

Abstract
Parkinson’s disease (PD) is characterized by a deficiency in motor cortex modulation due to degeneration of pigmented dopaminergic neurons of the substantia nigra projecting to the striatum. These neurons are particularly susceptible to oxidative stress, perhaps because of their dopaminergic nature. Like all catecholamines, dopamine is easily oxidized, first to a quinone intermediate and then to dopaminochrome (DAC), a 5-dihydroxyindole tautomer, that is cytotoxic in an oxidative stress-dependent manner. Here we show, using the murine mesencephalic cell line MN9D, that DAC causes cell death by apoptosis, illustrated by membrane blebbing, Annexin V, and propidium iodide labeling within 3 h. In addition, DAC causes oxidative damage to DNA within 3 h, and positive TUNEL fluorescence by 24 h. DAC, however, does not induce caspase 3 activation and its cytotoxic actions are not prevented by the pan-caspase inhibitor, Z-VAD-fmk. DAC-induced cytotoxicity is limited by the PARP1 inhibitor, 5-aminoisoquinolinone, supporting a role for apoptosis inducing factor (AIF) in the apoptotic process. Indeed, AIF is detected in the nuclear fraction of MN9D cells 3h after DAC exposure. Taken together these results demonstrate that DAC induces cytotoxicity in MN9D cells in a caspase-independent apoptotic manner, likely triggered by oxidative damage to DNA, and involving the translocation of AIF from the mitochondria to the nucleus.
Keywords: Parkinson’s disease, dopaminochrome, neurodegeneration, apoptosis, PARP1, AIF
Introduction
The loss of modulation of the motor cortex in Parkinson’s disease (PD) is due to degeneration of dopaminergic neurons projecting from the substantia nigra pars compacta (SNpc) to the striatum. While the effects of PD are clearly defined (an inability to initiate voluntary movements, bradykinesia, and a resting tremor) its cause(s) remain elusive. The SNpc consists primarily of dopaminergic neurons that are pigmented due to the accumulation of neuromelanin in the cell bodies. Neuromelanin is formed as a result of the polymerization of an oxidized form of dopamine, dopaminochrome (DAC), within these cells (Graham 1978, Galzigna et al. 1999, Sulzer et al. 2000). Indeed, a hallmark of PD is the loss of the pigment in the SN as the dopaminergic neurons degenerate (Zecca et al. 2002).
Intracellular dopamine is oxidized at physiological pH to a transient quinone that autocyclizes to form the relatively stable DAC, a 5-dihydroxyindole tautomer (Graham 1978). While the existence of neuromelanin is empirical evidence for the formation of DAC in vivo, little is known about the effects of the unpolymerized molecule. The transient quinone form of dopamine has been implicated in the fibrilization of α-synuclein that is frequently associated with the formation of Lewy bodies, a hallmark of PD (Bisaglia et al. 2007). We, and others, have also shown that oxidized dopamine is cytotoxic to both MN9D and PC12 cell lines in a manner consistent with stimulating oxidative stress (Linsenbardt et al. 2009, Liu et al. 2005). Oxidized dopamine has also been shown to participate in protein modification and subsequent cell damage in rat substantia nigra (Wang et al. 2011, Van Laar et al. 2009). Finally, we have demonstrated that the formation of DAC is increased in striatal samples from an oxidative-stress dependent mouse model of PD (Mallajosyula et al. 2008). Taken together these data demonstrate that oxidized dopamine is detrimental to cell viability both in vitro and in vivo, but the mechanisms involved in that cytotoxicity are not well defined.
Although the cause of dopaminergic cell loss in the SNpc is poorly understood, the neurodegeneration is accompanied by an increase in oxidative stress and inflammation (Whitton 2007, Andersen 2004). The neurodegeneration has also been confirmed to be apoptotic in nature as evidenced by the presence of markers such as membrane blebbing, DNA laddering, damage to organelles and cell membranes (Mills et al. 1998, Galluzzi & Kroemer 2008).
Classical apoptosis commonly involves a series of cysteine proteases known as caspases, enzymes that are activated in a cascade ultimately resulting in the degradation of DNA, although apoptosis has also been observed in a variety of cell types in the absence of caspase activation (Chu et al. 2005, Yu et al. 2002, Yu et al. 2003). For example, it has been shown that direct oxidative damage of DNA can lead to the over-activation of poly(ADP)ribose polymerase (PARP), a DNA repair protein that produces poly(ADP)ribose (PAR) (Daugas et al. 2000, Cregan et al. 2004). It is thought that these PAR or PAR-conjugated proteins migrate to the mitochondria where they cause a release of caspase-independent factors including apoptosis inducing factor (AIF) and/or endonuclease G (EndoG). Once released from the mitochondria, these proteins translocate to the nucleus where they cause large-scale DNA strand breaks directly, without the need for ATP or caspase activation (Cande et al. 2002, Hong et al. 2004).
We previously reported that DAC increases superoxide in the mesencephalic cell line, MN9D, that is correlated with a loss of glutathione in the cells, indicative of high oxidative stress (Linsenbardt et al. 2009). In the present study, we show that DAC causes MN9D cell death in a caspase-independent apoptotic manner that involves oxidative damage to DNA, is PARP1 dependent and involves the translocation of AIF from the mitochondria to the nucleus.
