

Abstract
Background
The ubiquitous crosslinking enzyme tissue transglutaminase (TG2) has been implicated in irreversible collagen stabilization in liver fibrosis, although functional evidence is lacking. We studied the contribution of TG2 to hepatic fibrotic matrix stability, as well as liver fibrosis progression and regression in TG2-deficient mice.
Methods
Advanced liver fibrosis was induced by carbon tetrachloride (CCL4) or thioacetamide (TAA) in TG2−/− mice and their wild-type littermates to study fibrosis progression and its spontaneous regression for up to 36 weeks. Pattern and extent of fibrosis were analyzed by histology and hepatic hydroxyproline quantification. Dynamic changes in hepatic matrix crosslinking were assessed by stepwise collagen extraction. Expression of 7 transglutaminases and of fibrosis-related genes were determined by quantitative reverse transcription PCR.
Results
Transglutaminase activity was increased in fibrosis, and level of TG2 mRNA correlated with expression of fibrosis-related genes. Biochemical analysis revealed progressive collagen stabilization, with an up to 6-fold increase in the highly crosslinked, pepsin-insoluble fraction (26%). In TG2−/− mice, hepatic transglutaminase activity was significantly decreased, but chronic administration of CCL4 or TAA led to a comparable extent and pattern of liver fibrosis, as in wild-type mice. In TG2−/− mice, the composition of hepatic collagen fractions and levels of fibrosis-related transcripts were unchanged, and fibrosis reversal was not facilitated.
Conclusions
TG2 and transglutaminase activity are upregulated during hepatic fibrosis progression, but do not contribute to fibrogenesis or stabilization of the collagen matrix. TG2 deletion does not promote regression of liver fibrosis. TG2-independent collagen crosslinking is a remarkable feature of progressing hepatic fibrosis and represents an important therapeutic target for liver fibrosis.
Keywords: carbon tetrachloride, cirrhosis, collagen, crosslink, thioacetamide, liver fibrosis reversal, cirrhosis, liver disease, mouse model, fibrosis gene expression
INTRODUCTION
Liver cirrhosis as a consequence of many forms of chronic liver diseases is associated with a high morbidity and mortality1. Causal treatment for advanced fibrosis and cirrhosis is unsatisfactory or inefficient. Therefore, antifibrotic therapies to treat and reverse fibrosis are urgently needed2. While most antifibrotic strategies are directed against hepatic stellate cell/myofibroblast (collectively termed HSC) proliferation and profibrogenic activation3, only a few smaller studies have targeted extracellular matrix (ECM) stabilization, mainly the cross-linking of collagen.
Targeting the process of fibrotic ECM stabilization therapeutically would indeed be a very attractive approach to induce fibrosis regression or even reversibility, e.g. after successful treatment of the underlying cause of a given chronic liver disease. In vivo, stabilization of the fibrotic ECM is achieved by cross-linking of the abundant (fibrillar) collagens type I, III and V, which can represent more than 50% of the fibrotic liver tissue4 and of other ECM molecules, such as elastin and fibronectin. Stabilization of the collagenous and elastic ECM can occur via three distinct processes: by the enzymatic action of lysyl oxidase or transglutaminase (mainly tissue transglutaminase, TG2), or driven by age and hyperglycemia via non-enzymatic glycation and lipid peroxidation. Lysyl oxidase (LO, EC1.4.3.13, encoded by the LOX gene5) oxidizes the side chain of peptidyl lysine, resulting in residues of α-aminoadipic-δ-semialdehyde, which permits the covalent cross-linking of the component chains of collagen and elastin6. The family of transglutaminases (EC2.3.2.13) consists of 8 family members (TG1-7 and factor XIII7), of which TG2 is the most ubiquitously expressed and abundant enzyme. TG2 and most other family members catalyze the formation of ε-(γ-glutamyl) lysine isopeptide cross-links between a donor protein with a particular glutamine residue and a more general acceptor protein. TG2- catalyzed isopeptide bond formation has been described for some ECM glutamine-donor proteins, namely several collagens (via their telopeptide extensions), procollagen type III (via the N-propeptide), nidogen and fibronectin8. Transglutaminases, especially TG2, are involved in several human diseases9, 10, in wound healing and fibrosis11, and represent promising pharmacological targets.
TG2 is overexpressed in human liver fibrosis and cirrhosis of various etiologies, and transglutaminase-catalyzed ε-(γ-glutamyl) lysine isopeptide cross-links can be detected in hepatic scar tissue12. These findings are consistent with descriptive in vivo data in CCL4-induced liver fibrosis in rats, which suggested that the limited reversibility of advanced fibrosis after toxin withdrawal can be explained by TG2-mediated collagen cross-linking13. Moreover, in vitro TG2-mediated cross-linking can render collagen type I resistant to the action of collagenases. Thus, cleavage of collagen type I secreted by dermal fibroblasts by interstitial collagenase (MMP-1) was reduced by 50% in the presence of recombinant TG214, and cross-linking of rat tail collagen type I with TG2 increased its resistance to proteolytic degradation by MMP-1 and MMP-215.
However, while involvement of TG2 in promoting fibrosis and limiting its reversibility is plausible, the contribution of TG2 to fibrotic ECM stabilization or to reversibility of hepatic fibrosis in vivo has not been addressed in functional studies. Since tranglutaminase inhibitors, including agents specific for TG2, have already been developed as treatments of celiac disease16, validation of TG2 as a relevant antifibrotic drug target for patients with fibrotic liver diseases is of high interest 3.
In this study, we focused on the role of TG2 in stabilization of fibrotic ECM and reversibility of liver fibrosis in vivo. First, we characterized TG2 expression and activity in relation to fibrosis progression and stage, collagen accumulation and ECM turnover in two models of murine liver fibrosis in vivo. Second, we quantitatively assessed the dynamic changes in ECM stability using stepwise collagen extractions. Finally, we studied how TG2 deficiency affects ECM stability and fibrosis reversibility by comparing TG2−/− mice with their wildtype littermates.
