

Abstract
Initiation of a successful immune response requires a working set of sensors that detect any noxious agent within the cellular microenvironment and molecular platforms that process this signal to trigger an appropriate effector response. Pattern recognition receptors can engage different signalling cascades that lead to proinflammatory gene expression. At the same time, transcription-independent events such as activation of proteases and/or phagocytosis are also initiated. The inflammasome pathway constitutes a signalling platform that leads to the activation of so-called inflammatory caspases, most notably caspase-1, which plays a pivotal role in the cleavage and thus maturation of proinflammatory cytokines, but also in the induction of pyroptosis, a special type of cell death. In this review we elaborate on the currently known inflammasome complexes with a special focus on the mechanism behind their activation. Understanding these mechanisms could provide important information regarding the potential signalling nodes that might be targeted for therapeutic intervention.
Keywords: caspase-1, IL-1β, inflammasome
OTHER THEMES PUBLISHED IN THIS IMMUNOLOGY IN THE CLINIC REVIEW SERIES
Allergy, Host Responses, Cancer, Type 1 diabetes and viruses, Metabolic diseases.
Introduction
Survival of multi-cellular organisms depends on the ability to distinguish between self and non-self, the maintenance of the delicate balance between symbiotic and pathogenic organisms and the existence of systems that enable a competent, efficient and swift response to different types of noxious challenges. Working sets of sensors that detect the presence of harmful stimuli in the extra and/or intracellular milieu are one of the key components of the response system. A whole gamut of pattern recognition receptors (PRRs) has been identified that are activated by various injurious stimuli. In general, these PRRs can be classified into four major families that can, either individually or co-operatively, detect danger signals: the Toll-like receptor system (TLRs), the retinoic-acid-inducible protein (RIG)-like receptors (RLRs), the C-type lectin receptors (CLRs) and the nucleotide-binding domain leucine-rich repeats (NLRs). These PRRs can be broadly divided further into membrane-associated PRRs such as TLRs and CLRs, and cytosolic PRRs such as RLRs, and NLRs. These PRRs detect distinct microbial ligands, known as microbe-associated molecular patterns (MAMPs), and orchestrate the subsequent innate and adaptive immune response. At the same time, these receptors can also be activated by endogenous signals that arise during tissue damage or cell stress. In analogy to MAMPs, these ligands are commonly referred to as danger associated molecular patterns (DAMPs). Some of these PRR pathways are also activated in certain sterile inflammatory conditions that present in the absence of microbial infection and without adaptive immune responses. This disease entity, also known as autoinflammation, is driven in large part by the proinflammatory cytokine interleukin (IL)-1β, which requires post-translational activation by the so-called inflammasome pathway. Here, a chronic or periodic inflammatory response is probably triggered by a spontaneously activated or hyper-activatable inflammasome system. This can be due to gain of function mutations within inflammasome components themselves, loss of function mutations in negative regulators and also mutations that lead to a constant release of DAMPs (for a review see [1]). In this review we want to give an overview on currently known inflammasome complexes and focus particularly on their known mechanisms of activation. In this respect, we will concentrate on ligand-dependent modes of activation, whereas mutations leading to autoinflammatory diseases are discussed by L. Franchi and R. Goldbach-Mansky in separate reviews in this issue [2,3].
Inflammasome: an overview
The inflammasome is a large multimeric procaspase-1 activating platform that is essential for the processing and thus activation of the proinflammatory cytokines IL-1β and IL-18. Initial biochemical characterization by the pioneering work of Jürg Tschopp's group led to the term ‘inflammasome’, which highlighted its role in inflammation and described the fact of a large protein complex forming (σῶµα = soma; ancient Greek for body). Activation of inflammasomal sensors, such as nucleotide-binding domain and leucine-rich repeat containing family pyrin (NLRP) proteins or absent in melanoma 2 (AIM2), triggers the cleavage of procaspase-1 to active caspase-1 that then processes, among a variety of other targets, the highly proinflammatory cytokine pro-IL-1β to mature IL-1β. In the case of IL-1β, three distinct events are required for secretion of the bioactive cytokine: first, activation of the cell by PRRs, like TLRs, leads to transcription of pro-IL-1β. Secondly, pro-IL-1β is cleaved into biologically active IL-1β by an active caspase-1 complex. Thirdly, mature IL-1β is secreted into the extracellular space. The inflammasome complexes play an important role in the second step and are probably also instrumental in initiating the third and final step of secretion. Next to its prominent role in processing of proinflammatory cytokine targets, caspase-1 also cleaves many other cytosolic proteins, the role of those is only starting to be understood [4].
Activation of procaspase-1 can also trigger a special type of cell death, which is commonly referred to as pyroptosis. This caspase-1-dependent cell death was first identified in Salmonella-infected macrophages and was then termed pyroptosis to differentiate it from the caspase-3-dependent apoptosis and also necrosis [5]. A unique feature of pyroptosis is its association with the secretion of inflammatory cytokines caused by the rapid loss of cell membrane integrity and release of cytosolic contents [6]. It was always suspected that pyroptosis was part of the defence mechanism employed by the immune system to promote the removal of the infectious agent. Although certain mechanistic aspects of pyroptosis had been characterized in vitro, its function in vivo remained unknown. However, very recently it was demonstrated that macrophages infected with mutated Salmonella undergo inflammasome/caspase-1-dependent, yet IL-1β and IL-18-independent pyroptosis, thereby releasing the bacteria that are subsequently phagocytosed and degraded by neutrophils [7]. Thus inflammasome-triggered pyroptosis can be considered as an additional and independent effector mechanism that limits the spread of infectious agents by promoting death of the infected immune cells.
While procaspase-1 activation is an important downstream consequence of inflammasome activation, several studies have alluded to inflammasome-dependent, but caspase-1-independent outcomes upon inflammasome activation. John Bertin's group first reported that over-expression of ASC resulted in strong nuclear factor (NF)-κB activation, that was augmented synergistically by addition of NLRP3 [8]. This phenomenon of ASC-triggered NF-κB activation has been employed successfully ever since to characterize and identify putative upstream activators of ASC using transient expression assays [9]–[11]. However, ASC-deficient mouse macrophages display normal production of proinflammatory cytokines upon tumour necrosis factor (TNF)-α and lipopolysaccharide (LPS) activation [12,13]. Nevertheless, in studies undertaken by Jenny Ting and colleagues in humans cells infected with Porphyromonas gingivalis, a reduction in several key NF-κB-regulated cytokines such as IL-6, IL-8, IL-10 and TNF-α in an ASC-dependent manner was observed [14]. The same group expanded further on their results and identified DUSP10, a dual-specificity phosphatase as a downstream target of ASC, and showed that DUSP10 modulated post-translational phosphorylation of mitogen–activated protein kinase (MAPK) [15]. These ASC-dependent effects were, however, independent of NLRP3 or NLRC4, which alludes to an NLR-independent role of ASC in proinflammatory signalling. Nevertheless, at this point it cannot be ruled out that redundant, but still NLR-dependent, mechanisms are at play. Moreover, the indirect effects of IL-1β, IL-18 and cell death on the activation of NF-κB needs to be taken into consideration when interpreting these results.
