

Abstract
The disease-based discovery of the molecular basis for autoinflammatory diseases has led not only to a rapidly growing number of clinically and genetically identifiable disorders, but has unmantled key inflammatory pathways such as the potent role of the alarm cytokine interleukin (IL)-1 in human disease. Following its initial failures in the treatment of sepsis and the moderate success in the treatment of rheumatoid arthritis, IL-1 blocking therapies had a renaissance in the treatment of a number of autoinflammatory conditions, and IL-1 blocking therapies have been Food and Drug Administration (FDA)-approved for the treatment of the autoinflammatory conditions: cryopyrin-associated periodic syndromes (CAPS). CAPS and deficiency of the IL-1 receptor antagonist (DIRA), both genetic conditions with molecular defects in the IL-1 pathway, have provided a pathogenic rationale to IL-1 blocking therapies, and the impressive clinical results confirmed the pivotal role of IL-1 in human disease. Furthermore, IL-1 blocking strategies have shown clinical benefit in a number of other genetically defined autoinflammatory conditions, and diseases with clinical similarities to the monogenic disorders and not yet identified genetic causes. The discovery that IL-1 is not only triggered by infectious danger signals but also by danger signals released from metabolically ‘stressed’ or even dying cells has extended the concept of autoinflammation to disorders such as gout, and those that were previously not considered inflammatory, such as type 2 diabetes, coronary artery disease, obesity and some degenerative diseases, and provided the conceptual framework to target IL-1 in these diseases. Despite the tremendous success of IL-1 blocking therapy, the use of these agents in a wider spectrum of autoinflammatory conditions has uncovered disease subsets that are not responsive to IL-1 blockade, including the recently discovered proteasome-associated autoinflammatory syndromes such as chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperatures (CANDLE), Japanese autoinflammatory syndrome with lipodystrophy (JASL), Nakajo–Nishimura syndrome (NNS) and joint contractures, muscle atrophy, panniculitis induced lipodystrophy (JMP), and urge the continued quest to characterize additional dysregulated innate immune pathways that cause autoinflammatory conditions.
Keywords: CANDLE, CAPS, DIRA, neonatal disorder, NOMID
OTHER THEMES PUBLISHED IN THIS IMMUNOLOGY IN THE CLINIC REVIEW SERIES
Allergy, Host Responses, Cancer, Type 1 diabetes and viruses, Metabolic diseases.
Introduction
The concept of autoinflammatory diseases was initially suggested by Dan Kastner in 1999 after the discovery of the genetic causes for two familial fever syndromes, familial Mediterranean fever (FMF) [1,2] and tumour necrosis factor (TNF) receptor-associated periodic syndrome or (TRAPS) [3]. He suggested a novel group of immunological disorders characterized by ‘seemingly unprovoked’ episodes of fever and inflammation without evidence of autoantibodies or antigen-specific T cells, implying the importance of dysregulation in the innate immune system. Over the last decade the advent of powerful genetic tools has accelerated the rate of genetic discoveries and led to the genetic characterization of many other inflammatory conditions with early onset disease and episodes of systemic and organ-specific inflammation (Table 1) and has validated the usefulness of the concept of autoinflammation beyond its initial scope. The discovery of mutations in an intracellular ‘danger receptor’, initially termed CIAS1 (now NLRP3) by Hal Hoffman as the genetic cause of the two autosomal dominant syndromes, familial cold-induced autoinflammatory syndrome (FCAS) and Muckle–Wells syndrome (MWS) [4] and the sporadic and clinically severe disease neonatal-onset multi-system inflammatory disease/chronic infantile neurological, cutaneous and articular syndrome (NOMID/CINCA) [5,6], led to the recognition of a disease spectrum of cryopyrin-associated periodic syndromes (CAPS) that has provided us with a pathogenic model for autoinflammatory diseases as triggered by danger signals and dysregulated responses to such stimuli. The discovery that interleukin (IL)-1 secretion in response to Toll-like receptor stimulation is mediated through co-operation with the nucleotide-binding domain and leucine-rich repeat containing family pyrin domain containing 3 (NLRP3) inflammasome [7], and the recognition that the NLRP3/CIAS1 containing NLRP3 inflammasome is an intracellular receptor that is triggered not only in response to microbial, ‘exogenous’ but also to ‘endogenous stress molecules’ by Jorg Tschopp and others [8] and co-ordinates processing and IL-1 secretion through caspase-1 activation [9], have provided a molecular understanding of the mechanism of IL-1-mediated inflammation and the episodic nature of the inflammatory response. Furthermore, the shared activation pathways for endogenous and exogenous danger signals in the regulation of the potent proinflammatory cytokine IL-1 provide further evidence that the immune system has evolved to recognize danger, regardless of whether it is exogenous or endogenous [10]. The successful treatment of a patient with early-onset pustulosis and osteolytic lesions led to the discovery of another IL-1-mediated disorder, deficiency of the IL-1 receptor antagonist (DIRA) [11,12] and the successful treatment of CAPS patients with IL-1 blocking agents has encouraged the exploration of IL-1 inhibition in a number of other monogenic autoinflammatory diseases, including FCAS 2 [13], FMF [1,2], hyperimmunoglobulin D syndrome (HIDS) [14,15], Majeed syndrome [16], TRAPS [3] and pyogenic arthritis, pyoderma gangrenosum and acne syndrome (PAPA) [17]–[20] (Table 1), and provided new treatments for a group of autoinflammatory disorders in whom the genetic cause of the disease is still unknown, including systemic-onset juvenile idiopathic arthritis (SOJIA), adult-onset Still's disease (AOSD) and periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis (PFAPA). The observation that metabolic substrates that accumulate in target tissues such as monosodium urate in gout, islet amyloid polypeptide (IAPP) and oxidized low-density lipoprotein (oxLDL) in diabetes, ceramide and others in obesity, and cholesterol crystals in atherosclerosis, can stimulate the NLRP3 inflammasome to release IL-1β has led to studies of the use of IL-1 blocking agents in some of these conditions (Table 2). However, the use of IL-1 blocking agents in autoinflammatory disease has also revealed subsets of patients who do not respond to IL-1 blocking therapies, and recent discoveries of the cause of these diseases provide a glimpse into additional key inflammatory pathways involved in innate immune recognition and regulation ([21]–[27] and Table 1).