MATERIALS AND METHODS
Animal experiments
TG2-deficient mice were generated and characterized as described previously in detail17 and backcrossed onto the C57Bl/6J background (Jackson Labs, Bar Harbor, ME) for 12 generations.
Animals were housed on a 12h dark and light cycle and fed a standard rodent chow and tap water ad libitum. Wildtype age- and sex-matched littermates from the TG2−/− colony, and wildtype C57Bl/6J mice purchased from Jackson Labs (Bar Harbor, ME), were used as controls. No difference in fibrosis susceptibility was found between controls from our colony and from Jackson (not shown). Animal experiments had been approved by the institutional IACUC (BIDMC, protocol #158-2008).
Hepatotoxin-induced liver fibrosis was induced in 7-8 weeks old mice by chronic injections of carbon tetrachloride (CCL4) or thioacetamide (TAA) and spontaneous recovery was monitored short-term (4 weeks) and long-term (8-36 weeks) after withdrawal of the hepatotoxin. First, optimal dosing regimens for fibrosis induction (CCL4 and TAA) were established in a series of pilot experiments by comparing the effect of i/p injections versus oral gavage, and constant versus escalating dose protocols on hepatic collagen accumulation. Constant hepatotoxin doses (1.75ml/kg for CCL4 and 200mg/kg for TAA) and less than 3× weekly injections were found to be insufficient to induce significant liver fibrosis in wildtype C57Bl/6 mice, resulting in a less than 2-fold increase in liver hydroxyproline (not shown). Both gavage and i/p injections of escalating doses of CCL4 led to advanced liver fibrosis in 6 weeks (with a ≥4-fold increase in liver hydroxyproline, comparable to the rat model18), but the i/p route was abandoned due to frequent peritoneal adhesions, accumulation of vehicle (oil) and inflammatory infiltrates in the peritoneal cavity of mice after 6 weeks of treatment. CCl4 was given in mineral oil via oral gavage three times a week for up to 12 weeks according to an escalating dose protocol (first dose, 0.875ml/kg; week 1-3, 1.75ml/kg; week 4-6, 2.5ml/kg; week 7-12, 3.25ml/kg). Alternatively, fibrosis was induced by escalating the dose of i/p TAA for 6 weeks (first dose, 100mg/kg, week 1-2, 200mg/kg; week 3-4, 300mg/kg; week 4-6, 400mg/kg). Mice were sacrificed always 3 days after the last CCL4 or TAA application, unless specified otherwise. Livers and spleens were weighed at sacrifice and livers snap-frozen for further analysis.
Hepatic collagen content was determined as relative hydroxyproline (μg/g liver) in 300-400mg liver samples from two different lobes after hydrolysis in 6N HCl for 16h at 110°C as described18. Total hydroxyproline (mg/whole liver) was calculated based on individual liver weights and the corresponding relative hydroxyproline content18, 19.
Extracellular matrix stability was assessed biochemically by complete collagen fractionation via serial extractions from snap frozen livers. Solubility of collagens in the ECM is inversely correlated to the extent of cross-linking 20. To characterize and quantify the stability of the collagenous ECM, we employed stepwise collagen solubilization from livers using neutral salt (freshly secreted collagens and procollagens), acetic acid (more mature collagens), and acid pepsin (fibrillar, moderately cross-linked collagens) as described by us previously21, 22 and summarized in Fig.2A. Briefly, 1g of snap frozen liver (3 samples per group, each pooled from pieces of 2-3 individual livers) was homogenized in 20ml of neutral salt buffer (0.5M NaCl, 0.05M Tris, pH 7.5, with Complete™ protease inhibitor, Roche) and incubated at 4°C overnight on a rotary shaker. After centrifugation at 24.000g for 30min, the supernatant was collected for collagen determination (fraction A: neutral salt-soluble collagen). The resulting pellet was extracted with 20ml 0.5M acetic acid (fraction B: acid-soluble collagen), followed by pepsin (2mg/ml in 0.5M acetic acid, fraction C: pepsin-soluble collagen). The remaining insoluble fraction D represents mature, highly cross-linked collagen. Each fraction was hydrolyzed in 6N HCl, concentrated 10-fold by evaporation under nitrogen and used to quantify the collagen content via hydroxyproline determination (see above). Efficient extraction was confirmed by a repeated extraction with fresh buffer, which yielded less than 2% of collagens extracted in the first step. The percentage of collagen in each fraction was calculated as fraction of the total hydroxyproline determined in all fractions. The recovery of collagens in all fractions was routinely greater than 90% of the total tissue content as determined in parallel samples.
Figure 2. Analysis of collagen solubility indicates increased cross-linking of the fibrotic ECM during progression of liver fibrosis.

A) Liver collagens were solubilized from 1g of liver tissue via sequential overnight extractions with neutral salt, acetic acid and pepsin, and collagen content in each fraction was quantified biochemically via hydroxyproline determination, as described in detail in M&M(0=healthy liver; 1,3,6,12=fibrotic livers 1, 3, 6, 12 weeks after CCL4 treatment, respectively). Data represent means±SEM from 3 samples per bar (each pooled from 2-3 individual livers for extractions and analysis). ***, p values >0.0001 analyzed using ANOVA with Dunnet’s post-test comparing to healthy controls.
Tissue transglutaminase activity was determined in liver homogenates using the biotinamidopentylamine incorporation assay as previously described in detail23, with modifications. Briefly, a 96-well plate was coated with N,N-dimethylcasein (Sigma C9801; 10μg/ml in 50mM Tris, 0.15M NaCl, 5mM EDTA, pH 7.5) for 16h, 40°C and followed by 3 washing steps with PBS/0.1% Tween. Human recombinant TG2, expressed and purified as described previously24 (1-100ng), or liver homogenate (5μg/well) was added to the reaction buffer (50mM Tris, 0.15M NaCl, 5mM CaCl2, 5mM DTT, 0.1% Tween, pH 7.5), immediately followed by the addition of 4μM biotinamidopentylamin (Pierce Nr 21345) and incubation at RT for 2-4h. Incorporation of biotinamidopentylamine into dimethylcasein was quantified using 50μl horseradish peroxidase-conjugated streptavidin (Sigma S 2438, in PBS/0.1% Tween) and 50μl tetramethylbenzidine substrate and detection at 630nm.