Methods
Growth and Differentiation of MN9D Cells
MN9D cells, a kind gift from Drs. Al Heller and Lisa Won at the University of Chicago, were grown in high-glucose (4500 mg/L) Dulbecco’s modified Eagle’s Medium (DMEM, Sigma-Aldrich, St. Louis, MO) with L-glutamine, penicillin and streptomycin solution (Cellgro, Manassas, VA), and 10% fetal bovine serum (Hyclone, Logan, UT) in a humidified 5% CO2 atmosphere at 37 °C. When passaged, cells were washed with PBS and treated with Trypsin (Fisher, Fairlawn, NJ), followed by deactivation of the Trypsin with DMEM containing 20% fetal bovine serum. Cells were then counted and seeded into flasks or onto plates according to experimental conditions. For all experiments, cells were differentiated for 5 days with 1 mM sodium butyrate (Sigma-Aldrich, St. Louis, MO) and seeded onto collagen coated 6-well or 96-well plates, or onto Petri dishes for fluorescence microscopy.
Dopaminochrome Preparation
Dopamine was oxidized by reacting with NaIO4 for 45 min at 37 °C in a 2:1 ratio in ddH2O as described previously (Graham 1978, Ochs et al. 2005, Linsenbardt et al. 2009). All compounds were obtained from Sigma-Aldrich (St. Louis, MO). After the reaction was complete, all samples were tested using HPLC-ED (Ochs et al. 2005) to verify the purity of our samples, and that the conversion to dopaminochrome from dopamine yielded no remaining intermediates.
Cell Viability Assay
All cell viability measurements were carried out using the MTS assay (CellTiter96 AQ, Promega, Madison, WI). In brief, cells were grown on collagen coated 96 well plates, and then each well was administered 20 μL of reagent at the specified time points for 3 hours at 37 °C and 5% CO2 atmosphere. The wells were then evaluated for colorimetric absorption on a Powerwave X plate reader (Biotek instruments; Winooski, VT) and the calculation of sample value was analyzed by KC Junior Software (Biotek instruments).
TUNEL Analysis
Cells were evaluated for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) using the Dead-End Fluorometric TUNEL System (Promega, Madison, WI, USA) and measured via flow cytometry (BD FACSCalibur; BD Biosciences, Mississauga, ON, Canada) according to the manufacturer’s instructions. Briefly, cells were washed in phosphate buffered saline (PBS), fixed in 1% methanol-free formaldehyde, and permeablized in 70% ethanol. After centrifugation and washing in PBS, cells were incubated in Equilibration Buffer for 5 min followed by treatment with rTdT Incubation Buffer for 60 min. After termination of the reaction with EDTA (20 mM) and resuspension of cells in PBS, cells were analyzed via flow cytometry for fluoroscein-12-dUDP fluorescence at 520 nm and interpreted via CellQuest software (BD Biosciences, Mississauga, ON, Canada).
Caspase 3 Colorometric Assay
MN9D cells were exposed to either peroxynitrite or DAC, then harvested, lysed and processed according to the Caspase 3 Assay Kit, Colorometric (Sigma-Aldrich, St. Louis). In brief, cells were pelleted, washed, then suspended in 1x Lysis buffer and incubated on ice. Cells were then centrifuged and supernatant transferred to new tubes. Lysates were analyzed immediately on 96-well plates after addition of 1x Assay Buffer and incubated for 90 min at 37 °C. Plates were read at 405 nm on a Powerwave X plate reader (Biotek instruments; Winooski, VT) and the calculation of sample value was analyzed by KC Junior Software (Biotek instruments). Values of caspase 3 activation were determined by comparing experimental trials with a calibration curve of p-nitroaniline.
8-hydroxy-2-deoxyguanosine Assay
MN9D cells exposed to DAC were evaluated using the 8-OHdG Assay (Cayman Chemical, Ann Arbor, MI). Briefly, DNA was purified using a commercially available kit and quantified using a Nanodrop. The pH of the samples was adjusted to 5.2 using sodium acetate. DNA was denatured by boiling for ten minutes, digested with nuclease P1 and treated with alkaline phosphatase. Samples were boiled again to inactivate the alkaline phosphatase, diluted as appropriate with EIA Buffer and used directly in the immunoassay. A standard curve was established by serial dilution of 8-hydroxy-2-deoxy guanosine between 10.3 and 3,000 pg/mL using EIA Buffer as the matrix. The concentration of each sample was calculated from a logistic four-parameter fit of the standard concentrations versus % Bound/Maximum Bound (%B/B0).
Annexin-V Studies
Cells were grown on collagen coated 24-well tissue culture plates (Corning, Lowell, MA) in DMEM, treated with Annexin V-FITC [50 μg/mL; Sigma-Aldrich, St. Louis, MO] and propidium iodide [100 μg/mL; Sigma-Aldrich, St. Louis, MO], then visualized by fluorescence microscopy using a GFP filter (excitation = 488 nm, emission = 595 nm) and a Texas Red filter (excitation = 596 nm, emission = 615 nm). Five images were taken per experiment carried out, chosen randomly across each well. Cells were quantified using ImageJ software, normalized to the total number of cells in each field of view, and averaged across all experiments.