Table 1.
‘Monogenic’ autoinflammatory syndromes
| Inflammatory cause | Disease (MIM #) | Gene | Protein | Inheritance pattern/year of discovery/Refs | Disease onset | Flare/fever pattern | Specific organ inflammation | Treatment |
|---|---|---|---|---|---|---|---|---|
| IL-1β | CAPS | |||||||
| FCAS #120100 | CIAS1 (1q44) | Cryopyrin | AD/(2001)/[4] | First 6 months of life, cold-induced | <24 h | Skin, eyes, joints, systemic | IL-1 blockade | |
| MWS #191900 | CIAS1 (1q44) | Cryopyrin | AD/(2001)/[4] | Infancy to adolescence | 24–48 h | Skin, eyes, joints, inner ears, meninges (mild), systemic | IL-1 blockade | |
| NOMID #607115 | CIAS1 (1q44) | Cryopyrin | AD/de novo/(2002)/[5,6] | Neonatal or early infancy | Continuous with flares | Skin, eyes, joints, inner ears, meninges, bony epiphyseal hyperplasia, systemic | IL-1 blockade | |
| IL-1β and IL-1α | DIRA #612852 | IL1RN (2q14) | IL-1 receptor antagonist | AR/(2009)/[11] | Neonatal or early infancy | Continuous with flares | Skin, bones, lungs (rare), vasculitis (rare) | Anakinra |
| IL-1+ 0ellip; | FCAS2 #611762 | NLRP12 (19q13) | NLRP12 | AD/(2008)/[13] | Neonatal or early infancy | Continuous with flares | Skin, eyes, joints, systemic | IL-1 blockade |
| IL-1+ 0ellip; | FMF #249100 | MEFV (16p13) | pyrin | AR/(1997)/[1,2] | 80% of the cases occur before the age of 20 | 1–3 days | Skin, joints, peritoneum, pleura | Colchicine, rarely |
| IL-1 and TNF blockade or thalidomide if colchicine-resistant | ||||||||
| IL-1+ 0ellip; | HIDS #260920 | MVK (12q24) | Mevalonate kinase | AR/(1999)/[14,15] | Median age at onset 6 months | 3–7 days | Skin, eyes, joints, prominent lymph nodes | NSAIDS, corticosteroids, TNF and IL-1 blockade |
| IL-1+ 0ellip; | Majeed's Syndrome #609628 | LPIN2 (18p11) | Lipin2 | AR/(2005)/[16] | Early infancy (1–19 months) | Weeks–months | Bones, periosteum, anaemia | NSAIDS, corticosteroids, interferon-α, possibly IL-1 blockade |
| IL-1+TNF 0ellip; | TRAPS #191190 | TNFRSF1A (12p13) | TNF receptor 1 | AD/(1999)/[3] | Median age at onset 3 years | 1–4 weeks | Skin, eyes, joints, peritoneum, pleura | TNF blockade, steroids, IL-1 blockade, colchicine is ineffective |
| IL-1+TNF 0ellip; | PAPA #604416 | CD2BP1 (15q24) | PSTPIP1 | AD/(2002)/[17] | Early childhood | Common | Skin, joints | Local and systemic corticosteroids, TNF or IL-1 blockade |
| Other pathways | ||||||||
| TNF, IL-1+ 0ellip; | PGA* #186580 | NOD2 (16q12) | Nod2 | AD/de novo (2001,2005)/[18,19] | Early childhood | Uncommon | Skin, eyes, joints | NSAIDS, corticosteroids, methotrexate, cyclosporin, TNF or IL-1 blockade |
| TNF+ 0ellip; | Cherubism #118400 | SH3BP2 (4p16) | SH3BP2 | AD/(2001)/[20] | Childhood, spontaneous remission by 3rd decade | Uncommon | Jaws, eyes (rare) | NSAIDS, TNF inhibition, interferon-α, azithromycin, bisphosphonates |
| Lack of IL-10 signalling | Early-onset inflammatory bowel disease #613148 | IL10RA (11q23) | IL-10 receptor, IL10RB also forms IL-22, -26, -28-29 receptors | AR/(2010)/[21] | Neonatal or early infancy | Continuous with flares | Colitis with fistula formation, folliculitis in patients with IL10RB mutations | Bone marrow transplantation |
| IL10RB (21q22) | ||||||||
| IL-36α, IL-36β, IL-36γ (role of IL-1 unknown but unlikely complete) | DITRA # 614204 | IL36RN/IL1F5 (2q13-14) | IL-36 | AR/ 2011)/[22,23] | Carriable to adulthood, early onset at 2 weeks was reported in some cases | Flares of generalized and palmoplantar pustulosis | Skin | Acitretin |
| Topical steroids | ||||||||
| Limited use of biologics, i.e. adalimumab | ||||||||
| Increase in IFN signalling? | JMP, NNS, JASL, CANDLE #613732 | PSMB8 (6p21) | Inducible β5 subunit of the immunoproteasome | AR/(2011)/[24]–[27] | Neonatal or early infancy | Continuous with flares | Skin, joints, lipodystrophy and muscle atrophy | Partial response to steroids, TNF inhibition, IL-6R inhibition |
CAPS: cryopyrin-associated periodic syndromes; FCAS: familial cold autoinflammatory syndrome; MWS: Muckle–Wells syndrome; NOMID: neonatal-onset multi-system inflammatory disease; DIRA: deficiency of the interleukin (IL)-1 receptor antagonist; DITRA: deficiency of the IL-thirty-three receptor antagonist; FMF: familial Mediterranean fever; HIDS: hyperimmunoglobulin D syndrome; PAPA: pyogenic arthritis, pyoderma gangrenosum and acne syndrome; TRAPS: tumour necrosis factor receptor-associated periodic syndrome; PGA: paediatric granulomatous arthritis, *including Blau syndrome (MIM 186580) and early-onset sarcoidosis (MIM 609464); JMP: joint contractures, muscle atrophy, panniculitis-induced lipodystrophy; NNS: Nakajo–Nishimura syndrome; JASL: Japanese autoinflammatory syndrome with lipodystrophy; CANDLE: chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperatures; AR: autosomal recessive; AD: autosomal dominant; NSAID: non-steroidal inflammatory drug; TNF: tumour necrosis factor.