Current evidence points to the existence of at least six different inflammasome sensors, namely NLRP1, NLRP3, NLRC4, NLRP6, Pyrin and AIM2 (Fig. 1). Even though good genetic evidence confirms the functional existence of these inflammasomes, for none of these complexes has a precise mode of activation been elucidated and even more, for most of these proteins, it is unclear whether they are direct receptors. In the next sections of this review we attempt to put together the general principles involved in inflammasome activation and subsequently elaborate on the individual inflammasome complexes and their mechanism of activation.
Fig. 1.
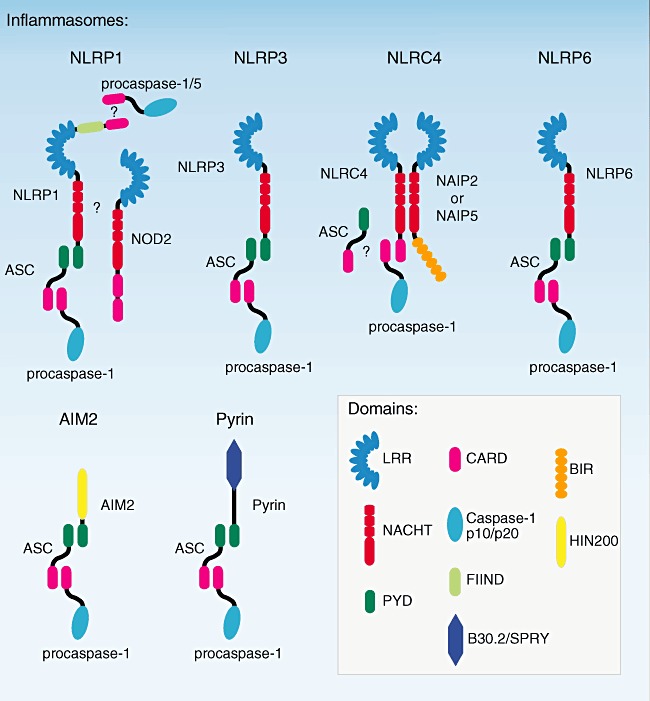
Known inflammasomes. The six known inflammasomes are depicted. Nucleotide-binding domain and leucine-rich repeat containing family (NLRP3) NLRP1, NLRP3, NLRP6, AIM2 and Pyrin all recruit and activate procaspase-1 indirectly through the effector molecule apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). Although NLRC4 has been shown to activate caspase-1 independent of ASC, there are several reports that stress the requirement for ASC. Apart from this controversy, the exact role of nucleotide oligomerization domain (NOD2) and caspase-5 in the NLRP1 inflammasome function has to be elucidated.
Shared structural principles of inflammasome sensors
NLRs, the biggest group of inflammasome-forming complexes, have high structural and functional homology to R genes in plants [16], which detect the presence of pathogens and trigger MAPK activation and eventually cell death. In the human system, all these proteins are characterized by three structural domains: first, an N-terminal effector domain, which can be a pyrin domain (PYD), a caspase recruitment domain (CARD) or a baculovirus inhibitor of apoptosis protein repeat (BIR) domain. According to the recently proposed nomenclature, NLR proteins containing PYD domain belong to the NLRP family, CARD domain to the NLRC family and BIR domain to the NLRB family [17]; secondly, the intermediate NACHT [NBS; nucleotide oligomerization domain (NOD)] domain, which is essential for the activation of the NLRs by oligomerization and formation of the core structure of the inflammasome; and thirdly, the C-terminal leucine-rich repeat (LRR) domain. The LRR domain is considered to confer specificity and to play a regulatory function in the inflammasome activation, but for many NLRs it is not clear whether it interacts directly with ligands or if some other intermediate ligand-sensing molecules are required.
The N-terminal PYD domain and the role of ASC in procaspase-1 activation
In 1999 Sagara's group identified ASC (also known as TMS1 or Pycard), employing an elegant cDNA cloning approach searching for the target of a monoclonal antibody that was raised against the insoluble fraction of an apoptotic promyelocytic leukaemia cell line [18]. This 22 kDa protein could be visualized in large, singular speck-like structures within the cytoplasm of the cells undergoing apoptosis and, moreover, these early studies already indicated that ASC speck formation might be more than just an epiphenomenon of the observed apoptosis process. ASC has a peculiar domain architecture with two domains belonging to the death domain superfamily: a N-terminal pyrin domain (PYD or DAPIN) and a C-terminal caspase recruitment domain (CARD). Both these domains are involved in protein–protein complex formation through homotypic PYD–PYD or CARD–CARD domain interactions, respectively. The similarity of the N-terminal PYD domain of ASC with its name-giving protein Pyrin was soon confirmed [19] and soon thereafter, three more ASC-binding proteins were identified through their N-terminal domain homology: PYPAF1 (now known as NLRP3) [8], PYPAF7 (now known as NLRP12) [9] and PYPAF5 (now known as NLRP6) [10]. In addition, the C-terminal CARD domain of ASC was soon been identified to interact with procaspase-1, resulting in its activation and processing and leading to the subsequent maturation of pro-IL-1β[9,20]. With these data at hand, a concept of ASC being a bridging molecule that links proteins with an N-terminal PYD domain to procaspase-1 was developed. In fact, it appears that all known inflammasome pathways employ ASC for procaspase-1 activation. This even seems to be true for NLRC4, which does not have an N-terminal PYD domain. Here, recent evidence suggests that ASC is also required for productive procaspase-1 cleavage [21].
The NACHT domain and inflammasome core structure formation
NACHT domains are part of P-loop NTPases, which have predicted nucleotide triphosphatase activity and are related closely to the oligomerization module found in AAAC ATPases [22]–[24]. The nucleotide oligomerization domain (NOD) family contains more than 20 proteins in human cells that play an important role in apoptosis and innate immunity. NOD proteins are members of the AAA+ superfamily of ATPases [22,25], and usually contain a conserved nucleotide binding domain (NBD) and a small helical domain (HD1) that binds nucleotides (Fig. 2a) [26,27]. However, in contrast to other members of the AAA+ superfamily, NOD proteins also contain an additional winged helix domain (WHD) that is involved in oligomerization, followed by a second helical domain (HD2) [28].
Fig. 2.
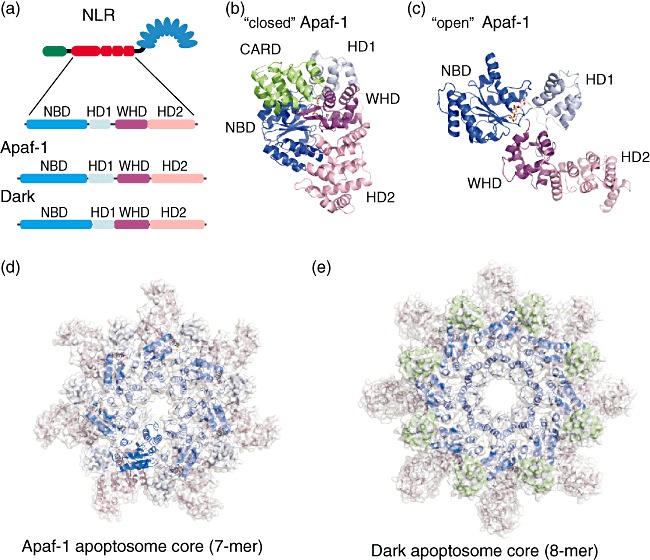
Structural organization of the apoptosome core-forming NACHT domain of human apoptotic protease-activating factor-1 (Apaf-1) and Drosophila Dark in inhibited free and active apoptosome forming stages. (a) Subdomain organization of the NACHT domain of a nucleotide-binding domain and leucine-rich repeat containing family (NLR), Apaf-1 and Dark. The NACHT domain can be subdivided into four subdomains – conserved nucleotide binding domain (NBD) and a small helical domain (HD1) that binds nucleotides, a winged helix domain (WHD) that is involved in oligomerization and a second helical domain (HD2). (b) Structure of human Apaf-1 without regulatory domains [caspase recruitment domain (CARD) and NACHT only] in the ‘close’ inactive conformation with adenosine diphosphate (ADP) bound (PDB ID 1z6t). (c) Structure of the human Apaf-1 NACHT domain with adenosine triphosphate (ATP) bound in ‘open’ active conformation capable to oligomerize (this structure was extracted from Apaf-1 apoptosome, PDB ID 3izA, chain A). (d,e) The apoptosome core structure formed by seven human Apaf-1 and 8 Drosophila Dark active NACHT domains, respectively. All subdomains are coloured according to the colouring in (a).