Subcelluar Fractionation and Western Immunoblot Analysis
Cells, grown and differentiated on collagen coated 6-well tissue culture plates, were exposed to DAC for 1 or 3h. Immediately following the experiment cells were harvested and subjected to subcellular fractionation in order to attain purified mitochondrial and nuclear fractions. A commercially available kit (ProteoExtract Cytosol/Mitochondria fractionation kit, EMD Chemicals, Gibbstown, NJ) was used for subcellular fractionation of mitochondria from other cellular components. The nucleus-containing fraction obtained from the kit process was purified by sucrose gradient centrifugation. The nuclear fraction was resuspended in 0.25 M sucrose buffer (with 100mM MgCl2 and 100mM HEPES, pH 7.5) and then pipetted in a layer over 0.35M sucrose buffer (with 5mM MgCl2 and 100mM HEPES, pH 7.5) The resulting pellet was then lysed in a nuclear lysis buffer to liberate nuclear proteins.
Western Analysis was performed on both the mitochondrial and nuclear fractions. Protein from mitochondrial or nuclear fractions was resolved by SDS-PAGE, transferred onto PVDF membranes (BioRad, Hercules, CA) and probed with the following primary antibodies all obtained from Abcam (Cambridge, MA): anti-AIF (1:500 dilution), anti-EndoG (1:750 dilution), anti-COX IV (1:1000 dilution) and anti-H2B (1:2000 dilution). The secondary antibody was goat anti-rabbit (1:10,000 dilution) obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Immunoreactivity was detected using an ECL-detection system (Thermo Fisher Scientific, Waltham, MA). Autoradiography films were exposed at multiple time-points to ensure non-saturation of images. Images were acquired from the photographic films using the Fujifilm LAS-3000 imaging system and densitometric analysis was carried out using Image Reader LAS-3000 v2.2 software.
Statistical Analysis
Results are expressed as mean ± s.e.m. for (n) wells. Statistical differences between treatments were determined by Student’s t-test; by one-way analysis of variance followed by Student-Newman-Keuls test; or by two-way analysis of variance followed by Bonferroni Post-Hoc test, depending on the experimental conditions. Statistical differences were accepted when P<0.05.
Results
Dopaminochrome induces early signs of apoptosis
MN9D cells, grown and differentiated in DMEM, were exposed to DAC [175 μM]. Over a period of 3 h, the cells (in a humidified incubator maintained at 37 °C with 5% CO2) were observed for ultrastructural changes in the cell membrane using phase contrast microscopy (Figure 1). Membrane blebbing, a classic apoptotic sign, was clearly present by 3 h (Figure 1B; arrows).
Figure 1. DAC induces membrane blebbing in MN9D cells.

MN9D cells were exposed to DAC [175 μM] and visualized by phase contrast microscopy over 3 h. Panel A shows cells at 1h post DAC exposure. Panel B depicts the same cells at 3 h post DAC. The arrows represent sites of membrane blebbing (B). These images are representative of 3 individual experiments. Scale bars represent 100 μm visualized at 20x magnification.
In separate experiments, the presence of phosphatidylserine (PS) on the cell surface, a sign of early-stage apoptosis, was investigated. MN9D cells in DMEM were incubated for 3h in the presence or absence of DAC [175 μM] and then labeled with Annexin V conjugated to a FITC fluorophore [50 μg/mL], along with propidium iodide [PI; 100 μg/mL]. MN9D cells exposed to DAC exhibited significant increases in both Annexin V labeling and PI labeling at 3 h (Figure 2).
Figure 2. DAC induces Annexin V and propidium iodide labeling in MN9D cells.

MN9D cells (Control; Ai–Aiii) or exposed to DAC [175 μM; Bi–Biii] for 3 h) were visualized by phase contrast microscopy (i), or fluorescence microscopy for Annexin V–FITC (ii) and propidium iodide (iii). Cells were quantified using ImageJ software from 25 individual images across 5 separate experiments (C). *** P < 0.0001 compared with control cells. Scale bars represent 100 μm visualized at 10x magnification.
Dopaminochrome induces DNA damage
MN9D cells were exposed to DAC [100–200 μM] for 24 h, fixed with paraformaldehyde, and then treated for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Control cells (Figure 3A) evaluated by flow cytometry exhibited no increase in TUNEL fluorescence, while the positive control, 6-hydroxydopamine [6-OHDA; 100 μM] increased the number of TUNEL positive cells by 5-fold (Figure 3B). Exposure of MN9D cells to 100 μM DAC (Figure 3C) yielded no significant increase in TUNEL fluorescence, however 200 μM DAC increased labeling by nearly 4-fold (Figure 3D - E). Overall, the data revealed a significant increase in TUNEL fluorescence in cells treated with 200 μM DAC for 24 h (Figure 3E).
Figure 3. DAC induces DNA laddering in MN9D cells.

MN9D cells were left untreated (A), exposed to 6-OHDA [100 μM; B], or exposed to DAC [100 μM and 200 μM; (C) and (D), respectively] for 24h after which they were fixed, processed for TUNEL and analyzed by flow cytometry. Panel E depicts the analysis of flow cytometry results for 3 separate experiments. * P < 0.05 compared to untreated (control) cells.
Dopaminochrome is cytotoxic in a caspase-independent manner
In order to address the participation of caspases in DAC-induced cytotoxicity, we analyzed the direct activation of caspase 3, one of the primary effector proteases in the caspase system. MN9D cells were exposed to DAC [50 - 200 μM] or, as a positive control, peroxynitrite [ONOO−; 0.5-1.0 mM] and assessed for caspase 3 activation. ONOO− increased caspase 3 activation by nearly two-fold within 16 hrs of treatment compared with untreated control cells, whereas DAC had no effect on caspase 3 activation within the same time frame (Figure 4).