Quantitative Real-Time RT-PCR
Relative mRNA levels were quantified by real-time RT-PCR on a LightCycler 1.5 instrument (Roche, Mannheim, Germany) using the TaqMan and SYBR Green methodology as described by us in detail 18, 19, 25. TaqMan probes (dual-labeled with 5′-FAM and 3′-TAMRA) and primers (summarized in Suppl. Tab.1) were designed and validated as described18, 19, 25.
Statistical analyses
Data are expressed as means±SEM, and statistical analyses were performed using Microsoft EXCEL and GraphPad Prism version 5.00 (GraphPad Software, San Diego, CA). Multiple comparisons were performed by one-way analysis of variance (ANOVA). Two planned comparisons were performed to each of the control groups: healthy non-fibrotic mice (sham) and mice at peak of fibrosis (PF) in fibrosis reversal experiments using the Dunnett’s post-test. Differences among selected experimental groups with p values lower than 0.05 were considered significant.
RESULTS
Increase in TG2 expression and activity parallels progression of experimental liver fibrosis in mice
Previous cross-sectional studies indicated an increase in TG2 mRNA and activity in humans with liver fibrosis12. We further characterized the dynamic changes in TG2 gene expression and activity in relation to both fibrosis stage and activity of fibrogenesis in a model of progressive liver fibrosis due to chronic CCL4 injection in C57Bl6 mice. An optimized protocol of CCL4 administration with an escalating dose regimen (see Methods) led to robust collagen deposition and progressive fibrosis detectable both biochemically as an increase in hydroxyproline and histologically as an increase in fibrosis stage. Fibrosis was characterized as incipient at week 1 (mean stage F1 according to Metavir: 1.98-fold collagen increase), moderate at week 3 (mean stage F2: 3.44-fold collagen increase), advanced at week 6 (stage F2-F3: 4.56-fold increase in collagen) and early cirrhosis at week 12 (F3-F4: 6.21-fold collagen increase) (Fig.1A,B and Tab. 1). Splenomegaly, an indirect measure of portal hypertension, developed starting at week 6 (spleen weight 98.6±0.5mg), reaching 144.7±1.2mg at 12 weeks compared to 76.4±0.3mg in healthy controls (p<0.0001). TG2 mRNA was elevated 3-fold after 1 week and remained elevated 2-fold up to 12 weeks (Fig.1C). Total transglutaminase (transamidating) activity was moderately elevated at any time point tested during fibrosis induction (up to 1.5-fold compared to normal controls at week 3, Fig.1D). Moreover, there was a strong correlation of TG2 mRNA to commonly used markers of fibrogenic activity (p>0.001), such as transcripts for procollagen α1(I) (COL1A1) mRNA (the major collagen chain present in scar tissue, r=0.8735 in Pearson test, Fig.1E), TGFβ1 (the major profibrogenic cytokine, r=0.8639, not shown) and α-SMA (a hepatic stellate cell activation marker, r=0.8954, not shown).
Figure 1. Tranglutaminase expression and activity is elevated in progressive CCL4-induced liver fibrosis and correlates with fibrogenesis activity.

A) Total hepatic collagen content progressively increases at 1, 3, 6 and 12 weeks after CCL4 treatment in C57Bl/6 mice as determined biochemically via hepatic hydroxyproline. B) Low magnification images of connective tissue stain (Sirius Red) of representative sections from livers at 1, 3, 6 and 12 weeks after CCL4 treatment (original magnification, x50). C) Time-course of TG2 mRNA expression, which D) strongly correlates with COL1A1 (procollagen type 1) mRNA (Pearson’s test).Hepatic transcript levels were determined by quantitative RT-PCR, and are expressed as arbitrary units (fold increase vs. controls) after normalization to β2MG mRNA. (E) Total transglutaminase activity in liver homogenates. All data are means±SEM(n=6-8 for each bar) *, p<0.05 as compared to vehicle treated controls (Ctrl) (ANOVA).
Table 1.
Changes in liver/spleen weight and hepatic collagen content in C57Bl/6 mice treated with an escalating dosing regimen of CCL4 via gastric gavage, as described in M&M. Liver and spleen weights were measured at sacrifice and collagen deposition was determined biochemically as relative (μg/g) hydroxyproline (HYP) content in 250mg liver samples from two different lobes. Total HYP(mg/whole liver) was calculated from individual liver weights and respective relative HYP values. Ctrl: non-fibrotic control group (n=6) that received vehicle (mineral oil) only; CCL4: fibrotic mice treated with CCL4 for 1, 3, 6, and 12 weeks. Data are expressed as means ±SEM.
| Group | Liver weight, g | Spleen weight, mg | Relative HYP, μg/100mg |
Total HYP, μg/liver |
|---|---|---|---|---|
| Ctrl,n=6 | 1.30±0.04 | 76.38±3.08 | 13.50±0.96 | 178±17.34 |
| CCl4, 1w,n=7 | 1.60±0.05* | 90.86±5.60 | 22.15±0.37* | 353±10.29* |
| CCl4, 3w,n=7 | 1.77±0.11* | 88.43±4.06 | 34.87±1.59* | 612±36.36* |
| CCl4, 6w,n=7 | 1.78±0.04* | 98.57±4.79* | 45.57±0.92* | 811±28.32* |
| CCl4, 12w,n=6 | 2.05±0.06* | 144.70±11.50* | 54.00±2.48* | 1107±58.46* |
, p<0.05 as compared to Ctrl (ANOVA followed by Dunett’s post-test).