A similar nucleotide-binding motif, called the nucleotide-binding ARC domain (NB-ARC), is found in the Apaf-1 (apoptotic protease-activating factor-1) protein. In an inactive state NBD, HD1, WHD and HD2 domains of the Apaf-1 NACHT domain form a ‘closed’ compact structure with the CARD domain also bound to the NBD domain (Fig. 2b) [28,29]. Activation upon binding of deoxyadenosine triphosphate (dATP) and cytochrome-c results in a conformational change that leads to the formation of the ‘open’ NACHT domain structure (Fig. 2c). This initiates the release of the CARD domain, oligomerization of the NACHT domains and formation of the core structure of the Apaf-1 apoptosome (Fig. 2d), which then results in activation of procaspase-9 [30].
A similar oligomeric structure is formed by Drosophila Apaf-1 related killer (Dark) [31] (Fig. 2e) and CED-4, the Caenorhabditis elegans homologue of the human Apaf-1 [32]. In these complexes (Dark and CED-4) apoptosome core structures are formed by eight NACHT domains (human Apaf-1: seven NACHT domains). Taking into account that NACHT domains of all inflammasomes belong to the family of AAA+ ATPases, it is logical to assume that the core structure of all NLR inflammasomes is also formed by seven or eight NACHT domains and that this core structure formation plays a key role in the formation of an active NLR inflammasome which results in procaspase-1 activation and processing of pro-IL-1β and pro-IL-18. In fact, the formation of such core structure has been shown upon electronmicroscopy analysis of NLRP1 inflammasomal complexes [33]. In addition, as human Apaf-1 requires ATP for the formation of the active apoptosome [28], NACHT domain of NLRs also utilize ATP for proper inflammatory signalling [34].
The LRR domain and its regulatory function
LRRs are 22–28 amino acid motifs that form non-globular, horseshoe-like structures and are found in a number of proteins with diverse functions and cellular locations [35]. LRR domains are usually involved in protein–protein interactions and mediate macromolecular interactions in diverse immune processes [35]. Crystal structures have revealed that in three-dimension these repeats take up a crescent shape and the particular function of each LRR crescent is specified by different residues arranged in an appropriate orientation on the surface of the structurally conserved three-dimensional fold. In contrast to TLRs, where the LRR domains constitute the domain structures that bind directly to ligands, for NLRs of the inflammasome family no analogous function has yet been found. It has been reported that the deletion of the LRR domain results in a constitutively active form of the molecule by removing a possible autoinhibitory function of the LRR [36]. Mayor et al. showed a direct physical interaction between the LRR domain of NLRP3 and NLRC4 and the chaperone proteins heat shock protein 90 (HSP90) and ubiquitin ligase-associated protein (SGT1) [37]. These proteins prevented the proteasomal degradation of free NLRP3 and upon activation facilitated the conformational change of NLRP3 protein promoting the formation of the inflammasomal complex.
The mechanism of inflammasome activation
As mentioned above, NLR proteins have similarity, on one hand, to apoptosome-forming sensors such as human Apaf-1 or Drosophila Dark, and on the other hand to the plant R-proteins [30,31,38]–[40]. Taking this into account, two possible mechanisms of inflammasome activation could be imagined and have been proposed in the literature (Fig. 3) [17,39,41,42]. The main and critical difference between these two models is the implementation of the activation signal. The first mechanism is based on apoptosome formation, where Apaf-1 is activated upon direct binding of the activator molecule cytochrome c (Fig. 3a). According to this model, the NLR would be present in the cell in an ‘off’ state in a self-inhibitory conformation complex in which the regulatory LRR domain is bound to the PYD–NACHT domains, therefore preventing nucleotide exchange and structural rearrangement that would lead to the formation of an ‘open’ active state. Direct binding of a DAMP/MAMP or their complex with adapter molecules to the regulatory LRR domain of the NLR would then result in disruption of this self-inhibitory NLR complex and initiate a conformational change leading to the subsequent formation of an NLR protein that is capable to oligomerize (Fig. 3c). The second hypothetical mechanism of NLR inflammasome activation is based on the findings on R-proteins in plants that are kept in a ground inactive form by host guard proteins and become active upon release of their guarding complex (Fig. 3b) [38,39]. According to this model, the NLR is present in the cell in the ground ‘off’ state bound to a host guard complex, which not only protects the NLR from proteasomal degradation but also keeps it in an inactive ‘off’ conformation. In this complex the regulatory LRR domain could interact with the PYD domain of the NLR so that it is protected from interaction with the PYD domain of ASC. Recent studies by J. Tschopp's group proposed that this guarding complex could be formed by mammalian homologues of plant HSP90 and SGT1 [37]. Direct or indirect activation of this guarding complex by a DAMP/MAMP would lead to its complete or partial dissociation from the inactive NLR. In both models the NLR could then undergo a conformational change that is necessary for nucleotide exchange on the NACHT domain [29], and subsequently form an activated NLR in which the PYD domain of the NLR could now interact with the PYD domain of ASC (Fig. 3c). This could then lead to a core inflammasome structure (seven as in Apaf-1 or eight as in Dark apoptosome) (Fig. 3d). Of note, a lot more experimental evidence needs to be obtained before the above-described hypotheses can be confirmed.
Fig. 3.