Table 2.
Autoinflammatory diseases responsive to interleukin (IL)-1 blockade
| Syndrome | Studies |
|---|---|
| Monogenic disorders other than CAPS and DIRA | |
| Other diseases with IL-1 response | |
| Familial Mediterranean fever (FMF) | [78,119]–[125] |
| TNF receptor-associated periodic syndrome (TRAPS) | [126]–[129] |
| Hyper immunoglobulin (Ig)D syndrome (HIDS) | [130,131] |
| PAPA syndrome† | [132,133] |
| Polygenic disorders‡ | |
| SOJIA§ | [71,72,134] |
| AOSD¶ | [135]–[141] |
| PFAPA | [73] |
| Behçet's disease | [77] |
| Schnitzler syndrome | [74]–[76] |
| ‘Metabolic’ autoinflammatory diseases | |
| Gout | [81]–[83] |
| Diabetes type 2 | [84] |
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome.
These probable polygenetic diseases do not yet have any genetic mutations or polymorphisms identified but are believed to be caused by genetic predispositions.
Only studies with more than 10 patients are listed.
Only studies with more than five patients are listed. DIRA: deficiency of the interleukin (IL)-1 receptor antagonist; CAPS: cryopyrin-associated periodic syndromes; PAPA: pyogenic arthritis, pyoderma gangrenosum and acne syndrome; FPAPA: periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis; SOJIA: systemic onset juvenile idiopathic arthritis; AOSD: adult-onset Still's disease; TNF: tumour necrosis factor.
The impact of the understanding of the NLRP3-inflammasome on shaping the concept of autoinflammatory diseases
The discovery of CIAS1/NLRP3 as the genetic cause for CAPS has not only provided a molecular understanding of the pathogenesis of autoinflammatory diseases, but also pointed to an important role of IL-1 in innate immune regulation, as an important mediator triggered by microbial and endogenous danger signals [28]. Structural similarities between NLRP3 and pathogen-resistance receptors found in plants, particularly the presence of a leucin-rich repeat (LRR) domain [29], a motif known to act as a pathogen-binding domain, and a regulatory NACHT domain have suggested that NLRP3 may be involved in the recognition of pathogen-specific signals [30] and fuelled the interest in NLRP3 as a human intracellular danger ‘receptor’. NLRP3 and three other NLR family members can complex with the adaptor protein, apoptosis-associated speck-like protein containing a CARD domain (ASC), and the protease, procaspase-1, and form an IL-1 activating platform termed ‘inflammasome’[31,32]. The inability to crystallize the NLRP3/cryopyrin protein has hampered the investigation of molecules that can bind directly to the LRR domain of NLRP3. However, a growing number of stimuli can activate the NLRP3 inflammasome, including microbial stimuli such as lipopolysaccharide (LPS), nucleic acids, muramyl dipeptide (MDP), toxins (i.e. nigericin, maitotoxin), environmental large inorganic crystallinic structures, such as asbestos and silica, and adjuvants including aluminium hydroxide (alum), and endogenous cellular ‘danger’ molecules such as adenosine triphosphate (ATP), uric acid crystals, hyaluronan, heparan sulphate, amyloid-β fibrils (reviewed in [33], as well as metabolic triggers [28,34] such as glucose, free fatty acids, oxidated LDL, ceramide [28,35,36], islet amyloid polypeptide IAPP [37] and cholesterol crystals [38], all molecules that are released when tissue is stressed or cells are dying) (Fig. 1). Although exposure to cold and air conditioning are reliable triggers of inflammatory attacks in FCAS patients, and cold challenges have even been used in patients to evaluate the pathogenesis of FCAS [39], the molecular triggers that fuel the inflammatory response in MWS and NOMID are still not known. It remains to be determined whether fluctuations in the concentrations of the known endogenous stimuli of the inflammasome are the physiological triggers of the ongoing baseline inflammation and the acute disesase exacerbations in patients with MWS and NOMID that are typically not triggered by cold exposure.
Fig. 1.
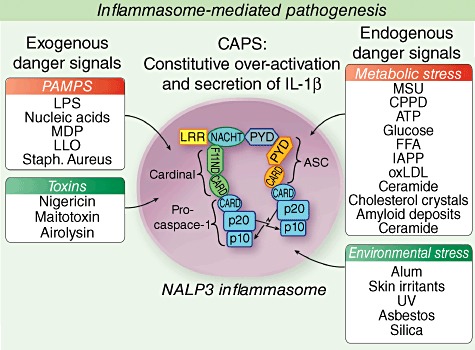
Triggers of the inflammasome.