Analysis of hepatic fibrotic matrix in vivo reveals progressive collagen stabilization
To study dynamic changes in the ECM stability in liver fibrosis, we performed a comprehensive analysis of the extractability of collagens from livers along the progression of CCL4-induced hepatic fibrosis. Liver tissue from each group of mice was homogenized and subjected to a series of sequential collagen extractions using neutral salt, acetic acid and pepsin to solubilize the collagens from the hepatic ECM. The amount of collagen extracted at each step (as well as the collagen remaining in the insoluble fraction) was directly quantified by hydroxyproline determination after complete hydrolysis of each fraction. In normal murine liver, salt-soluble and acid-soluble fractions, representing newly synthesized collagens, amounted to 3.17% and 1.16% of total collagen, respectively. The pepsin-soluble (modestly to moderately cross-linked) fraction represented the vast majority of collagens (91.14%), while 4.53% of collagens were not solubilized by any of the above mentioned extraction procedures. During fibrosis progression, the salt- and acid soluble fractions did not change significantly at any time point compared to healthy liver, suggesting a constant proportion of freshly deposited collagen. However, the insoluble collagen fraction increased dramatically with fibrosis progression, from 4.5% in control livers to almost 27% (6-fold) at week 12 of CCl4 administration (Fig.2A). This increase in highly-cross-linked, insoluble collagen apparently occurred at the expense of the pepsin-soluble fraction, which decreased from ~91% in controls to ~70% in mice treated with CCL4 for 12 weeks (Fig.2B).
Genetic deletion of TG2 decreases total tranglutaminase activity in the normal and fibrotic liver
TG2, which is prominently expressed in the liver, was hypothesized to play a role in fibrotic matrix stabilization, thus limiting fibrosis reversibility12, 13, but it remained to be shown if lack of TG2 (or pharmacologic inhibition of its activity) would lead to 1) reduced fibrosis due to facilitated matrix degradation and removal during fibrogenesis, and 2) a less stable fibrotic ECM that would be more susceptible to proteolytic degradation in the recovery (reversal) phase. By using mice deficient in TG2 we showed that the deletion of TG2 was indeed associated with a significant (2-fold) decrease in total transglutaminase activity in the normal and fibrotic liver in vivo. Reduced transglutaminase activity failed to increase in fibrotic TG2−/− livers, indicating that no compensation occurred from other TG isoforms (see Suppl. Fig.1 and supplementary material for further details). Therefore, TG2−/− mice represent a reliable model to study the functional consequences of deficient transglutaminase activity (or its pharmacological inhibition) in fibrotic ECM turnover in the liver, and were employed in the subsequent experiments.
TG2 deficiency does not facilitate collagen removal during short-term recovery from hepatotoxin-induced liver fibrosis
Next, we addressed the question of how significantly the lack of TG2 expression and activity affects the properties of the fibrotic ECM during spontaneous recovery, and whether this matrix will be more prone to degradation and removal once the profibrogenic stimuli are eliminated. After 6 weeks of CCl4 treatment as established previously (Fig.1), the “peak of fibrosis” group and the healthy controls (Ctrl, receiving vehicle only) of both TG2−/− and TG2+/+ mice were sacrificed, and the extent of fibrosis evaluated. CCL4 injections were stopped in another group of fibrotic mice, which were allowed to recover (“recovery” group) for an additional 4 weeks without any further treatment. Wildtype and TG2−/− mice developed advanced bridging fibrosis after 6 weeks of CCL4 treatment, with TG2−/− mice displaying a slightly higher (10%) total collagen content (not significant), as determined by biochemical quantification and histological assessment (Fig.3, Suppl. Tab.2), in agreement with histological evidence in an earlier report26. This trend of more collagen deposited in the TG2−/− mice was confirmed in the mechanistically different model of thioacetamide (TAA)-induced liver fibrosis (a 15% higher relative and total collagen content compared to wildtype mice, not significant, Suppl. Tab.2). After 4 weeks of recovery in both TG2−/− and WT mice, inflammatory infiltrates had disappeared almost completely (not shown), and profibrogenic gene expression had normalized (Fig.3C). This was associated with a notable improvement of liver histology, characterized mainly by hepatocyte regeneration and recovery from toxic damage (reduced vacuolization and eosinophilic necrosis) (Fig.3B,E), in accordance with prior observations by us and others in rats and mice13, 18. The number of α-SMA positive cells, representing largely activated myofibroblasts, increased dramatically at the peak of fibrosis and returned to nearly normal as early as after 4w of recovery, without significant difference between wildtype and TG2−/− mice and the fibrosis models (Suppl. Fig. 2). Notably, there was no decrease in total collagen content in either wildtype or TG2−/− mice recovering from CCL4- or TAA-induced fibrosis for 4 weeks (Fig.3A,D).
Figure 3. Lack of spontaneous fibrosis regression after short-term recovery in wildtype and TG2−/− mice with advanced liver fibrosis induced with CCL4 or TAA.

Total hepatic collagen content at the peak of fibrosis induced by CCL4 (A) or TAA (D) for 6 weeks, and after 4 weeks of recovery in TG2−/− (open bars) mice and their wildtype littermates (closed bars), as determined biochemically via hepatic hydroxyproline. B) Low magnification images of connective tissue stain (Sirius Red) of representative sections from livers at 6w of CCL4 (B) or TAA (E) treatment and after 4 weeks of spontaneous recovery (original magnification, ×50). C) Normalization of profibrogenic gene expression after 4w of recovery. Hepatic transcript levels of procollagen type 1(COL1A1), TGFβ1 and TIMP-1 were determined by quantitative RT-PCR, and expressed in arbitrary units (fold increase vs. controls) after normalization to β2MG mRNA. All data are means±SEM(n=6-8 for each bar) *, p<0.05 vs. The vehicle controls (Ctrl) of the respective genotype. #, p<0.05 vs. the fibrotic controls (ANOVA).