Figure 4. DAC does not induce caspase 3 activation in MN9D cells.

MN9D cells were untreated (control; open bars) exposed to peroxynitrite [ONOO−; 0.5 or 1.0 mM; closed bars] or DAC [50–100 μM; shaded bars] for 16 h after which they harvested and evaluated for caspase 3 activity. (n = 3) * P < 0.05 compared to control cells.
In order to assess whether other caspases are involved in DAC-induced cytotoxicity, MN9D cells were pre-treated with Z-VAD-fmk [50 μM], a general caspase inhibitor, before being exposed to DAC [175 μM] or tumor necrosis factor-alpha [TNF-α; 50 μM] as a positive control for caspase-induced apoptosis. At 24h the cells were analyzed for cell viability by the MTS assay (Figure 5). TNF-α reduced cell viability to 75% of control cell values, and this was prevented by the presence of Z-VAD-fmk. In contrast, Z-VAD-fmk had no protective effect on DAC-induced cytotoxicity (~30% of control).
Figure 5. The pan-caspase inhibitor Z-VAD-fmk does not protect against DAC-induced cytotoxicity in MN9D cells.

MN9D cells were pre-treated with vehicle (Control Treatment) or Z-VAD-fmk [50 μM] for 30 min before being exposed to either DAC [175 μM] or TNF-α [50 μM]. Cells were evaluated at 24 h for viability. (n = 24 wells from 3 individual experiments) *** P < 0.0001 compared to control treated cells.
DAC Increases Oxidative DNA Damage
Despite the absence of caspase activity, apoptosis is clearly occurring in DAC-induced MN9D cell toxicity, therefore a caspase-independent mechanism must be in place. One such caspase-independent pathway involves the activation of PARP1 in response to oxidative damage to DNA. Since DAC is capable of increasing oxidative stress within MN9D cells (Linsenbardt et al. 2009) we assessed the effect of DAC on the oxidation of DNA using an 8-hydroxy-2-deoxyguanosine (8-OHdG) assay. MN9D cells were exposed to DAC [175 μM] and compared to control cells. After 1 h there was no significant change in oxidized DNA in cells exposed to DAC, but by 3 h, a significant increase in 8-OHdG had occurred (Figure 6).
Figure 6. DAC increases 8-OHdG in MN9D cells.

MN9D cells were untreated (Control) or exposed to DAC [175 μM] for 1 h or 3 h and then assayed for the presence of 8-hydroxy-2-deoxyguanosine (8-OHdG), a marker for oxidized DNA. 8-OHdG was normalized to total DNA within the samples. (n = 3) * P < 0.05 compared with control cells.
MN9D cytotoxicity of DAC involves PARP1 activation
MN9D cells were pre-treated with the PARP1 inhibitor, 5-aminoisoquinolinone [AIQ; 50–100 μM], for 30 min before being exposed to DAC [175 μM]. After 24 h, MN9D cells exposed to DAC alone exhibited a 50% reduction in viability. Pre-treatment with AIQ limited this reduction to a 30% decrease in viability (Figure 7).
Figure 7. Aminoisoquinolinone protects against DAC-induced cytotoxicity.
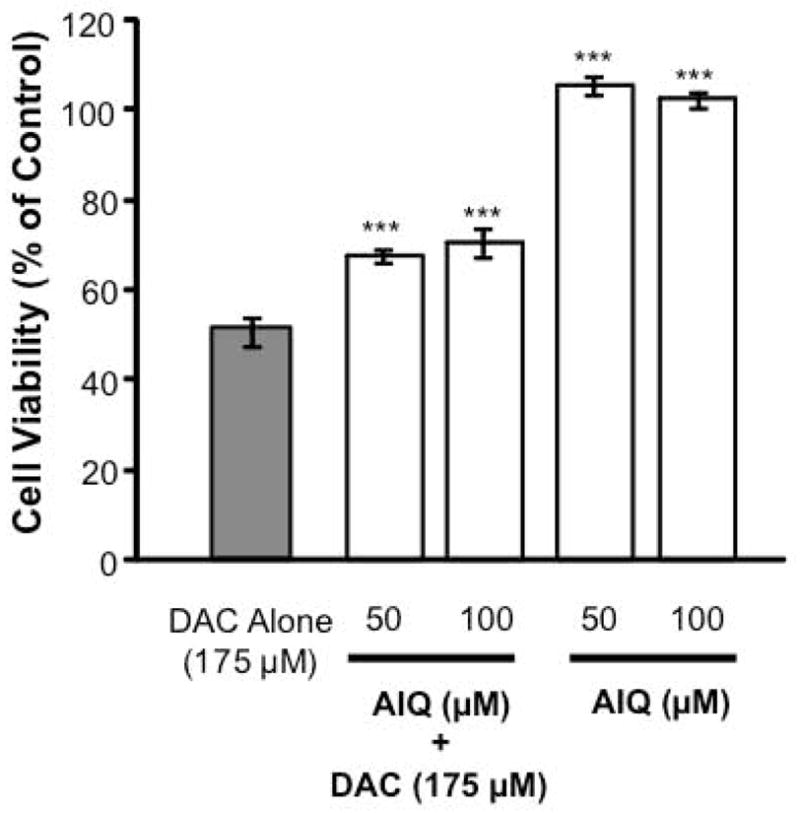
MN9D cells were pre-treated with 5-Aminoisoquinolinone [AIQ; 50–100 μM] for 30 min, then exposed to DAC [175 μM]. After 24 h, cell viability was evaluated. (n = 32 wells from 4 individual experiments) *** P < 0.0001 compared with DAC alone.