The diverse type of NLRP3 inflammasome triggers led to the search of a common NLRP3 inflammasome-activating pathway and the increase of reactive oxygen species (ROS), which are released under many conditions causing cell stress, have been postulated as an attractive mechanism. Recently, data suggesting a role for mitochondrial ROS in inflammasome activation have been published. Blockade of complexes in the respiratory chain in the mitochondria lead to the accumulation of ROS and subsequent inflammasome activation. ROS generation and inflammasome activation can be suppressed when mitochondrial membrane channels, the voltage-dependent anion channels (VDAC), are blocked, suggesting that the NLRP3 inflammasome senses various degrees of mitochondrial dysfunction and may be primed by the mitochondrial ROS to produce IL-1β[40]. Other studies have proposed that macrophages lacking autophagy keep producing increased levels of IL-1β, which suggests that autophagy may reduce inflammasome activation by eliminating ROS, producing damaged mitochondria [40]–[42]. The role of increased inflammasome activity as a result of loss of dysfunctional autophagy has been implicated in the pathogenesis of Crohn's disease (reviewed in Tschopp [43]). Recently, data from CAPS patients suggest that the balance of ROS and the anti-oxidant production within the macrophages is shifted towards an increase in intracellular ROS and anti-oxidant production in cells carrying a disease causing CIAS1 mutation, suggesting that increased cellular ROS production may contribute to the constitutive release of IL-1β in CAPS patients [44].
In summary, the NLRP3 inflammasome illustrates that IL-1 is released in response to danger from exogenous and endogenous sources and provides a model of chronic activation and stimulation that cannot be down-regulated in patients with CAPS due to the intrinsic activating mutations. In metabolic diseases the continued stimulation is thought to be due to high local concentrations of the inflammasome-activating metabolic trigger.
Monogenic autoinflammatory diseases responsive to IL-1 blockade
CAPS
The disease spectrum of CAPS includes FCAS at the least severe, MWS and NOMID, also called CINCA at the most severe end of the clinical disease spectrum.
Pathogenesis of CAPS/NOMID
CAPS are caused by autosomal dominant gain of function mutations in NLRP3 (also NALP3, CIAS1 or PYPAF1). While most patients with FCAS and MWS constitute familial cases, the mutations in most NOMID patients are sporadic. About 30–50% of patients with the clinical diagnosis of NOMID/CINCA who respond favourably to IL-1 blocking therapy are ‘negative for mutations’ in NLRP3/CIAS1 based on Sanger-sequencing of DNA prepared from peripheral blood, which raised the questions of additional genetic mechanisms that cause disease, including the search for somatic mutations. While somatic NLRP3 mosaicism was suggested in earlier studies [45,46], a recent study assessed the frequency of somatic mutations in mutation-negative NOMID patients and identified somatic mutations in 18 of 26 patients (69·2%). Estimates of the level of mosaicism ranged from 4·2% to 35·8% [47]. Although questions about the kind of disease relevant ‘mosaic’ cell populations and how low-level mosaicism in the blood can cause a severe disease phenotype remain unresolved, mosaicism seems a plausible mechanism to explain disease in the Sanger mutation-negative patients.
It had been well established that cryopyrin mutations in CAPS are gain of function mutations leading to caspase-1 activation and increased activation and secretion of the mature form of IL-1β[48]. However, recently the administration of the anti-IL-1β antibody canakinumab allowed quantification of IL-1β–canakinumab complexes and the approximate basal IL-1β production in healthy controls was estimated to be around 6 ng/dl. In FCAS/MWS patients the level was about 31 ng/dl [49], and these levels are even higher in NOMID/CINCA patients (unpublished observations), suggesting a correlation of disease severity and organ involvement with systemic levels of IL-1 production.
The clinical phenotype of CAPS and NOMID and treatment
FCAS, MWS and NOMID/CINCA form a disease spectrum of CAPS, with FCAS on the mildest and NOMID on the severe end of the disease spectrum. The systemic inflammatory disease manifestations in all CAPS patients include episodes of fever, urticarial rash, joint pain and elevations in acute phase reactants which are triggered by exposure to cold in patients with FCAS. However, with increasing disease severity of CAPS the inflammatory involvement of additional organs and the resulting organ damage and disability differ widely. While the long-term outcome in FCAS is generally favourable, the functional impact of the disease in NOMD/CINCA patients is significant [50,51]. The disability seen in NOMID patients is a consequence of the duration, the extent and the severity of the inflammatory organ involvement that, if untreated, leads to permanent organ damage and disability. The ability to achieve resolution of systemic and organ inflammation with IL-1 blocking treatment indicates the importance of early initiation of treatment before permanent organ damage and disability have developed [51]. In patients with NOMID, chronic severe systemic inflammation causes growth retardation, osteopenia/osteoporosis and MWS, and NOMID patients are at an increased risk to develop amyloidosis [52]. The skin lesions in CAPS are due to neutrophilic urticaria and in FCAS patients the rash is triggered by cold exposure. While skin lesions disappear 24–48 h after the cold challenge in FCAS patients, the skin lesions wax and wane chronically in MWS and NOMID patients. Conjunctivitis is the most common form of eye involvement and anterior uveitis, corneal infiltrates, papilloedema and rarely posterior uveitis can be seen in patients with MWS or NOMID. Band keratopathy, corneal clouding, retinal scarring and optic nerve atrophy develop as consequence of uncontrolled inflammation and lead to visual impairment [51,53]. Sensorineural hearing loss develops in most patients with NOMID/CINCA in the first decade of life; in contrast, in MWS patients, progressive neurosensory hearing loss often develops significantly later in life and is absent in patients with FCAS [54,55]. Hearing loss in NOMID is caused by cochlear inflammation which can be visualized on contrast-enhanced fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) [56]. Cochlear enhancement improves with treatment [56] and chronic inflammation may lead to damage of neuroepithelial Corti hair cells, as was suggested in a paper stating data from necrobiopsy of inner ears in MWS patients [57]. Central nervous system (CNS) manifestations can be the most devastating. Aseptic meningitis and increased intracranial pressure are common, and permanent brain damage includes ventriculomegaly due to increased intracranial pressure and brain atrophy. The degree of cognitive impairment in patients with NOMID can vary widely from normal to severely impaired [50,56,57]. Interestingly, MRI findings seen in many NOMID patients have not been found in patients with milder disease [58], which is consistent with more severe IL-1-mediated inflammation in the brain in NOMID patients. The ‘bony lesions’ in NOMID are tumour-like protrusions originating from the growth plate. Physical disability in patients with NOMID is also caused by limb length discrepancies and joint contractures secondary to reduced longitudinal growth of the affected bone, which can lead to significant loss of mobility and severe growth retardation [59].