Lack of TG2 does not facilitate collagen removal during long-term recovery from hepatotoxin-induced liver fibrosis
In order to exclude the possibility that 4 weeks of recovery were insufficient to unequivocally demonstrate a lack of advanced fibrosis resolution, we extended the recovery phase up to 36 weeks in mice with CCL4-induced fibrosis. Long-term recovery was accompanied by a significant decrease in relative hepatic (amount per g of tissue) collagen content beginning at 12 weeks of recovery (~26% in TG2−/− mice at 12 weeks and ~23% in WT mice at 24 weeks, compared to the peak of fibrosis). However, when total collagen content was determined factoring in individual liver weights, the reduction was not significant, irrespective of genotype and of the duration of recovery up to 36 weeks (Fig.4C). Thus the average total collagen content per liver, even after 36 weeks of recovery, was reduced only by ~12.7% and ~7.4% in WT and TG2−/− mice, respectively, not reaching statistical significance in any group. This is in line with our prior observations in the rat model of TAA-induced fibrosis when livers were examined at 8 weeks of recovery18. Therefore, the reduction in relative collagen content was evidently due to hepatocyte regeneration and a subsequent increase in liver mass after cessation of the injury (Fig.4) and not due to true fibrolysis, since the 23-26% reduction in relative collagen content was paralleled by a corresponding 18-20% increase in liver weights (Fig.4C, Suppl. Tab.3). This was confirmed microscopically by a thorough examination of connective tissue staining, which demonstrated the persistence of fibrillar collagen deposits throughout the hepatic parenchyma after 36 weeks of recovery, which were now more loosely associated within the former locations of the fibrotic septa (Fig.4D). Interestingly, a frequent finding was that “invading” hepatocytes caused dissipation and widening of the fibrotic septa, splitting these into multiple strands of thinner fibrils as also demonstrated by morphometric analysis (Fig. 4E,F). This was evident as early as 4 weeks of fibrosis induction in both genotypes, further increasing up to 36 weeks after CCL4 withdrawal (Fig.4E), and closely resembled features of fibrotic liver remodeling described in advanced human fibrosis 27.
Figure 4. Lack of significant collagen removal after long-term recovery up to 36 weeks in wildtype and TG2−/− mice with advanced CCL4-induced liver fibrosis.

Relative (A) and total (B) hepatic collagen content at the peak of fibrosis induced with CCL4 for 6 weeks, and after 8, 12, 24 and 36 weeks of recovery in wildtype (closed bars) and TG2−/− (open bars) C57Bl/6 mice, as determined biochemically via hepatic hydroxyproline. C) Compensatory increase in liver volume during recovery coincides with a decrease in relative, but not total, hepatic collagen content as measured by liver weight at sacrifice. All data are means±SEM(n=6-10 for each bar). D) High magnification images of collagen (Sirius Red) of representative sections from livers at 6 weeks of CCL4 treatment and after 36 weeks of spontaneous recovery (original magnification, ×200). E) Hepatocytes invading fibrotic septa are a characteristic feature observed during long-term recovery (WT mice after 8 weeks of recovery shown, original magnification as indicated). F) Morphometric analysis of scar tissue remodeling during recovery demonstrates widening of septa and splitting of condensed collagen bundles into thinner fibrils. Measurements were performed at ×200 magnification, using an ocular net micrometer. Thickness of septa and number of fibrils was assessed at outer boundaries of the middle third of at least 10 randomly selected complete fibrotic septa in specimens from a right and a left liver lobe and from 4 individual mice/group at peak fibrosis and at 4 weeks of resolution). *, p<0.05 as compared to vehicle controls (Ctrl) of the respective genotype. #, p<0.05 vs. the fibrotic controls (ANOVA).
Stability of the fibrotic matrix is not affected by the lack of TG2 during progression or recovery
Finally, we quantitatively assessed the effect of TG2 deficiency on fibrotic matrix stabilization during progression and recovery from CCL4-induced liver fibrosis using fractional collagen extractions as described above. At none of the several time points of fibrosis progression did collagen extractability differ between wildtype and TG2−/− mice, including the insoluble, highly cross-linked scar collagen. Interestingly, after 36 weeks of recovery, the percentage of insoluble collagen tended to be slightly higher in both wildtype and TG2−/− mice (24.3% and 21.4%, respectively), compared to the peak of fibrosis (19.69% and 16.69%, respectively). This may indicate that the process of ECM stabilization does not stop (or reverse) after elimination of the fibrogenic stimulus, but even continues, although at a much slower pace, even in the absence of TG2 (Fig.5) Additional studies are needed to confirm this observation.
Figure 5. Genetic deletion of tTG does not affect solubility of collagen in mice without fibrosis, and mice with advanced fibrosis or after long-term recovery.
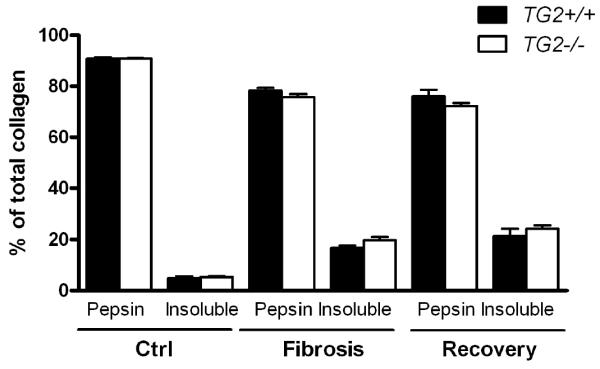
Fibrotic ECM solubility was determined in TG2−/− mice (open bars) and wildtype controls (closed bars) by serial extraction of liver tissue and quantification of extracted collagen in each fraction as described in M&M. Only the quantitatively relevant pepsin-soluble and insoluble fractions are shown. Ctrl, vehicle-treated, nonfibrotic controls. “Fibrosis”, fibrotic mice at the peak of fibrosis (6 weeks of CCL4 treatment). “Recovery”, CCL4 treatment was stopped, and fibrotic mice were allowed to recover for an additional 36 weeks. No differences between genotypes or treatments was found in the salt- and acid-soluble fractions, which amounted to less than 5% of total collagen combined (not shown). Data represent means±SEM from 3 samples per bar (each pooled from 2-3 individual livers for extractions and analysis).