AIF detected in nucleus of MN9D cells after exposure to dopaminochrome
MN9D cells were exposed to DAC (200 μM) for 1 or 3 h. The cells were harvested, subjected to subcellular fractionation, and protein samples from nuclear and mitochondrial fractions were assessed by Western immunoblot analysis for the presence of AIF and EndoG. No evidence of nuclear AIF was observed in cells exposed to DAC for 1 h, however by 3 h AIF was present in nuclear fractions in significant quantities (Figure 8A-B). In contrast, EndoG was not detected in the nuclear fractions. The presence of both AIF and EndoG was clearly evident in DAC treated mitochondrial fractions at 1 h and 3 h. However, the amount of AIF present in the DAC treated mitochondrial fraction decreased between 1 and 3 h, consistent with the translocation of AIF to the nucleus at this time-point (Figure 8D). Untreated cells showed significant amounts of EndoG and AIF in the mitochondrial fractions at both 1 and 3 h (Figure 8C), but no appearance of AIF in the nuclear fractions at either time-point (Figure 8A).
Figure 8. DAC induces AIF but not EndoG translocation to the nucleus of MN9D cells.

MN9D cells were left untreated (control) or exposed to DAC (200 μM) for 1 or 3 h. After subcelluar fractionation and nuclear purification, Western Analysis was performed for the detection of AIF and EndoG within A) nuclear or C) mitochondrial fractions. Nuclear (B) and mitochondrial AIF levels (D), normalized to the nuclear marker H2B and mitochondrial marker COXIV respectively, were quantified and expressed as arbitrary units ± s.e.m. (n=3) * P < 0.05 compared to levels at 1 h. Death by dopaminochrome; a role for AIF but not caspase Dopaminochrome (DAC) may play a role in neurodegeneration observed in Parkinson’s disease (PD). Using MN9D cells we show that DAC causes caspase-independent apoptosis through activation of poly(ADP)ribose polymerase 1 (PARP1), and subsequent translocation of apoptosis inducing factor (AIF) from the mitochondria to the nucleus. These results provide a mechanism by which DAC could contribute to neurodegeneration in PD.
Discussion
The mechanisms by which dopaminergic neurons of the SNpc are lost during the course of PD are not clearly understood. Mutations in genes such as Parkin and LRRK2 (Kitada et al. 1998, Zimprich et al. 2004) are known to be causes of the neurodegeneration associated with genetic forms of PD, but the causes of idiopathic PD (>80% of PD cases) remain unclear, although mitochondrial dysfunction and increased neuronal oxidative stress are involved (Olanow 2007, Sulzer 2007, Keeney et al. 2006). Interestingly most, if not all, genetic forms of PD also ultimately involve mitochondrial dysfunction and/or oxidative stress giving rise to the idea that PD is essentially a mitochondrial-driven disease (Dodson & Guo 2007, Shadrina et al. 2010, Branco et al. 2010, Martin et al. 2011, Schapira & Gegg 2011).
The hallmark symptoms of PD typically present after most neurons along the nigrastriatal tract have been lost, suggesting that the disease slowly develops over time (Chen et al. 2008, Wang et al. 2011). As previously stated, these neurons produce dopamine that oxidizes to dopamine quinones and then DAC, before polymerizing to form neuromelanin (Graham 1978, Galzigna et al. 1999, Sulzer et al. 2000) the hallmark pigment of the substantia nigra. Thus, dopamine derived oxidants make attractive candidates for the role of endogenous toxic factor(s) that contribute to neurodegenerative processes in PD. Indeed, dopamine quinones have been shown to not only accumulate in the midbrain of aged rats, but also modify key mitochondrial proteins that could contribute to oxidative stress and apoptosis (Van Laar et al. 2009, Wang et al. 2011). DAC is clearly detrimental to neuronal cells, although the exact signaling mechanism by which this toxicity occurs is not yet clear (Linsenbardt et al. 2009, De Iuliis et al. 2008).
Inflammatory pathways, antioxidant imbalance, a lack of growth factors or nutrients, or exposure to toxins have all been implicated in neurodegeneration (Kostrzewa & Segura-Aguilar 2003, Beal 1992, Kaushal & Schlichter 2008). Cells affected by neurodegeneration undergo necrosis, apoptosis, or sometimes a mode of toxicity that resembles both (Galluzzi & Kroemer 2008, Bras et al. 2005). Necrosis involves early permeabilization of the cell membrane, swelling of the membrane, DNA damage and organelle dysfunction (Golstein & Kroemer 2007). Apoptosis, on the other hand, is marked by cell shrinkage, membrane blebbing, activation of cellular proteins to degrade DNA, and extracellular signals to attract inflammatory cells to phagocytose the apoptotic cell debris (Bras et al. 2005). There has been a great deal of research into the pathology of PD in post-mortem human brains yielding multiple hallmarks of apoptosis and necrosis including DNA nick-end labeling and increases in nuclear translocation of NFκB, a transcription factor associated with oxidative stress (Kostrzewa 2000). In addition, nuclear condensation is observed in the MPTP treated rat, an animal model of PD (Banerjee et al. 2007). Furthermore, in cell culture models of PD, neurotoxins like MPP+ and rotenone cause apoptotic signals such as membrane blebbing and caspase 3 activation (Raza & John 2006, Liu et al. 2005, Segura Aguilar & Kostrzewa 2004). Overall, our data show that MN9D cells treated with DAC exhibit many of the hallmark traits of apoptosis, including positive labeling of phosphatidylserine on the membrane surface by Annexin V, DNA laddering by TUNEL, and membrane blebbing but not caspase activation.