Clinical trials in CAPS and safety of IL-1blockade
The current standard of care to treat CAPS patients is lifelong treatment with IL-1 blocking therapies based on the dramatic responses to IL-1 blockade alone without the need of additional disease-modifying anti-rheumatic drugs (DMARDs) or steroids. Drug withdrawals of the IL-1 targeting therapies lead to subsequent disease flares, and three agents that block IL-1 have been used to treat these patients; trials are summarized in Table 2. Anakinra (Kineret®) was Food and Drug Administration (FDA)-approved in the United States for the treatment of rheumatoid arthritis in 2001, and although it has been the first IL-1 blocking agent to be administered to CAPS patients it is not yet FDA-approved for that indication [39,56,60,61]. Anakinra doses adminiatered in clinical studies ranged from 1 to 10 mg/kg/day with good tolerability of the treatment [57]. Two longer-acting IL-1-blocking medications that have subsequently been used in CAPS patients have been FDA-approved for the treatment of FCAS and MWS under the orphan drug programme: rilonacept (Arcalyst®, IL-1 Trap), recombinant human IL-1 receptor–immunoglobulin (Ig) fusion protein with a half-life of 34–57 h was FDA-approved in February 2008 and canakinumab (Ilaris®), and a humanized anti-IL-1β antibody with a half-life of 21–28 days in June 2009. Weekly subcutaneous injections of rilonacept were efficacious in the treatment of FCAS and MWS [62,63]. Subcutaneous injections of canakinumab every 8 weeks were given to 35 patients in a randomized withdrawal study [64]. Several long-term outcome studies with the different agents suggest sustained responses [57,63,65].
All three currently approved IL-1 blocking agents are generally well tolerated, and no increase in opportunistic infections or malignancies in CAPS have been reported. The most common side effect with anakinra is an injection site reaction within the first 4 weeks of therapy. Most patients continued on study drug and typically the injection reacations resolve [66]. The most commonly reported adverse reaction associated with rilonacept was also an injection site reaction, which was mild. In 360 patients treated with rilonacept and 179 treated with placebo, the incidence of infections was 34% versus 27% for rilonacept and placebo respectively. One Mycobacterium intracellulare infection after bursal injection in a patient with Still's disease and a death from Streptococcus pneumoniae meningitis occurred [67]. In the canakinumab studies injection site reactions occurred in up to 9% of patients and up to 14% of patients developed vertigo with the injections [68].
DIRA
The clinical phenotype of DIRA
DRA is an autosomal recessive disease caused by mis-sense and non-sense mutations in IL1RN, the gene encoding the IL-1-receptor antagonist (IL-1Ra) protein. The mutations lead to either absence of protein expression [11,12] or expression of a non-functional protein [69]. The mutations described are founder mutations in Puerto Rico, the Netherlands, Newfoundland, Palestine/Lebanon and Brazil [69] and in a compound heterozygote patient of northern European ancestry, who also carried the Dutch mutation p.E77X on the maternal allele [70]. Non-expression or expression of a non-functional protein leads to unopposed IL-1 receptor activation and an increased response to IL-1α and IL-1β stimulation [11,12,69]. Patients with DIRA present in the neonatal period. Perinatal comorbidities and need for instrumentation including the placement of central catheters seem to contribute to the perinatal morbidity and mortality risk. Most patients develop skin pustulosis in the perinatal period that can be localized or generalized. Joint swelling, painful osteolytic lesions, periosteitis affecting the distal ribs and the long bones and heterotopic bone formation, mainly around the proximal femur, are seen typically. Patients can develop fevers, but the height of the fever has been modest in context with the high elevation of acute-phase reactants during disease flares. A striking feature is the pathergy response that develops in areas of mechanical skin trauma, but the formation of thrombi around catheters and around areas of irritation such as G-tubes are also observed, and indicate an exaggerated inflammatory response of the vascular endothelium and the surrounding tissue to a mechanical trigger.
Patients with DIRA respond incompletely to high doses of prednisone; however, a rapid and dramatic response to the recombinant IL-1 receptor antagonist anakinra has been seen in all children treated so far. Interestingly, the genomic deletion observed in the Puerto Rican patients includes the locus for IL-1RN and five additional genes: IL1F6, IL1F8 and IL1F9 encoding the receptor ‘agonists’ IL-36α, IL-36β and IL-36γ; IL1F5 encoding the IL-36 receptor antagonist (Fig. 2); and IL1F10. However, three patients with this deletion had complete responses to anakinra suggesting no additional contribution of the deleted IL-36 receptor agonists and antagonists on the inflammatory phenotype of these patients (unpublished observations).