DISCUSSION
Tissue transglutaminase (TG2) has been proposed to play an important role in fibrosis due to its (in vitro) activity to cross-link collagen within the fibrotic matrix, thus limiting fibrosis reversibility. In vivo evidence was based on the detection of ε-(γ-glutamyl) lysine isopeptide cross-links in fibrotic livers of humans12 and rodents13. In our study, we characterized dynamic changes in ECM (collagen) stability in progressive liver fibrosis and directly addressed the functional contribution of TG2, the predominant hepatic transglutaminase isoform, to collagen stabilization during fibrosis progression and fibrosis reversal in vivo using mice with genetic deletion of TG2. Analysis of fibrotic ECM stability via sequential collagen extractions revealed that irreversible collagen stabilization is a characteristic feature of progressive fibrosis, with a striking stage-dependent increase in the pepsin-insoluble fraction, which was accompanied by an increase in TG2 activity and mRNA expression. However, although TG2 expression and activity increased during fibrosis progression and strongly correlated with transcript levels of fibrogenic genes, TG2 deficiency had no measurable effect on the stability of the fibrotic ECM or on reversibility of hepatic fibrosis in vivo in two hepatotoxin-induced models. Thus, contrary to what was widely believed, TG2-mediated cross-linking does not seem to functionally contribute to collagen stabilization and irreversibility of fibrosis in the liver.
Targeting ECM stability is an attractive strategy to treat fibrosis, and to increase its reversibility once the underlying cause has been eliminated3. Although this field is largely unexplored, there are isolated reports supporting a potential benefit of blocking post-translational collagen modifications. Thus, earlier studies suggest that administration of beta-aminopropionitrile (BAPN), which irreversibly inhibits lysyl oxidase-dependent collagen cross-linking, might mitigate the development of cirrhosis28, and partially prevent the increase in liver stiffness determined by direct rheometry in fibrotic rats subjected to chronic CCL4 administration29. Interestingly, fibrotic matrix stiffness itself, which is believed to depend on collagen cross-linking, has been shown to directly activate primary hepatic myofibroblasts (the major fibrogenic effector cell in the liver), when they are cultured on artificial matrices of increasing stiffness30. Moreover, it was demonstrated that mice lacking ADAMTS2, the protease which permits lateral alignment and cross-linking of freshly secreted triple helical collagen to collagen fibrils by cleaving the N-terminal procollagen propeptide from procollagens type I, II, III, and V, had a slower rate of collagen deposition during chronic CCL4 administration, and a higher rate of fibrosis reversal after CCL4 withdrawal31. These data, coupled with the observation that fibrous septa in ADAMTS2-/- mice were made of thinner and irregular collagen fibers, is consistent with the concept that collagen maturation and stabilization is an important therapeutic target in hepatic fibrosis.
In vivo studies on collagen stabilization are difficult, requiring the direct quantification of various collagen cross-links after complete hydrolysis of the liver tissue with multiple internal standards and expensive equipment6, and are hampered by the lack of reagents to detect cross-links in situ. Indeed, we were not able to reliably detect the transglutaminase-generated ε-(γ-glutamyl) lysine isopeptide in terminal proteolytic digests of normal or fibrotic liver homogenates by HPLC and mass spectrometry-based methods. This is likely due both to the low abundance of this crosslink in vivo and an inhibitory “matrix effect” in complex biological samples (see Suppl. Fig. 3&4 and supplementary material for details). With regard to TG2-mediated cross-links, erroneous results may have been produced in the past by using commercial antibodies that show unspecific binding in the context of native proteins32. We therefore used mice with genetic deletion of TG2 and compromised transamidating activity as the major approach, coupled with quantification of the degree of overall collagen cross-linking using sequential collagen extraction and the model of spontaneous fibrosis reversal as a major functional end-point.
We clearly demonstrated that progressive collagen cross-linking is a central characteristic during liver fibrosis progression in vivo, which to our knowledge had not been shown directly in models of experimental hepatic fibrosis before. We developed a simple and highly reproducible methodology to assess ECM stability via sequential collagen extractions, which will be useful for future studies on fibrotic matrix stabilization and the evaluation of antifibrotic agents targeting this mechanism. It is plausible that this process, which we show to be TG2-independent, limits the reversibility of hepatic fibrosis, even once the fibrogenic stimuli are eliminated. Surprisingly, no quantitative reduction in total liver collagen was observable neither in wildtype nor in TG2−/− mice during long-term recovery for up to 36 weeks, indicating that collagen degradation and removal does not occur during the recovery phase. This finding contradicts a prior report which suggested that mice bearing a mutation in the procollagen I gene, which confers resistance to collagenase, demonstrate a lack of spontaneous collagen degradation after recovery from CCL4-induced fibrosis33. While the aforementioned study reports a 43% decrease in relative hydroxyproline levels in wildtype controls and impaired hepatocyte proliferation in r/r mice, changes in liver volume were not considered and total hepatic collagen content was not determined. Our present and prior studies18, 34 indicate that an increase in hepatocyte mass during regeneration results in changes in relative collagen levels during recovery even in the absence of collagen degradation. Importantly, 36 weeks of recovery in mice is equivalent to roughly one third of their life-span, comparable to 20-30 years in humans. From the clinical perspective, since most patients are diagnosed with advanced fibrosis or cirrhosis in their 4th to 6th decade, it is tempting to speculate that even if the cause of hepatic fibrosis is eliminated, e.g., due to successful pharmacological treatment of hepatitis B or C, fibrolytic therapy which actively reverses advanced fibrosis/early cirrhosis may still be required.