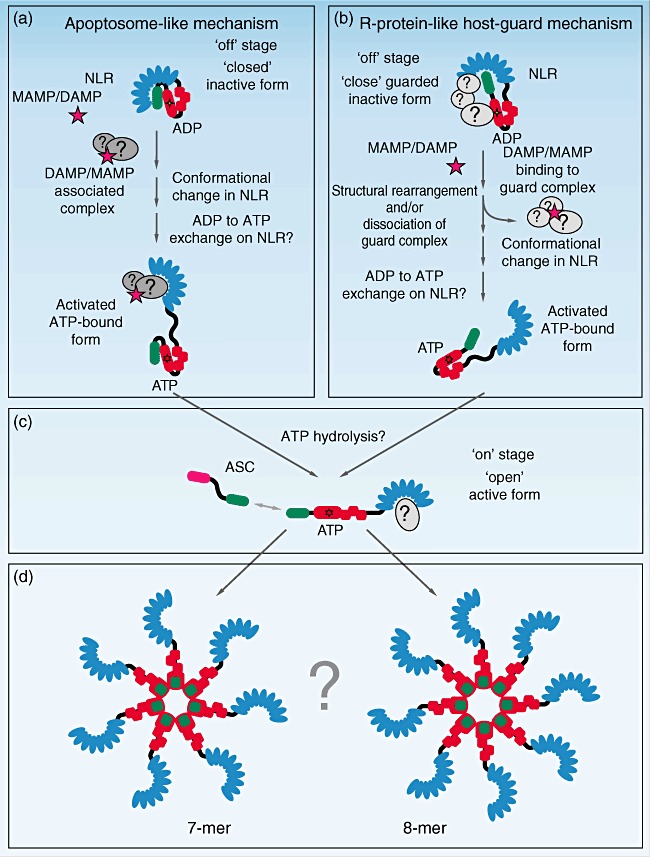
Possible mechanisms of nucleotide-binding domain and leucine-rich repeat containing family (NLR) inflammasome activation. (a) Mechanism based on apoptosome formation: in ground ‘closed’ state the NLR protein forms a self-inhibitory complex in which the regulatory leucine-rich repeat (LRR) domain is bound to a pyrin domain (PYD)–NACHT domain complex, therefore preventing nucleotide exchange and structural rearrangement leading to the formation of an ‘open’ active state. Upon binding of a microbe-associated molecular patterns/danger associated molecular patterns (MAMP/DAMP) or a MAMP/DAMP-associated complex to the regulatory LRR domain it is released from the PYD–NACHT complex and allows exchange of adenosine diphosphate (ADP) to adenosine triphosphate (ATP). This leads to a conformational change and the formation of an activated NLR capable to oligomerize (c). (b) Host-guarded NLR mechanism based on the plant R-protein model. Initially, a ‘close’ inactive complex is kept by the bound host guard complex. In this complex the regulatory LRR domain could interact with the PYD domain of the NLR and protects it from the interaction with the PYD domain of ASC. Cell stress leads to the activation of the guard complex, which results in complete or partial loss of interaction of the guarding complex with the NLR. This results in release of the PYD domain, which could now interact with the PYD domain of ASC, exchange of ADP to ATP, conformational change and formation of an oligomerization complex (c). (d) The net result would be an apoptosome-like core structure [e.g. seven, as in apoptotic protease-activating factor-1 (Apaf-1) or eight as in the Dark apoptosome].
Activation of specific inflammasome complexes
NLRP3 inflammasome
The NLRP3 inflammasome is one of the most extensively studied inflammasomes, and for this pathway a large array of physiochemically diverse activators have been described. This ranges from ionophoric compounds over cytosolic RNA to large crystalline material of diverse origin (for a detailed review see [43]). This high promiscuity in sensing is most likely due to the fact that NLRP3 activation constitutes a common response mechanism to perturbation of cellular integrity in the broadest sense. Despite tremendous research efforts, no common denominator upstream of NLRP3 could be identified so far that could explain this apparently highly versatile sensing by the NLRP3 inflammasome. Various modes of activation have been proposed that are, in part, complementary, yet not always in line with each other.
Priming and subsequent activation of NLRP3 inflammasome
As opposed to other inflammasomes, the activity of the NLRP3 inflammasome is tightly regulated by involving an additional regulatory step at the transcriptional level [44,45]. Unlike NLRP1, NLRC4 or AIM2, NLRP3 is expressed at limiting amounts under resting conditions and a proinflammatory signal is required to induce its expression to a level that allows its activation. Thus, the mechanism of activation of NLRP3 can be broken down into two stages: first, a ‘priming step’ wherein the amount of NLRP3 protein expression is augmented and secondly, the presence of a ‘licensing stimulus’ that promotes formation of the inflammasome complex itself. These findings explained the long-known phenomenon that ATP by itself does not activate caspase-1 in macrophages [46], yet requires an additional signal such as LPS. Of note, this unique requirement for priming also explains some confusion in the field regarding the elucidation of the mechanisms of NLRP3 inflammasome activation. In many studies small molecule inhibitors, RNAi knock-downs or knock-outs have been employed to study NLRP3 inflammasome activation, yet in most cases their influence on NLRP3 expression was not assessed. This, of course, complicates the interpretation of results, in particular with regard to their specific impact on the activation of NLRP3 itself.
Currently, three models exist to explain NLRP3 inflammasome activation: (a) lysosomal disintegration and release of its content by phagocytosed material; (b) induction of reactive oxygen species (ROS) production at mitochondrial membranes; and (c) potassium efflux by membrane channels or ionophoric compounds. It is possible that some of these processes act in concert or need to be involved sequentially, and there could also be a significant overlap between the different mechanisms of activation.
Lysosomal disintegration and release of its content
Uric acid crystals that have already been known for their IL-1-dependent proinflammatory activity [47] were one of the first compounds to be identified as potent NLRP3 activators [48]. Subsequently a number of crystalline or particulate substances were reported to activate the NLRP3 pathway [49]–[51]. Upon phagocytosis macrophages activate a number of enzymatic cascades in order to degrade the phagocytosed material within lysosomal compartments that are safely shielded away from the cytosol. However, as a result of unproductive phagocytosis, disintegration of lysosomal membranes and leakage of lysosomal contents into the cytoplasm can occur. In the case of NLRP3, it was observed that lysosomal rupture coincided with inflammasome activation and, moreover, it was shown that inhibition of lysosomal enzymes, most potently of cathepsin B, prevented lysosomal rupture and thus NLRP3 inflammasome activation [49,51]. Phagocytosis per se was not required to trigger inflammasome activation, as adding a chemical compound that specifically disrupts lysosomes was sufficient on its own to activate NLRP3 inflammasome activation. Of note, ionophoric NLRP3 activators or ATP were not blocked by cathepsin B inhibitors or other lysosome inhibitors (e.g. Bafilomycin A), indicating that these two pathways represent independent routes upstream of NLRP3. In addition, cathepsin B deficiency only partially affects NLRP3 inflammasome activation by phagocytic material, indicating that other, redundant mechanisms are at play upstream of NLRP3 inflammasome activation [51]. There is no doubt that further studies are needed to understand the exact mechanism of how lysosomal rupture activates the components of the NLRP3 inflammasome complex.
Activation by ROS
ROS is a term used to describe oxygen intermediates, including singlet oxygen, superoxides, peroxides, hydroxyl radical and hypochlorous acid [52]. Produced predominantly by the phagocytes, including macrophages and neutrophils, these highly reactive agents facilitate rapid inactivation of the invading microorganisms. However, unregulated production of ROS can also cause oxidative damage to the nucleic acids, proteins and lipids. Cruz et al. first proposed a link between ROS production and procaspase-1 activation [53]. The authors showed that treatment of macrophages with ATP resulted in ROS production that initiated the phosphatidylinositol 3-kinase (PI3K) pathway leading to procaspase-1 activation and increased IL-1β secretion. Petrilli et al. extended these studies, and observed that pretreatment of human acute monocytic leukaemia cell line (THP-1) cells with ROS inhibitor (N-acetyl-cysteine) diminished R837- and MSU-induced IL-1β secretion [54]. A series of subsequent studies expanded the link between ROS and inflammasome activation by showing that phagosomal ROS production was required for silica and asbestos-triggered NLRP3 activation [50]. Moreover, J. Tschopp's group identified TXNIP [thioredoxin (TRX)-interacting protein], a protein that regulates the anti-oxidant function of thioredoxin, to bind directly to NLRP3 in a ROS-sensitive manner [55]. Nevertheless, patients suffering from the primary immunodeficiency disease CGD (chronic granulomatous disease), a condition resulting from mutations in any of the components of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits (e.g. gp91phox), present with sterile inflammation [56]–[58]. These patients fail to generate phagosomal ROS, yet release copious amounts of IL-1β. In line with these contradictory findings, the ROS model was recently refined further by showing that mitochondria and not phagosomes were indeed the primary source for ROS production during inflammasome activation [59,60].