Fig. 2.
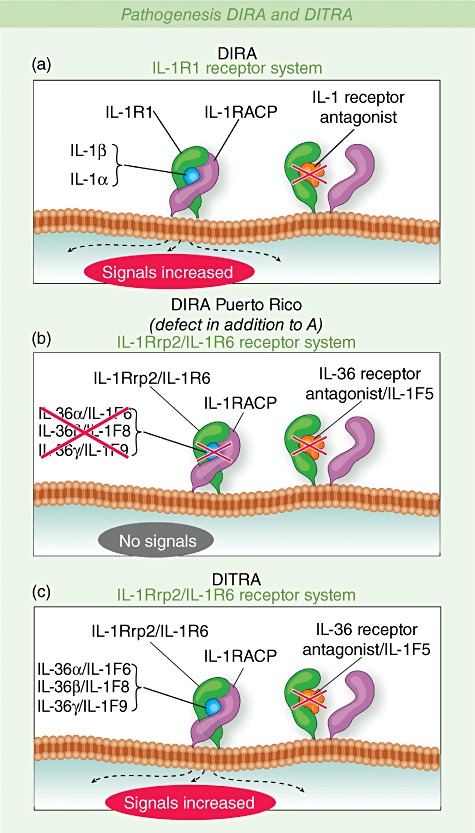
Pathogenesis deficiency of the interleukin (IL)-1 receptor antagonist (DIRA) and deficiency of the IL-thirty-three receptor antagonist (DITRA). Blockade of IL-1 signalling by IL-1 receptor antagonist. IL-1 receptor antagonist competes with IL-1 for binding to the IL-1 receptor preventing recruitment of the IL-1 accessory protein and downstream signalling.
Polygenic autoinflammatory diseases with clinical evidence of IL-1-mediated pathogenesis
Systemic inflammatory conditions with phenotypic similarities to monogenic autoinflammatory diseases responding to IL-1 inhibition
Among the polygenic or genetically undefined autoinflammatory diseases some respond to IL-1 blockade, including systemic onset Still's disease in children or adults [71,72], PFAPA, usually presenting in childhood [73], and Schnitzler syndrome [74]–[76] and Behçet's disease, presenting typically in adulthood [77,78].
The clinical bone manifestations of DIRA resemble the bone lesions seen in another autoinflammatory disease, chronic recurrent multi-focal osteomyelitis (CRMO) and in an animal model of CRMO, the cmo mouse; patients with CRMO can also present with pustular skin lesions. In the cmo mouse model high levels of IL-1 have been found in the bone lesions [79,80] but clinical trials with IL-1 blocking agents have not been performed.
‘Metabolic’ polygenic autoinflammatory conditions and IL-1
A group of clinically diverse disorders that all present with typically minimal systemic inflammation have been referred to recently as autoinflammatory diseases. Interestingly, these disorders share a metabolic defect that leads to accumulation of endogenous molecules that can activate the NLRP3 inflammasome (Fig. 1), hence triggering an IL-1 response. In some of these postulated autoinflammatory disorders the use of IL-1 blocking agents has validated that concept. IL-1 blockade is efficacious in the treatment of gout [81]–[83] and possibly diabetes [84], where insulin resistance and glycosylated haemoglobin were significantly lower in the treatment group compared to the placebo group. In an animal model of Alzheimer's disease, amyloid β fibrils were able to stimulate IL-1β release in an inflammasome-dependent fashion [85] and in a model of asbestos-induced lung fibrosis [86] inflammasome-dependent IL-1 release was also observed, suggesting a possible role for IL-1 in these conditions.
Novel autoinflammatory diseases suggesting additional pathogenic pathways beyond IL-1
The discovery of the role of IL-1 in NOMID and DIRA was spearheaded by clinical trials using targeted IL-1 inhibition that clinically confirmed the role of IL-1. Unresponsiveness to IL-1 blockade on the other hand have helped to distinguish and clinically characterize novel autoinflammatory diseases.
Deficiency of the IL-thirty-six [37] receptor antagonist (DITRA)
The clinical phenotype and the pathogenesis of DITRA
Recently, another gene among the IL-1 family members was implicated in the development of generalized or palmoplantar pustulosis. Two groups identified three missense mutations in IL36RN that lead to generalized or plantopalmar pustular psoriasis of variable onset reaching from perinatal, as early as 2 weeks of age, to well into adulthood [22,23]. One mutation, p.Leu27Pro (L27P), was seen in nine Tunisian families and two mutations in three unrelated patients of European ancestry, with two being heterozygous for a p.Ser113Leu (S113L) mutation and in a third patient a compound heterozygote for the p.Ser113Leu and a p.Arg48Trp (R48W) missense mutation. The clinical phenotype of DITRA is confined to the skin; some patients have mucosal findings of a migratory glossitis and nail dystrophies, and a few patients also had oligoarthritis. In mice, Il1f6 over-expression (the mouse orthologue of IL-36α) leads to abnormal IL-36 signalling and causes skin inflammation characterized by acanthosis, hyperkeratosis and a mixed-cell infiltrate containing neutrophils [87]. When crossed with Il1f5 (the mouse orthologue of IL-36RN)-deficient mice, the skin phenotype became enhanced with extensive pustule formation [87]. The response of this condition to IL-1 blocking therapy is currently not known but when the conditional IL-36 knock in mice were crossed with IL-1 receptor knock out mice, the skin disease was not abrogated suggesting that IL-36 can cause IL-1 independent inflammation and patients may likely not respond to I-1 blocking therapy.