While TG2 deficiency did not facilitate reversal of liver fibrosis, TG2−/− deficient mice seem to be slightly more susceptible to liver inflammation and fibrosis, consistent with a prior study26. One plausible explanation would be the role of TG2 in facilitating apoptosis and phagocytic removal of apoptotic cells35, 36, which are processes directly implicated in control of both inflammation and fibrosis progression37. Indeed, observed profibrogenic effect of TG2 deficiency that is unrelated to ECM cross-linking is consistent with inefficient macrophage phagocytosis. This may shift the balance towards fibrogenesis by limiting fibrolysis, since macrophages secrete fibrolytic MMPs upon apoptotic cell engulfment and play a critical role in (biliary) fibrosis reversal34. However, whatever the contribution of these and other activities of TG2 are, our approach clearly demonstrated that overall TG2 does not favor collagen (ECM) crosslinking, nor significantly affects (liver) fibrosis progression or regression.
In conclusion, we demonstrate that 1) TG2 is upregulated during fibrogenesis but is dispensable for the stabilization of the fibrotic liver ECM in vivo; 2) The fibrotic matrix formed in the absence of TG2 is not more easily degradable during spontaneous recovery from liver fibrosis than that of wildtype mice; 3) TG2-independent irreversible collagen cross-linking is a remarkable feature of progressive hepatic fibrosis, and represents a promising target for novel antifibrotic therapies.
Supplementary Material
Acknowledgements
We would like to thank Jessica Zaks (BIDMC) for her excellent technical assistance with the animal experiments, Prof. M. Griffin (Nottingham Trent University, UK) for sharing protocols and expert advice for performing terminal proteolytic liver digests for cross-link quantification, Mary Adams (Dana-Farber Cancer Institute, Boston) for help with HPLC and Suzanne White (BIDMC) for her valuable assistance with histology.
Grant support: This work was supported by an appointment Grant by the Beth Israel Deaconess Medical Center, and the National Institutes of Health (grants NIH 1 R21 DK076873-01A1 and 1 R21 DK075857-01A2) to D.S. Y.P. was recipient of a Sheila Sherlock fellowship of the European Association for the Study of the Liver.
Abbreviations used in this paper
- CCL4
carbon tetrachloride
- ECM
extracellular matrix
- HYP
hydroxyproline
- LOX
lysyl oxidase
- MMP
matrix metalloproteinase
- TG
transglutaminase
- TAA
thioacetamide
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors contributions: Study concept and design, analysis and interpretation of the data, wrote the manuscript - Yury Popov and Detlef Schuppan; performed experiments and data acquisition - Yury Popov, Deanna Y. Sverdlov, Anisha K Sharma, K. Ramakrishnan Bhaskar, Shaoyong Li, James Lee; contributed tools and reagents - Tobias L. Freitag, Walburga Dieterich, Gerry Melino; obtained funding - Detlef Schuppan.
Conflict of interest/financial disclosures: none to be declared
REFERENCES
- 1.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–51. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50:1294–306. doi: 10.1002/hep.23123. [DOI] [PubMed] [Google Scholar]
- 4.Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21:351–72. doi: 10.1055/s-2001-17556. [DOI] [PubMed] [Google Scholar]
- 5.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304–16. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robins SP. Biochemistry and functional significance of collagen cross-linking. Biochem Soc Trans. 2007;35:849–52. doi: 10.1042/BST0350849. [DOI] [PubMed] [Google Scholar]
- 7.Beninati S, Piacentini M. The transglutaminase family: an overview: minireview article. Amino Acids. 2004;26:367–72. doi: 10.1007/s00726-004-0091-7. [DOI] [PubMed] [Google Scholar]
- 8.Collighan RJ, Griffin M. Transglutaminase 2 cross-linking of matrix proteins: biological significance and medical applications. Amino Acids. 2009;36:659–70. doi: 10.1007/s00726-008-0190-y. [DOI] [PubMed] [Google Scholar]
- 9.Kim SY, Jeitner TM, Steinert PM. Transglutaminases in disease. Neurochem Int. 2002;40:85–103. doi: 10.1016/s0197-0186(01)00064-x. [DOI] [PubMed] [Google Scholar]
- 10.Elli L, Bergamini CM, Bardella MT, Schuppan D. Transglutaminases in inflammation and fibrosis of the gastrointestinal tract and the liver. Dig Liver Dis. 2009;41:541–50. doi: 10.1016/j.dld.2008.12.095. [DOI] [PubMed] [Google Scholar]
- 11.Telci D, Griffin M. Tissue transglutaminase (TG2)--a wound response enzyme. Front Biosci. 2006;11:867–82. doi: 10.2741/1843. [DOI] [PubMed] [Google Scholar]
- 12.Grenard P, Bresson-Hadni S, El Alaoui S, Chevallier M, Vuitton DA, Ricard-Blum S. Transglutaminase-mediated cross-linking is involved in the stabilization of extracellular matrix in human liver fibrosis. J Hepatol. 2001;35:367–75. doi: 10.1016/s0168-8278(01)00135-0. [DOI] [PubMed] [Google Scholar]
- 13.Issa R, Zhou X, Constandinou CM, Fallowfield J, Millward-Sadler H, Gaca MD, Sands E, Suliman I, Trim N, Knorr A, Arthur MJ, Benyon RC, Iredale JP. Spontaneous recovery from micronodular cirrhosis: evidence for incomplete resolution associated with matrix cross-linking. Gastroenterology. 2004;126:1795–808. doi: 10.1053/j.gastro.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 14.Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, Griffin M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13:1442–53. doi: 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- 15.Zhou X, Jamil A, Nash A, Chan J, Trim N, Iredale JP, Benyon RC. Impaired proteolysis of collagen I inhibits proliferation of hepatic stellate cells: implications for regulation of liver fibrosis. J Biol Chem. 2006;281:39757–65. doi: 10.1074/jbc.M605621200. [DOI] [PubMed] [Google Scholar]