Of note, commonly used ROS inhibitors potently block proinflammatory gene expression, including NLRP3 [61]. The fact that NLRP3 is unique in its requirement for priming explains the relatively specific blocking activity of these inhibitors for the NLRP3 inflammasome in macrophages [45]. This does not exclude a role for ROS in NLRP3 inflammasome activation in these cells, but positions ROS rather upstream of NLRP3 induction than its direct activation. However, the situation seems to be different in the often-studied monocytic leukaemia cell line THP1, in which ROS inhibitors have been shown to block NLRP3 activation when added after priming [50,54]. Again, further experiments are required to address the role of ROS in NLRP3 inflammasome activation.
Potassium flux as a common denominator
Early studies identified high extracellular concentrations of ATP (mM range) as a potent trigger for IL-1β processing and secretion. ATP-triggered activation involves potassium efflux from the cytosol and among the candidate purinergic receptors P2X7 turned out to play a non-redundant role in this activation step [62]. P2X7 activation leads to the recruitment of the hemichannel protein Pannexin-1, and it was suggested that this recruitment leads to the formation of large, non-selective pores allowing the crossing of activators into the cytosol [63]–[65]. However, recent data suggest that Pannexin-1 recruitment is indeed dispensable for inflammasome activation [66] yet, rather, functions to release ATP from dying cells. Ionophoric compounds such as Nigericin, Valinomycin or Gramicidin A also trigger potent NLRP3 inflammasome activation and in analogy to ATP, potassium efflux has been reported to be the critical trigger for this event. Inhibition of NLRP3 inflammasome activation is usually achieved by raising the extracellular potassium concentration around 130 mM K+, which effectively blocks K+ efflux [54]. Even though the NLRP3 inflammasome appears to be most sensitive to high extracellular potassium levels, other inflammasomes are also affected in their activity [67]–[69]. This raises the question of specificity for this phenomenon. In addition, these experiments cannot clarify whether potassium efflux is sufficient or simply required among other events for NLRP3 inflammasome activation.
NLRP1 inflammasome
NLRP1 was the first member of NLR family identified to form a large protein complex with caspase-1, caspase-5 and ASC to promote caspase-1-dependent IL-1β maturation [70]. This complex was then named the ‘inflammasome’, and as described above the term ‘inflammasome’ is now generally used to describe complexes that are initiated by various sensor proteins resulting in caspase-1 activation.
Anthrax lethal factor and muramyl dipeptide (MDP) are two well-defined triggers of NLRP1 activation. Even though the bacterial peptidoglycan constituent MDP strongly activates procaspase-1 through NLRP1, it is likely that MDP-triggered NLRP1 activation is indirect. In fact, recent work suggests a surprising role for NOD2 (NLRC2) – another NLR member – in the assembly of the NLRP1 inflammasome in response to both MDP and anthrax lethal toxin [71]. NOD2, containing two N-terminal CARDs, serves as an intracellular sensor for MDP and initiates activation of NF-κB and MAPK kinases via RIP2 [72]. Even though in a cell-free system NLRP1 is sufficient to activate procaspase-1 in response to MDP, NOD2 is needed for in vivo sensing of both MDP. It has been shown that NOD2 could interact directly with procaspase-1 and NLRP1 but not with NLRP3. Thus, taken together, it seems that NOD2 is important for both signals required for secretion of bioactive IL-1β: first, NF-κB-dependent induction of pro-IL-1β expression and secondly, activation of procaspase-1 to mature the procytokines into their active forms [71,73].
The second well-characterized stimulus for NLRP1 is cytosolic anthrax lethal factor (LF) that is part of the lethal toxin produced by the spore-forming bacterium Bacillus anthracis, the causative agent of anthrax disease. In mouse macrophages, LF causes rapid necrosis that is central to the pathology of systemic B. anthracis infections. Catalytically active LF cleaves cytosolic substrates by a mechanism involving proteasome activity and Ca2+ ion flux. Interestingly, catalytically inactive LF mutant, which shares a structure identical with active LF, fails to activate procaspase-1, making a model of direct recognition of LF as a ligand for NLRP1 unlikely. LF may instead cleave additional, as yet unknown, targets leading to the degradation of NLRP1 inhibitors or the production of activating factors that trigger NLRP1 inflammasome activation [67,74,75].
More recently, NLRP1 was implicated in the progression of Toxoplasma gondii infection [76]. NLRP1 silencing in human monocytic line not only attenuated progression of T. gondii infection but also reduced secretion of proinflammatory cytokines and resulted in accelerated host cell death and eventual cell disintegration. However, the exact parasite factors that interfere with the NLRP1 signalling pathway still need to be elucidated. With regard to the other NLRPs, additional mechanistic studies are required to delineate the mode of activation for this sensor.
NLRC4 inflammasome
The inflammasome-containing protein NLRC4 (also known as IPAF or Card12) is essential for initiating immune response against infection with a series of Gram-negative bacteria, including Salmonella typhimurium, Legionella pneumophila, Shigella flexneri and Pseudomonas aeruginosa[13,77]–[79]. Early reports already suggested that it was the flagellin component of these organisms that activated the NLRC4 inflammasome [13]. Indeed, delivery of purified flagellin into the host cell cytosol resulted in the activation of procaspase-1 that was dependent on NLRC4 [80]. Furthermore, Salmonella and Legionella mutant strains that lacked flagellin did not activate this inflammasome [81]. Procaspase-1 activation by means of NLRC4 was also shown to be independent of TLR-5, suggesting that flagellin is indeed the primary MAMP activating NLRC4 [80]. Of note, in order to interact with NLRC4, flagellin has to enter the cytosol which is in most, but not all, cases due to the translocation of bacterial types 3 or 4 secretion systems (T3SS/T4SS). Conversely, the flagellated Gram-positive bacterium Listeria monocytogenes can also trigger NLRC4 activation, even though these bacteria do not express the above-mentioned secretion system [82]. In this case, flagellin can gain access to the cytosol and thus NLRC4 when Listeria escapes the phagolysosome to replicate in the cytosol. Next to flagellin, the inner rod component of bacterial T3SS can also activate NLRC4, e.g. PrgJ in the case of Salmonella[83]. This recent finding clarifies some of the confusion regarding flagellin-independent activation of the NLRC4 inflammasome by Salmonella.