Signaling mechanisms for apoptosis may differ, but membrane blebbing is a classic morphological sign of the process (Borner & Monney 1999, Shiratsuchi et al. 2002). The causes of blebbing are poorly understood, but the process has been described in both caspase-dependent and caspase-independent forms of apoptosis (Shiratsuchi et al. 2002, Mills et al. 1998). This early sign of apoptosis is also frequently accompanied by the translocation of phosphatidylserine lipids from the inner membrane leaflet to the outer membrane of the cell, a shift that can be visualized by labeling phosphatidylserine with FITC-conjugated Annexin V, a protein that binds to phosphatidylserine specifically (Rimon et al. 1997). The transfer of phosphatidylserine from the inside of the cell membrane to the outside has been clearly described and is most frequently associated with apoptosis (Vermes et al. 1995).
We have shown that MN9D cells exposed to DAC present both membrane blebbing and Annexin V labeling, clearly signifying an apoptotic, rather than a necrotic, mode of cell death. Within 3 hrs of exposure to DAC, MN9D cells exhibit profound membrane blebbing. By the same time point, phosphatidylserine has translocated to the outer cell membrane, as indicated by positive Annexin V labeling. Along with Annexin V labeling, positive PI labeling of the nucleus is also observed, indicating that the cell membrane has permeablized. The double-labeling of cells with both Annexin V and PI typically indicates late-stage apoptosis: Annexin V labels phosphatidylserine relatively early in apoptosis, while PI tends to label nuclei after the cell membrane has permeablized in the latter portions of the apoptotic process (Martin et al. 1995). The combination of these factors are evidence that apoptosis occurs within 3 h of exposure of MN9D cells to DAC.
Part of the course of programmed cell death involves extensive degradation of nuclear DNA, either induced by caspase-activated DNase or by caspase-independent mechanisms (Susin et al. 2000). Accordingly, incubation of MN9D cells with DAC results in specific DNA damage that is measured by TUNEL fluorescence, again supporting an apoptotic pathway of cell death.
Apart from morphological indicators of apoptosis, other internal effectors of programmed cell death include the caspase system (Schulz & Gerhardt 2001). This series of cysteine proteases can be induced by a variety of extracellular and intracellular signals and results in the hallmark signals of apoptosis. Although many forms of apoptosis involve the caspase system, DAC-induced cell death did not display activation of one of the most important effector caspases, caspase 3, nor were the cells protected from DAC induced cytotoxicity when pre-treated with a pan-caspase inhibitor. These data suggest that while DAC indeed initiates typical apoptotic processes like membrane blebbing , Annexin V labeling and TUNEL in MN9D cells, it does so in a caspase-independent manner.
Damage to DNA is known to induce apoptosis, and oxidative damage to DNA specifically can cause apoptosis through a mechanism independent of caspase activation (Chu et al. 2005, Yu et al. 2002, Yu et al. 2003). We know that DAC increases intracellular oxidative stress, therefore it is possible that this in itself could cause enough damage to DNA to elicit an apoptotic response without activation of caspases (Linsenbardt et al. 2009, Hong et al. 2004). Indeed, we show here that MN9D cells exposed to DAC exhibit oxidative DNA damage as measured by the presence of 8-OHdG. Thus, we investigated the involvement of PARP1-induced DNA repair that has been implicated in caspase-independent apoptosis (Yu et al. 2002, Yu et al. 2003).
The protective effect of AIQ, an inhibitor of PARP1, against the toxicity induced by DAC implicates the actions of this enzyme in the apoptosis observed. In the event of extensive damage to a cell’s DNA, PARP1 will attempt to make repairs, producing poly(ADP)ribose polymers (PARs) as a byproduct of its DNA repair function. Should these PARs enter the cytosol they cause the release of AIF from the mitochondria (Daugas et al. 2000). AIF will then translocate to the nucleus where it carries out large-scale (>50 kb) DNA scission (Hong et al. 2004). Accordingly, we found that nuclear fractions isolated from MN9D cells exposed to DAC did indeed show evidence for AIF translocation by 3h.
It has been shown that neurodegeneration is associated with increased oxidative stress in vitro and in vivo (Ebadi et al. 1996, Foley & Riederer 2000, Kostrzewa 2000). Moreover, we have shown that DAC exposure results in depleted glutathione stores in MN9D cells and, therefore, increased oxidative stress (Linsenbardt et al. 2009). In the context of this current study, it follows that DAC causes caspase-independent apoptosis via oxidative DNA damage leading to activation of AIF, a protein released from the mitochondria as a result of oxidative DNA damage. Indeed, a recent report suggests that increased oxidative stress induced by injection of lipopolysaccharide to the midbrain of rats resulted in caspase 3 activation in mostly glial cells, but AIF translocation in nigral dopamine neurons (Burguillos et al. 2011). The authors extended these experiments and tested substantia nigral dopaminergic neurons from PD patients and observed similar results. While these authors did not identify a specific cause of AIF translocation in PD patients, it is possible that DAC could serve as a potential mediator.