Comparison of NOMID, DIRA and DITRA
Although the three disorders discussed above have some clinical similarities, there are important clinical differences that may provide us with a molecular understanding of the organ specific disease manifestations and shed light on downstream pathways that mediate a clinical phenotype.
NOMID is caused by IL-1β oversecretion, DIRA by uninhibited IL-1α and ILl-1β signalling and DITRA by uninhibited IL-36α, β and γ (all members of the IL-1 family) signalling; NOMID and DIRA can be treated effectively with IL-1 inhibition; IL-1 inhibition has not been used in DITRA patients. The rash in NOMID is urticaria-like with a neutrophilic infiltrate in the dermis; in contrast, in DIRA and DITRA, the rash is pustular and the neutrophils in the epidermis form subcorneal pustules mainly along hair shafts. All disorders present with systemic inflammation characterized by elevated acute-phase reactants.
Other organ manifestations in NOMID described in detail above include aseptic neutrophilic meningitis, progressive hearing loss, increased intracranial pressure and eye inflammation and bony hypertrophy seen in 40–50% of severe NOMID patients. In DIRA patients, a hallmark of the disease is the presence of osteolytic lesions and periosteitis; other manifestations include CNS vasculitis and interstitial lung disease. In DITRA, no other organ other than the skin have been reported so far (Table 3). Hearing loss and eye inflammation other than occasional mild conjunctivitis have not been observed in DIRA and DITRA. Based on the phenotypic differences one might speculate that IL-1α may contribute to the osteolytic bone lesions in DIRA, which are absent in NOMID and DITRA and are characterized by an inflammatory cell infiltrate. These lesions disappear on anakinra. The bone lesions in NOMID/CINCA, in contrast, lack the presence of inflammatory cells [59] and have clinical and histological similarities with bone lesions from patients with the non-inflammatory disorders fibrous dysplasia (FD) and osteochondro-myxomas (OCM). These clinical similarities prompted investigations of a shared pathway and increased protein kinase A (PKA) activity observed previously in OCM and FD led to inflammation-independent activation of caspase-1 via over-expression of the proto-oncogene (and early osteoblast transcription factor) Ets-1, and further increased cyclic (c) adenosine-monophosphate (cAMP)-mediated PKA activity and wnt signalling also in NOMID cartilage cells [88], suggesting an amplification loop that is IL-1 independent and could explain the lack of bone response to anakinra treatment [57,59].
Table 3.
A comparison of neonatal-onset multi-system inflammatory disease/chronic infantile neurological, cutaneous and articular syndrome (NOMID/CINCA) with deficiency of the interleukin (IL)-1 receptor antagonist (DIRA) and deficiency of the IL-thirty-three receptor antagonist (DITRA)
| NOMID/CINCA | DIRA | DITRA | |
|---|---|---|---|
| Gene | CIAS1/NLRP3 | IL1RN | IL36RN |
| Functional consequence | Increased inflammasome activation | Decreased inhibition of IL-1α and IL-1β | Decreased inhibition of IL-36α, IL-36β and IL-36γ |
| Disease onset | perinatal | perinatal | Variable, perinatal to adulthood |
| Skin | Urticarial rash | Generalized pustulosis | Generalized pustulosis, palmarplantar pustulosis |
| Bone | Epiphyseal overgrowth | Multi-focal osteomyelitis, periosteitis | None described |
| CNS | Aseptic meningitis, progressive vision and hearing loss | Central nervous system vasculitis in two patients but no ocular or inner ear inflammation | None described |
| Lung | No lung disease observed | Interstitial lung disease (rare) | None described |
In DITRA and DIRA the neutrophils are present in the epidermis and in NOMID the neutrophils are present only in the dermis; pustules have never been observed. IL1RN and IL36RN are both highly expressed in the epidermis and their loss would render keratinocytes more active to IL-1 and IL-36 stimulation, respectively. In contrast, the presence of high levels of IL-1Ra in the epidermis in normal skin and in NOMID that is absent in DIRA may prevent IL-1β-mediated epidermal recruitment of inflammatory cells.
Whether the similarity of the skin lesions in DIRA and DITRA is caused by a shared common pathway that may involve the up-regulation of IL-23 and IL-17 that is present in the skin in DIRA patients [11] and has been found in mice over-expressing IL-1F6 (the mouse orthologue of IL-36α) needs further evaluation [89]. IL-36 is a potent inducer of the proinflammatory cytokines IL-12, IL-1β, IL-6, TNF-α and IL-23 in bone marrow-derived dendritic cells and also induced the production of interferon (IFN)-γ, IL-4 and IL-17 by CD4+ T cells and cultured splenocytes [90], suggesting that IL-36 has a direct proinflammatory effect on these cells and that its effects are less likely to be mediated through IL-1. In contrast to IL-1, there is currently no clinical evidence that IL-36 has a role in bone inflammation.