Another puzzling finding regarding NLRC4 activation was the fact that the protein NAIP5 (NLR family, apoptosis inhibitory protein 5) was required in addition to NLRC4 for the activation of the inflammasome by L. pneumophila but not Salmonella or other sources of flagellin, thereby making it difficult to reconcile with the idea of NLRC4 being the sole sensor for flagellin [84,85]. Interestingly, it was recently discovered that NLRC4 utilizes NAIP proteins (NLR family, apoptosis inhibitory proteins) as additional upstream components to sense these different ligands [85,86]. NAIP5 was identified to directly bind the 35 amino acids spanning C-terminal region of Legionella flagellin [84]. Upon ligand binding, NAIP5 binds to NLRC4 and then initiates the formation of an inflammasome complex. Conversely, NAIP2, a closely related paralogue of NAIP5, specifically senses Salmonella PrgJ protein in conjunction with NLRC4 [86]. These data imply that NLRC4 is not a sensor itself, but rather functions as a signalling adapter to NAIP proteins that are the receptors themselves.
Apart from its ligand sensing modalities, NLRC4 is unique among the known inflammasome complexes as it can associate directly with procaspase-1 via the CARD–CARD domain interaction. In fact, this unique function allows NLRC4 to induce pyroptosis independent of ASC. Interestingly, productive caspase-1 processing and subsequent cleavage of IL-1β still requires the presence of ASC, indicating that inflammasome formation might assemble independent signalling platforms for distinct functions [13].
AIM2 inflammasome
AIM2, a protein belonging to the family of PYHIN (Pyrin and HIN200 domain-containing) is a bona fide receptor for dsDNA. Unlike the other members of its family, AIM2 is localized within the cytosol [87]. While the HIN200 domain of AIM2 binds double-stranded DNA preferentially, the activated AIM2 molecule recruits the adaptor molecule ASC via PYD–PYD domain interaction and initiates the formation of a procaspase-1 activation platform. AIM2 is unique among the known inflammasome complexes in two aspects. First, it binds directly to its ligand DNA and thus is a true receptor itself. Secondly, AIM2 does not contain a domain to initiate oligomerization and thus formation of the core structure of the inflammasome. Oligomerization is believed to be essential for the recruitment of ASC, so one can speculate that binding of multiple AIM2 proteins to a single molecule of dsDNA could lead to a process mimicking NLRs core structure formation. This hypothesis is supported further by experiments demonstrating a length dependency for dsDNA to trigger caspase-1 activation with short dsDNA being inactive ([11] and unpublished results).
Our current data suggest that AIM2 recognizes cytosolic dsDNA in a species-independent manner. Thus, both prokaryotic and eukaryotic DNA, including mammalian cells, can activate the AIM2 inflammasome ([11] and unpublished results). It is therefore tempting to speculate that AIM2 might participate in autoinflammatory immune responses involving cytosolic DNA. Since AIM2 functions as a proinflammatory PRR one could simply conclude that AIM2 activation drives autoimmune disease. However, AIM2 seems to regulate type I IFN production negatively through a yet ill-defined mechanism [11], and thus could therefore function to repress autoimmunity, once activated by DNA. Of note, another member of the murine PYHIN family, IFI202 (p202) has been reported to antagonize AIM2 function, potentially by inhibiting AIM2 inflammasome complex formation [88]. In addition, both AIM2 and IFI202 seem to regulate reciprocally each other's expression and function. Aim2 deficiency leads to increased IFN-β and IFI202 expression and furthermore potentiates the nuclear localization of the IFI202 protein [89].
NLRP6 inflammasome
NLRP6 was also first identified by John Bertin's group at Millenium Pharmaceuticals as a member of the then-called PYPAF family with a N-terminal PYD domain activating ASC [10]. While initial reports suggested a role for NLRP6 in NF-κB and procaspase-1 activation, for a long time the exact function of this protein remained unknown. This lack in progress is due in part to the fact that NLRP6 is not expressed in immune cells, but confined largely to epithelial cells of the intestine. Kemptster et al., while looking for genes regulating intestinal microbiota during fetal/neonatal development, observed that the levels of NLRP6 and one of its substrates, IL-18, were increased at E20 compared with E16 in fetal intestine and not in the lung [90]. Most recently there has been a spurt of experimental evidence detailing the role of this particular inflammasome in intestinal microbiota regulation and colonic tumours. Richard Flavell's group showed that NLRP6 inflammasome-dependent activation of IL-18 by intestinal epithelial cells positively regulates the composition of the gut microbiota [91]. Absence of NLRP6 or its downstream cascade (ASC, caspase-1 or IL-18) allowed the outgrowth of a colitogenic bacterial strain, namely members of the Prevotellaceae family that rendered the host more sensitive to colitis. Another study focusing on the role of NLRP6 in colitis-associated tumours revealed that although NLRP6 expression is lowered in diseased colon, Nlrp6-deficient mice are highly susceptible to experimental colitis [92]. Upon injury, NLRP6 deficiency deregulated regeneration of the colonic mucosa and processes of epithelial proliferation and migration. Consistently, the absence of NLRP6 accelerated colitis-associated tumour growth in mice [92]. Altogether, these studies indicate that NLRP6 plays a pivotal role in regulating IL-18 processing in gut epithelial cells.
Pyrin-dependent inflammasome
Pyrin, also known as MEFV, is the gene that is mutated in familial Mediterranean fever syndrome (FMF). Pyrin harbours a N-terminal PYD domain, a B-box domain, a bZIP basic domain, coiled-coil domains and a C-terminal B30·2/SPRY domain. More than 50 disease-causing mutations have been reported for Pyrin, a majority of which have been found in its B30·2/SPRY domain [93]. As Pyrin is expressed primarily in neutrophils, monocytes and dendritic cells, its function is thought primarily to be an immunological one [94,95]. Through its N-terminal PYD domain Pyrin binds to ASC [19], and just as for NLRP proteins it was shown that this interaction between Pyrin and ASC could lead to productive processing of caspase-1 [96]. Nevertheless, despite these findings it was initially thought that Pyrin is a negative regulator of other inflammasome pathways and not an inflammasome-forming molecule itself [97]. As such, it was proposed that Pyrin binds to NLRP3, ASC or caspase-1 and that this binding inhibits the function or assembly of these components [98]. This model proposed that the proinflammatory capacity of Pyrin mutations (as they are observed in FMF patients) was due to a less effective negative regulation of the NLRP3 inflammasome by the altered Pyrin protein. To study the role of Pyrin in vivo, mice expressing a truncated Pyrin molecule that still possessed the full pyrin domain were generated. Injection of LPS into these mice resulted in enhanced caspase-1 activity, increased IL-1β secretion and in increased lethality, and thereby the negative regulatory role of Pyrin appeared to be confirmed [98]. However, under resting conditions the severity of phenotype of these mice was nowhere near the autoinflammatory FMF phenotype seen in patients, thereby questioning that a loss in negative regulation was possibly involved in the pathogenesis of FMF. Subsequently, knock-in mice for human FMF mutations in the B30·2 domains were made [99]. These mice spontaneously demonstrated a profound autoinflammatory phenotype consistent with FMF. The effect of mutant Pyrin over-expression was gene dose-dependent, and Pyrin knock-out mice showed no overt phenotype. Furthermore, the enhanced IL-1β levels observed in these mice were ASC-dependent but independent of NLRP3-, AIM2- or NLRP1. These results strongly suggest that Pyrin is an inflammasome-forming and possibly ligand-dependent sensor itself, as opposed to a negative regulator of another inflammasome pathway. In this regard, it will be interesting to elucidate a ligand-dependent mode of activation.