The results presented in this work suggest a specific role for DAC inducing caspase-independent apoptosis, though dopamine itself activates caspases 3 and 7, as well as PARP, possibly through proteasomal dysfunction (Jeon et al. 2010). Furthermore, synthetic neuromelanin, derived from dopamine, and dopamine-quinone, have also been shown to deplete glutathione stores and induce mitochondrial-mediated apoptosis in SH-SY5Y cells, including activation of caspase 3 (Naoi et al. 2009). In contrast to dopamine and neuromelanin, our results show that the mode of cell death induced by DAC lacks caspase activity and favors AIF activation, supporting a distinct mechanism for this specific dopaminergic oxidation product.
In closing we have shown that MN9D cells exposed to DAC exhibit the hallmarks of PARP1 driven caspase-independent apoptosis and supports a role for the oxidation of dopamine in the neurodegeneration associated with PD.
Acknowledgments
We wish to thank Drs. Al Heller and Lisa Won from the University of Chicago for the kind gift of MN9D cells used in these experiments. We also wish to thank Dr. Jan Ryerse and Ms. Joy Eslick at Saint Louis University for help with microscopy and flow cytometry respectively. This work was supported in part by NIGMS008306.
References
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nature medicine. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Sreetama S, Saravanan KS, Dey SN, Mohanakumar KP. Apoptotic Mode of Cell Death in Substantia Nigra Following Intranigral Infusion of the Parkinsonian Neurotoxin, MPP(+) in Sprague-Dawley Rats: Cellular, Molecular and Ultrastructural Evidences. Neurochem Res. 2007 doi: 10.1007/s11064-007-9299-8. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mechanisms of excitotoxicity in neurologic diseases. Faseb J. 1992;6:3338–3344. [PubMed] [Google Scholar]
- Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. Journal of Neurochemistry. 1999;73:1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- Bisaglia M, Mammi S, Bubacco L. Kinetic and structural analysis of the early oxidation products of dopamine: analysis of the interactions with alpha-synuclein. J Biol Chem. 2007;282:15597–15605. doi: 10.1074/jbc.M610893200. [DOI] [PubMed] [Google Scholar]
- Borner C, Monney L. Apoptosis without caspases: an inefficient molecular guillotine? Cell Death Differ. 1999;6:497–507. doi: 10.1038/sj.cdd.4400525. [DOI] [PubMed] [Google Scholar]
- Branco DM, Arduino DM, Esteves AR, Silva DF, Cardoso SM, Oliveira CR. Cross-talk between mitochondria and proteasome in Parkinson's disease pathogenesis. Front Aging Neurosci. 2010;2:17. doi: 10.3389/fnagi.2010.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bras M, Queenan B, Susin SA. Programmed cell death via mitochondria: different modes of dying. Biochemistry (Mosc) 2005;70:231–239. doi: 10.1007/s10541-005-0105-4. [DOI] [PubMed] [Google Scholar]
- Burguillos MA, Hajji N, Englund E, Persson A, Cenci AM, Machado A, Cano J, Joseph B, Venero JL. Apoptosis-inducing factor mediates dopaminergic cell death in response to LPS-induced inflammatory stimulus: evidence in Parkinson's disease patients. Neurobiol Dis. 2011;41:177–188. doi: 10.1016/j.nbd.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W, Hastings TG, Kang UJ, Zhuang X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J Neurosci. 2008;28:425–433. doi: 10.1523/JNEUROSCI.3602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CT, Zhu JH, Cao G, Signore A, Wang S, Chen J. Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. Journal of neurochemistry. 2005;94:1685–1695. doi: 10.1111/j.1471-4159.2005.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Dawson VL, Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. Faseb J. 2000;14:729–739. [PubMed] [Google Scholar]
- De Iuliis A, Arrigoni G, Andersson L, Zambenedetti P, Burlina A, James P, Arslan P, Vianello F. Oxidative metabolism of dopamine: a colour reaction from human midbrain analysed by mass spectrometry. Biochimica et biophysica acta. 2008;1784:1687–1693. doi: 10.1016/j.bbapap.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17:331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Ebadi M, Srinivasan SK, Baxi MD. Oxidative stress and antioxidant therapy in Parkinson's disease. Progress in neurobiology. 1996;48:1–19. doi: 10.1016/0301-0082(95)00029-1. [DOI] [PubMed] [Google Scholar]
- Foley P, Riederer P. Influence of neurotoxins and oxidative stress on the onset and progression of Parkinson's disease. J Neurol. 2000;247(Suppl 2):II82–94. doi: 10.1007/pl00007766. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Galzigna L, Zanatta L, Esposito N. Toxicity of dopamine and dopaminochrome on cultured cells. Neurotox Res. 1999;1:149–152. doi: 10.1007/BF03033278. [DOI] [PubMed] [Google Scholar]
