOI] [PubMed] [Google Scholar]
- 11.Dorjbal B, Derse D, Lloyd P, Soheilian F, Nagashima K, Heidecker G. The role of ITCH in human T-cell leukemia virus type 1 release. J Biol Chem. 2011 doi: 10.1074/jbc.M111.259945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog. 2010;6:e1000788. doi: 10.1371/journal.ppat.1000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albritton LM, Tseng L, Scadden D, Cunningham JM. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell. 1989;57:659–666. doi: 10.1016/0092-8674(89)90134-7. [DOI] [PubMed] [Google Scholar]
- 14.Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon- optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Verhaegent M, Christopoulos TK. Recombinant Gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal Chem. 2002;74:4378–4385. doi: 10.1021/ac025742k. [DOI] [PubMed] [Google Scholar]
- 16.Curcio MJ, Garfinkel DJ. Single-step selection for Ty1 element retrotransposition. Proc Natl Acad Sci U S A. 1991;88:936–940. doi: 10.1073/pnas.88.3.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heidmann O, Heidmann T. Retrotransposition of a mouse IAP sequence tagged with an indicator gene. Cell. 1991;64:159–170. doi: 10.1016/0092-8674(91)90217-m. [DOI] [PubMed] [Google Scholar]
- 18.Heidmann T, Heidmann O, Nicolas JF. An indicator gene to demonstrate intracellular transposition of defective retroviruses. Proc Natl Acad Sci U S A. 1988;85:2219–2223. doi: 10.1073/pnas.85.7.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Reeves L, Sanburn N, Croop J, Williams DA, Cornetta K. Packaging cell line DNA contamination of vector supernatants: implication for laboratory and clinical research. Virology. 2001;282:186–197. doi: 10.1006/viro.2001.0826. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Reeves L, Cornetta K. Safety testing for replication-competent retrovirus associated with gibbon ape leukemia virus-pseudotyped retroviral vectors. Hum Gene Ther. 2001;12:61–70. doi: 10.1089/104303401450979. [DOI] [PubMed] [Google Scholar]
- 21.Printz M, Reynolds J, Mento SJ, Jolly D, Kowal K, Sajjadi N. Recombinant retroviral vector interferes with the detection of amphotropic replication competent retrovirus in standard culture assays. Gene Ther. 1995;2:143–150. [PubMed] [Google Scholar]
- 22.Sastry L, Cornetta K. Detection of replication competent retrovirus and lentivirus. Methods Mol Biol. 2009;506:243–263. doi: 10.1007/978-1-59745-409-4_17. [DOI] [PubMed] [Google Scholar]
- 23.Knouf EC, Metzger MJ, Mitchell PS, Arroyo JD, Chevillet JR, Tewari M, et al. Multiple Integrated Copies and High-Level Production of the Human Retrovirus XMRV (Xenotropic Murine Leukemia Virus-Related Virus) from 22Rv1 Prostate Carcinoma Cells. J Virol. 2009;83:7353–7356. doi: 10.1128/JVI.00546-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morner A, Bjorndal A, Albert J, Kewalramani VN, Littman DR, Inoue R, et al. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J Virol. 1999;73:2343–2349. doi: 10.1128/jvi.73.3.2343-2349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgenstern JP, Land H. A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res. 1990;18:1068. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rein A. Interference grouping of murine leukemia viruses: a distinct receptor for the MCF-recombinant viruses in mouse cells. Virology. 1982;120:251–257. doi: 10.1016/0042-6822(82)90024-1. [DOI] [PubMed] [Google Scholar]
- 27.Sanburn N, Cornetta K. Rapid titer determination using quantitative real-time PCR. Gene Ther. 1999;6:1340–1345. doi: 10.1038/sj.gt.3300948. [DOI] [PubMed] [Google Scholar]
- 28.Aloia AL, Sfanos KS, Isaacs WB, Zheng Q, Maldarelli F, De Marzo AM, et al. XMRV: a new virus in prostate cancer? Cancer Res. 2010;70:10028–10033. doi: 10.1158/0008-5472.CAN-10-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]