Of note, next to its role in activating caspase-1, it is possible that Pyrin also activates or modulates other proinflammatory pathways, for example at the transcriptional level (for a review see [1]). In this context it is notable that Pyrin contains a nuclear localization domain and that endogenous Pyrin protein is indeed seen within the nucleus. These observations are in line with data that implicate Pyrin as a positive regulator of NF-κB regulation [100].
Conclusions
The inflammasome, a procaspase-1 processing molecular platform, is essential for the secretion of the key proinflammatory cytokines IL-1β and IL-18 and also for the induction of caspase-1 driven cell death, known as pyroptosis. The currently known inflammasome complexes are activated by different sets of ligands, and only some of these inflammasome-forming sensors can be currently seen as true receptor molecules (e.g. AIM2 for DNA and NAIP5/NLRC4 for flagellin). For the NLRP inflammasomes, the mode of activation appears to be indirect, and while it is unlikely that these proteins are receptors themselves, they are most probably signalling molecules that are activated by yet-to-be-defined components further upstream. Moreover, for none of these complexes has a clearly defined mode of activation been described, even though a model resembling the apoptosome appears to be likely. There is no doubt that further studies need to be undertaken to dissect the exact mechanism of activation of the inflammasome complexes. This information will enable the appropriate targeting of the different inflammasome complexes in diverse clinical disorders.
Acknowledgments
V.H. is supported by grants from the German Research Foundation (SFB704 and SFB670) and the European Research Council (ERC–2009–StG 243046).
Disclosure
The authors declare no financial or commercial conflict of interest.
Reference
- 1.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozkurede VU, Franchi L. Immunology in the clinic review series; focus on autoinflammatory diseases: role of inflammasomes in autoinflammatory syndromes. Clin Exp Immun. 2012;167:382–90. doi: 10.1111/j.1365-2249.2011.04535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldbach-Mansky R. Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin Exp Immun. 2012;167:391–404. doi: 10.1111/j.1365-2249.2011.04533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–31. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 5.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 6.Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x. [DOI] [PubMed] [Google Scholar]
- 7.Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–42. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manji GA, Wang L, Geddes BJ, et al. PYPAF1, a PYRIN-containing Apaf1-like protein that assembles with ASC and regulates activation of NF-kappa B. J Biol Chem. 2002;277:11570–5. doi: 10.1074/jbc.M112208200. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Manji GA, Grenier JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002;277:29874–80. doi: 10.1074/jbc.M203915200. [DOI] [PubMed] [Google Scholar]
- 10.Grenier JM, Wang L, Manji GA, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 2002;530:73–8. doi: 10.1016/s0014-5793(02)03416-6. [DOI] [PubMed] [Google Scholar]
- 11.Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–8. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto M, Yaginuma K, Tsutsui H, et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells. 2004;9:1055–67. doi: 10.1111/j.1365-2443.2004.00789.x. [DOI] [PubMed] [Google Scholar]
- 13.Mariathasan S, Newton K, Monack DM, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004;430:213–8. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 14.Taxman DJ, Zhang J, Champagne C, et al. Cutting edge: ASC mediates the induction of multiple cytokines by Porphyromonas gingivalis via caspase-1-dependent and -independent pathways. J Immunol. 2006;177:4252–6. doi: 10.4049/jimmunol.177.7.4252. [DOI] [PubMed] [Google Scholar]
- 15.Taxman DJ, Holley-Guthrie EA, Huang MT, et al. The NLR adaptor ASC/PYCARD regulates DUSP10, mitogen-activated protein kinase (MAPK), and chemokine induction independent of the inflammasome. J Biol Chem. 2011;286:19605–16. doi: 10.1074/jbc.M111.221077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 17.Ting JP, Lovering RC, Alnemri ES, et al. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–7. doi: 10.1016/j.immuni.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masumoto J, Taniguchi S, Ayukawa K, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274:33835–8. doi: 10.1074/jbc.274.48.33835. [DOI] [PubMed] [Google Scholar]
- 19.Richards N, Schaner P, Diaz A, et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem. 2001;276:39320–9. doi: 10.1074/jbc.M104730200. [DOI] [PubMed] [Google Scholar]
- 20.Srinivasula SM, Poyet JL, Razmara M, Datta P, Zhang Z, Alnemri ES. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem. 2002;277:21119–22. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 21.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–83. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leipe DD, Koonin EV, Aravind L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J Mol Biol. 2004;343:1–28. doi: 10.1016/j.jmb.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 23.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Koonin EV, Aravind L. The NACHT family – a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem Sci. 2000;25:223–4. doi: 10.1016/s0968-0004(00)01577-2. [DOI] [PubMed] [Google Scholar]
- 25.Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3:371–82. doi: 10.1038/nri1086. [DOI] [PubMed] [Google Scholar]
- 26.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–63. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 27.Danot O, Marquenet E, Vidal-Ingigliardi D, Richet E. Wheel of life, wheel of death: a mechanistic insight into signaling by STAND proteins. Structure. 2009;17:172–82. doi: 10.1016/j.str.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Riedl SJ, Li W, Chao Y, Schwarzenbacher R, Shi Y. Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature. 2005;434:926–33. doi: 10.1038/nature03465. [DOI] [PubMed] [Google Scholar]
- 29.Reubold TF, Wohlgemuth S, Eschenburg S. Crystal structure of full-length apaf-1: how the death signal is relayed in the mitochondrial pathway of apoptosis. Structure. 2011;19:1074–83. doi: 10.1016/j.str.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Yuan S, Yu X, Topf M, Ludtke SJ, Wang X, Akey CW. Structure of an apoptosome–procaspase–9 CARD complex. Structure. 2010;18:571–83. doi: 10.1016/j.str.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan S, Yu X, Topf M, et al. Structure of the Drosophila apoptosome at 6·9 a resolution. Structure. 2011;19:128–40. doi: 10.1016/j.str.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi S, Pang Y, Hu Q, et al. Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell. 2010;141:446–57. doi: 10.1016/j.cell.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 33.Faustin B, Lartigue L, Bruey JM, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007;25:713–24. doi: 10.1016/j.molcel.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 34.Duncan JA, Bergstralh DT, Wang Y, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA. 2007;104:8041–6. doi: 10.1073/pnas.0611496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Truhlar SM, Komives EA. LRR domain folding: just put a cap on it! Structure. 2008;16:655–7. doi: 10.1016/j.str.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Liao KC, Mogridge J. Expression of Nlrp1b inflammasome components in human fibroblasts confers susceptibility to anthrax lethal toxin. Infect Immun. 2009;77:4455–62. doi: 10.1128/IAI.00276-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol. 2007;8:497–503. doi: 10.1038/ni1459. [DOI] [PubMed] [Google Scholar]
- 38.Shirasu K. The HSP90-SGT1 chaperone complex for NLR immune sensors. Annu Rev Plant Biol. 2009;60:139–64. doi: 10.1146/annurev.arplant.59.032607.092906. [DOI] [PubMed] [Google Scholar]