- 16.Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009;137:1912–33. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 17.De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol. 2001;21:148–55. doi: 10.1128/MCB.21.1.148-155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popov Y, Patsenker E, Bauer M, Niedobitek E, Schulze-Krebs A, Schuppan D. Halofuginone induces matrix metalloproteinases in rat hepatic stellate cells via activation of p38 and NFkappaB. J Biol Chem. 2006;281:15090–8. doi: 10.1074/jbc.M600030200. [DOI] [PubMed] [Google Scholar]
- 19.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)-/- mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43:1045–54. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 20.Oxlund H, Jorgensen PH, Ortoft G, Andreassen TT. Alterations in the cross-links of skin collagen of rats treated with biosynthetic growth hormone. Connect Tissue Res. 1991;26:65–75. doi: 10.3109/03008209109152164. [DOI] [PubMed] [Google Scholar]
- 21.Schuppan D, Cantaluppi MC, Becker J, Veit A, Bunte T, Troyer D, Schuppan F, Schmid M, Ackermann R, Hahn EG. Undulin, an extracellular matrix glycoprotein associated with collagen fibrils. J Biol Chem. 1990;265:8823–32. [PubMed] [Google Scholar]
- 22.Becker J, Schuppan D, Benzian H, Bals T, Hahn EG, Cantaluppi C, Reichart P. Immunohistochemical distribution of collagens types IV, V, and VI and of pro-collagens types I and III in human alveolar bone and dentine. J Histochem Cytochem. 1986;34:1417–29. doi: 10.1177/34.11.3772076. [DOI] [PubMed] [Google Scholar]
- 23.Dieterich W, Esslinger B, Trapp D, Hahn E, Huff T, Seilmeier W, Wieser H, Schuppan D. Cross linking to tissue transglutaminase and collagen favours gliadin toxicity in coeliac disease. Gut. 2006;55:478–84. doi: 10.1136/gut.2005.069385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dieterich W, Trapp D, Esslinger B, Leidenberger M, Piper J, Hahn E, Schuppan D. Autoantibodies of patients with coeliac disease are insufficient to block tissue transglutaminase activity. Gut. 2003;52:1562–6. doi: 10.1136/gut.52.11.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, Kolb A, Friess H, Schuppan D. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48:453–64. doi: 10.1016/j.jhep.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 26.Nardacci R, Lo Iacono O, Ciccosanti F, Falasca L, Addesso M, Amendola A, Antonucci G, Craxi A, Fimia GM, Iadevaia V, Melino G, Ruco L, Tocci G, Ippolito G, Piacentini M. Transglutaminase type II plays a protective role in hepatic injury. Am J Pathol. 2003;162:1293–303. doi: 10.1016/S0002-9440(10)63925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis. Morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med. 2000;124:1599–607. doi: 10.5858/2000-124-1599-ROHC. [DOI] [PubMed] [Google Scholar]
- 28.Fiume L, Favilli G. Inhibition of experimental cirrhosis by carbon tetrachloride following treatment with aminoacetonitrile. Nature. 1961;189:71–2. doi: 10.1038/189071a0. [DOI] [PubMed] [Google Scholar]
- 29.Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, Mick R, Janmey PA, Furth EE, Wells RG. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1147–54. doi: 10.1152/ajpgi.00032.2007. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–56. doi: 10.1002/hep.21792. [DOI] [PubMed] [Google Scholar]
- 31.Kesteloot F, Desmouliere A, Leclercq I, Thiry M, Arrese JE, Prockop DJ, Lapiere CM, Nusgens BV, Colige A. ADAM metallopeptidase with thrombospondin type 1 motif 2 inactivation reduces the extent and stability of carbon tetrachloride-induced hepatic fibrosis in mice. Hepatology. 2007;46:1620–31. doi: 10.1002/hep.21868. [DOI] [PubMed] [Google Scholar]
- 32.Johnson GV, LeShoure R., Jr Immunoblot analysis reveals that isopeptide antibodies do not specifically recognize the epsilon-(gamma-glutamyl)lysine bonds formed by transglutaminase activity. J Neurosci Methods. 2004;134:151–8. doi: 10.1016/j.jneumeth.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Issa R, Zhou X, Trim N, Millward-Sadler H, Krane S, Benyon C, Iredale J. Mutation in collagen-1 that confers resistance to the action of collagenase results in failure of recovery from CCl4-induced liver fibrosis, persistence of activated hepatic stellate cells, and diminished hepatocyte regeneration. Faseb J. 2003;17:47–9. doi: 10.1096/fj.02-0494fje. [DOI] [PubMed] [Google Scholar]
- 34.Popov Y, Sverdlov DY, Bhaskar KR, Sharma AK, Millonig G, Patsenker E, Krahenbuhl S, Krahenbuhl L, Schuppan D. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol. 2010;298:G323–34. doi: 10.1152/ajpgi.00394.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarang Z, Toth B, Balajthy Z, Koroskenyi K, Garabuczi E, Fesus L, Szondy Z. Some lessons from the tissue transglutaminase knockout mouse. Amino Acids. 2009;36:625–31. doi: 10.1007/s00726-008-0130-x. [DOI] [PubMed] [Google Scholar]
- 36.Szondy Z, Sarang Z, Molnar P, Nemeth T, Piacentini M, Mastroberardino PG, Falasca L, Aeschlimann D, Kovacs J, Kiss I, Szegezdi E, Lakos G, Rajnavolgyi E, Birckbichler PJ, Melino G, Fesus L. Transglutaminase 2-/- mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc Natl Acad Sci U S A. 2003;100:7812–7. doi: 10.1073/pnas.0832466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canbay A, Higuchi H, Bronk SF, Taniai M, Sebo TJ, Gores GJ. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology. 2002;123:1323–30. doi: 10.1053/gast.2002.35953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.