- Golstein P, Kroemer G. Cell death by necrosis: towards a molecular definition. Trends in biochemical sciences. 2007;32:37–43. doi: 10.1016/j.tibs.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Jeon SM, Cheon SM, Bae HR, Kim JW, Kim SU. Selective susceptibility of human dopaminergic neural stem cells to dopamine-induced apoptosis. Exp Neurobiol. 2010;19:155–164. doi: 10.5607/en.2010.19.3.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–1401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM. Review of apoptosis vs. necrosis of substantia nigra pars compacta in Parkinson's disease. Neurotoxicity research. 2000;2:239–250. doi: 10.1007/BF03033797. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM, Segura-Aguilar J. Novel mechanisms and approaches in the study of neurodegeneration and neuroprotection. a review. Neurotox Res. 2003;5:375–383. doi: 10.1007/BF03033166. [DOI] [PubMed] [Google Scholar]
- Linsenbardt AJ, Wilken GH, Westfall TC, Macarthur H. Cytotoxicity of dopaminochrome in the mesencephalic cell line, MN9D, is dependent upon oxidative stress. Neurotoxicology. 2009;30:1030–1035. doi: 10.1016/j.neuro.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HQ, Zhu XZ, Weng EQ. Intracellular dopamine oxidation mediates rotenone-induced apoptosis in PC12 cells. Acta Pharmacol Sin. 2005;26:17–26. doi: 10.1111/j.1745-7254.2005.00003.x. [DOI] [PubMed] [Google Scholar]
- Mallajosyula JK, Kaur D, Chinta SJ, Rajagopalan S, Rane A, Nicholls DG, Di Monte DA, Macarthur H, Andersen JK. MAO-B elevation in mouse brain astrocytes results in Parkinson's pathology. PLoS ONE. 2008;3:e1616. doi: 10.1371/journal.pone.0001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson's disease. Annu Rev Genomics Hum Genet. 2011;12:301–325. doi: 10.1146/annurev-genom-082410-101440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. The Journal of experimental medicine. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JC, Stone NL, Erhardt J, Pittman RN. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 1998;140:627–636. doi: 10.1083/jcb.140.3.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naoi M, Yi H, Maruyama W, Inaba K, Shamoto-Nagai M, Akao Y, Gerlach M, Riederer P. Glutathione redox status in mitochondria and cytoplasm differentially and sequentially activates apoptosis cascade in dopamine-melanin-treated SH-SY5Y cells. Neurosci Lett. 2009;465:118–122. doi: 10.1016/j.neulet.2009.08.082. [DOI] [PubMed] [Google Scholar]
- Ochs SD, Westfall TC, Macarthur H. The separation and quantification of aminochromes using high-pressure liquid chromatography with electrochemical detection. Journal of neuroscience methods. 2005;142:201–208. doi: 10.1016/j.jneumeth.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Olanow CW. The pathogenesis of cell death in Parkinson's disease--2007. Mov Disord. 2007;22(Suppl 17):S335–342. doi: 10.1002/mds.21675. [DOI] [PubMed] [Google Scholar]
- Raza H, John A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol Appl Pharmacol. 2006;216:309–318. doi: 10.1016/j.taap.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Rimon G, Bazenet CE, Philpott KL, Rubin LL. Increased surface phosphatidylserine is an early marker of neuronal apoptosis. Journal of neuroscience research. 1997;48:563–570. doi: 10.1002/(sici)1097-4547(19970615)48:6<563::aid-jnr9>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Gegg M. Mitochondrial contribution to Parkinson's disease pathogenesis. Parkinsons Dis. 2011;2011:159160. doi: 10.4061/2011/159160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Gerhardt E. Apoptosis: its relevance to Parkinson's disease. Clinical Neuroscience Research. 2001;1:427–433. [Google Scholar]
- Segura Aguilar J, Kostrzewa RM. Neurotoxins and neurotoxic species implicated in neurodegeneration. Neurotox Res. 2004;6:615–630. doi: 10.1007/BF03033456. [DOI] [PubMed] [Google Scholar]
- Shadrina MI, Slominsky PA, Limborska SA. Molecular mechanisms of pathogenesis of Parkinson's disease. Int Rev Cell Mol Biol. 2010;281:229–266. doi: 10.1016/S1937-6448(10)81006-8. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi A, Mori T, Nakanishi Y. Independence of plasma membrane blebbing from other biochemical and biological characteristics of apoptotic cells. J Biochem. 2002;132:381–386. doi: 10.1093/oxfordjournals.jbchem.a003233. [DOI] [PubMed] [Google Scholar]
- Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Bogulavsky J, Larsen KE, et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci U S A. 2000;97:11869–11874. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Daugas E, Ravagnan L, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laar VS, Mishizen AJ, Cascio M, Hastings TG. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol Dis. 2009;34:487–500. doi: 10.1016/j.nbd.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. Journal of immunological methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Wang N, Wang Y, Yu G, Yuan C, Ma J. Quinoprotein adducts accumulate in the substantia nigra of aged rats and correlate with dopamine-induced toxicity in SH-SY5Y cells. Neurochem Res. 2011;36:2169–2175. doi: 10.1007/s11064-011-0541-z. [DOI] [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. British journal of pharmacology. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis. 2003;14:303–317. doi: 10.1016/j.nbd.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zecca L, Fariello R, Riederer P, Sulzer D, Gatti A, Tampellini D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson's disease. FEBS letters. 2002;510:216–220. doi: 10.1016/s0014-5793(01)03269-0. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]