- 39.Kadota Y, Shirasu K, Guerois R. NLR sensors meet at the SGT1–HSP90 crossroad. Trends Biochem Sci. 2009;35:199–207. doi: 10.1016/j.tibs.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 40.Yuan S, Yu X, Asara JM, Heuser JE, Ludtke SJ, Akey CW. The holo-apoptosome: activation of procaspase-9 and interactions with caspase-3. Structure. 2011;19:1084–96. doi: 10.1016/j.str.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–90. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jha S, Ting JP. Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J Immunol. 2009;183:7623–9. doi: 10.4049/jimmunol.0902425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bauernfeind F, Ablasser A, Bartok E, et al. Inflammasomes: current understanding and open questions. Cell Mol Life Sci. 2011;68:765–83. doi: 10.1007/s00018-010-0567-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bauernfeind FG, Horvath G, Stutz A, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–91. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–7. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kahlenberg JM, Lundberg KC, Kertesy SB, Qu Y, Dubyak GR. Potentiation of caspase-1 activation by the P2X7 receptor is dependent on TLR signals and requires NF-kappaB-driven protein synthesis. J Immunol. 2005;175:7611–22. doi: 10.4049/jimmunol.175.11.7611. [DOI] [PubMed] [Google Scholar]
- 47.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 48.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 49.Hornung V, Bauernfeind F, Halle A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008;9:847–56. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–9. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 55.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 56.Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A. Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood. 2010;116:1570–3. doi: 10.1182/blood-2010-01-264218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Bruggen R, Koker MY, Jansen M, et al. Human NLRP3 inflammasome activation is Nox1-4 independent. Blood. 2010;115:5398–400. doi: 10.1182/blood-2009-10-250803. [DOI] [PubMed] [Google Scholar]
- 58.van de Veerdonk FL, Smeekens SP, Joosten LA, et al. Reactive oxygen species-independent activation of the IL-1beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci USA. 2010;107:3030–3. doi: 10.1073/pnas.0914795107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 60.Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bulua AC, Simon A, Maddipati R, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–33. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Solle M, Labasi J, Perregaux DG, et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–32. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- 63.Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–82. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanneganti TD, Lamkanfi M, Kim YG, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity. 2007;26:433–43. doi: 10.1016/j.immuni.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 65.Marina-Garcia N, Franchi L, Kim YG, et al. Pannexin-1-mediated intracellular delivery of muramyl dipeptide induces caspase-1 activation via cryopyrin/NLRP3 independently of Nod2. J Immunol. 2008;180:4050–7. doi: 10.4049/jimmunol.180.6.4050. [DOI] [PubMed] [Google Scholar]
- 66.Qu Y, Misaghi S, Newton K, et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol. 2011;186:6553–61. doi: 10.4049/jimmunol.1100478. [DOI] [PubMed] [Google Scholar]
- 67.Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci USA. 2008;105:4312–7. doi: 10.1073/pnas.0707370105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arlehamn CS, Petrilli V, Gross O, Tschopp J, Evans TJ. The role of potassium in inflammasome activation by bacteria. J Biol Chem. 2010;285:10508–18. doi: 10.1074/jbc.M109.067298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muruve DA, Petrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–7. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 70.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 71.Hsu LC, Ali SR, McGillivray S, et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci USA. 2008;105:7803–8. doi: 10.1073/pnas.0802726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–72. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 73.Ferwerda G, Kramer M, de Jong D, et al. Engagement of NOD2 has a dual effect on proIL-1beta mRNA transcription and secretion of bioactive IL-1beta. Eur J Immunol. 2008;38:184–91. doi: 10.1002/eji.200737103. [DOI] [PubMed] [Google Scholar]
- 74.Squires RC, Muehlbauer SM, Brojatsch J. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J Biol Chem. 2007;282:34260–7. doi: 10.1074/jbc.M705687200. [DOI] [PubMed] [Google Scholar]
- 75.Wickliffe KE, Leppla SH, Moayeri M. Anthrax lethal toxin-induced inflammasome formation and caspase-1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol. 2008;10:332–43. doi: 10.1111/j.1462-5822.2007.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Witola WH, Mui E, Hargrave A, et al. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun. 2011;79:756–66. doi: 10.1128/IAI.00898-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zamboni DS, Kobayashi KS, Kohlsdorf T, et al. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol. 2006;7:318–25. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 78.Suzuki T, Franchi L, Toma C, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007;3:e111. doi: 10.1371/journal.ppat.0030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc Natl Acad Sci USA. 2008;105:2562–7. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006;7:576–82. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 81.Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 2007;29:275–88. doi: 10.1007/s00281-007-0078-z. [DOI] [PubMed] [Google Scholar]
- 82.Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J Immunol. 2008;180:7558–64. doi: 10.4049/jimmunol.180.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA. 2010;107:3076–80. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lightfield KL, Persson J, Brubaker SW, et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol. 2008;9:1171–8. doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lightfield KL, Persson J, Trinidad NJ, et al. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. 2011;79:1606–14. doi: 10.1128/IAI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kofoed EM, Vance RE. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature. 2011;477:592–5. doi: 10.1038/nature10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hornung V, Latz E. Intracellular DNA recognition. Nat Rev Immunol. 2010;10:123–30. doi: 10.1038/nri2690. [DOI] [PubMed] [Google Scholar]
- 88.Roberts TL, Idris A, Dunn JA, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–60. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 89.Panchanathan R, Shen H, Duan X, et al. Aim2 deficiency in mice suppresses the expression of the inhibitory Fcgamma receptor (FcgammaRIIB) through the induction of the IFN-inducible p202, a lupus susceptibility protein. J Immunol. 2011;186:6762–70. doi: 10.4049/jimmunol.1003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kempster SL, Belteki G, Forhead AJ, et al. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am J Physiol Gastrointest Liver Physiol. 2011;300:G253–63. doi: 10.1152/ajpgi.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–57. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Normand S, Delanoye-Crespin A, Bressenot A, et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci USA. 2011;108:9601–6. doi: 10.1073/pnas.1100981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Touitou I, Lesage S, McDermott M, et al. Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum Mutat. 2004;24:194–8. doi: 10.1002/humu.20080. [DOI] [PubMed] [Google Scholar]
- 94.Centola M, Wood G, Frucht DM, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223–31. [PubMed] [Google Scholar]
- 95.Diaz A, Hu C, Kastner DL, et al. Lipopolysaccharide-induced expression of multiple alternatively spliced MEFV transcripts in human synovial fibroblasts: a prominent splice isoform lacks the C-terminal domain that is highly mutated in familial Mediterranean fever. Arthritis Rheum. 2004;50:3679–89. doi: 10.1002/art.20600. [DOI] [PubMed] [Google Scholar]
- 96.Yu JW, Wu J, Zhang Z, et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13:236–49. doi: 10.1038/sj.cdd.4401734. [DOI] [PubMed] [Google Scholar]
- 97.Chae JJ, Wood G, Masters SL, et al. The B30·2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA. 2006;103:9982–7. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chae JJ, Komarow HD, Cheng J, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. doi: 10.1016/s1097-2765(03)00056-x. [DOI] [PubMed] [Google Scholar]
- 99.Chae JJ, Cho YH, Lee GS, et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity. 2011;34:755–68. doi: 10.1016/j.immuni.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chae JJ, Wood G, Richard K, et al. The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1 and activates NF-kappaB through its N-terminal fragment. Blood. 2008;112:1794–803. doi: 10.1182/blood-2008-01-134932. [DOI] [PMC free article] [PubMed] [Google Scholar]