Early-onset enterocolitis (IBD)
The balance of IL-10 and inflammatory bowel disease
Early-onset IBD, caused by autosomal recessive mutations in the IL-10 receptor, was first described in 2009. In four patients, three distinct homozygous mutations in the genes IL10RA and IL10RB, encoding the IL-10R1 and IL-10R2 proteins which form a heterotetramer to make up the IL-10 receptor have been reported to cause a neonatal disease presenting with severe inflammatory bowel disease and folliculitis [21]. These ‘lack of function’ mutations result in the loss of IL-10 signalling, as demonstrated by deficient signal transduction and activator of transcription-3 (STAT3) phosphorylation after stimulation with IL-10 and proves that loss of signalling of the anti-inflammatory cytokine IL-10 is sufficient to cause enterocolitis as a main clinical feature. Levels of TNF-α and other inflammatory cytokines, including IL-1α, IL-1β and IL-6, were also increased in these patients. One of the patients underwent an allogeneic stem cell transplant with sustained resolution of symptoms at 1 year following transplant. Early-onset inflammatory bowel disease (IBD) mimics a disease found in knock-out (KO) mice for IL10rb (crf2-4), a subunit of the IL-10 receptor, that is essential for IL-10-mediated effects, mainly on gut inflammation [91]. These disorders point to the key role of IL-10 in complex human disorders associated with gut inflammation. Single nucleotide polymorphisms (SNPs) associated with decreased IL-10 production were identified in genome-wide association studies in inflammatory bowel disease (Crohn's and ulcerative colitis) [92,93], as well as an association with IL-10 promoter polymorphisms producing low IL-10 levels in Behçet's disease, which also has an increased risk for the development of gut inflammation [94,95].
CANDLE and other proteasome associated disorders
Syndromes associated with immunoproteasome dysfunction cause an autoinflammatory phenotype
The independent discovery that autosomal recessive mutations in PSMB8, which encodes the inducible proteasome subunit beta type-8 (PSMB8) component of the immunoproteasome complex (i-proteasome), causes a number of syndromes that have been described in Japan since the 1980s as Nakajo–Nishimura syndrome (NNS) [96]–[98], also recently named JASL [26], and in the West as joint contractures, muscle atrophy and panniculitis-induced lipodystrophy (JMP) syndrome [99,100]; and chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperatures (CANDLE) [101,102], is illustrating the clinical spectrum of proteasome-associated autoinflammatory syndromes. Three different homozygous mutations have been identified so far in PSMB8, p.T75M [24,27] in patients of European and Hispanic decent, p.C135X [27] in a Jewish patient and the same mutation G197V [25,26] by two different investigator groups in Japan.
Although most reported patients are homozygous for mutations in PSMB8, one patient with the disease was negative for mutations in PSMB8 and two were heterozygous, suggesting genetic heterogeneity [27]. Clinical features include a variable onset of repeated attacks of erythematous and violaceous, annular cutaneous plaques, lasting for a few days or weeks and leaving residual purpura. Most patients developed disease before the age of 6 months and other prominent features include persistent periorbital erythema and oedema, finger or toe swelling and hepatomegaly with variable elevation of acute phase reactants, progressive lipodystrophy and stigmata of chronic inflammation such as failure to thrive, lymphadenopathy and hypochromic or normocytic anaemia. An atypical neutrophilic infiltrate is seen in the dermis and marked elevation of IFN-γ-induced protein (IP-10) [27]. Once lipodystrophy has developed, the disease outcome is critical; two of our patients died before reaching adulthood [101]. The development of severe lipodystrophy, joint contractures and the development of muscle atrophy [99] and cardiomyopathy [103] show the severe outcome of untreated disease. The high elevation of IP-10 levels [24,25,27] and the prominent IFN signature that distinguish this disorder from the IL-1-mediated disorders persisted upon treatment with TNF, IL-1 and IL-6 inhibition and may point to a pathway that constitutes a rational target for therapeutic intervention [27]. Recent data in psmb8/lmp7 KO mice [104] suggest an important additional role of the proteasome in maintaining cell homeostasis by removing accumulating proteins marked for degradation from the cells and failure to process/degrade protein results in the formation of ubiquitin-rich cytoplasmic aggregates or inclusions that consequently increase cellular sensitivity to apoptosis [105], suggesting that the inability of the mutant i-proteasome to process and degrade the up-regulated proteins may cause a vicious circle of abnormal IFN responses that may contribute to at least some of the inflammatory disease manifestations.
Common theme of an interferon stress response?
Some disease manifestations in patients with CANDLE and the other PSMB8-associated syndromes are reminiscent of clinical features in patients with dermatomyositis and lipodystrophy; interestingly, a prominent IFN signature has been described in blood and skin samples from these patients [106]–[109]. Other patients with prominent IFN signatures and some phenotypic similarities, including basal ganglion calcifications, include disorders with complement deficiencies [110]–[112], Aicardi–Goutières syndrome (AGS) [113,114], a disorder of defective endonucleases processing of DNA, and the recently identified disease spondyloenchondrodys-plasia (SPENCD), a disease caused by deficiency of a tartrate-resistant form of acid phosphatase (TRAP) which leads to accumulation of phosphorylated osteopontin [115,116] and possibly monogenic systemic lupus erythematosis associated with C1q deficiency [117,118] (Table 4). It is intriguing to speculate that analogous to IL-1, which is an alarm signal to a number of microbial and intracellular danger signals, type 1 IFNs may be pivotal alarm cytokines in response to not only viral infections but a number of endogenous triggers, including those accumulated in IFN-associated disorders [118]. Whether the IFN signature in these disorders provides a pathogenic link that can point to an effective treatment strategy awaits further studies.
Table 4.
Diseases with prominent interferon (IFN) (mainly type 1) signatures and phenotypic similarities to proteasome-associated disorders
| Syndrome | Gene | Enzyme defect | Studies indicating IFN signature |
|---|---|---|---|
| Monogenic disorders | |||
| Systemic lupus erythematosis (with immunodeficiencies) | C1QA, C1QB, C1QC | Complement defect, C1q deficiency | [117,118] |
| Aicardi–Goutières syndrome (AGS) | TREX1→ | 3′ to 5′ DNA exonuclease digests DNA : RNA hybrids putative nuclease | [113,114,142] |
| RNaseH2→ | |||
| SAMHD1→ | |||
| Spondyloenchondrodysplasia (SPENCD) | ACP5 | Tartrate-resistant acid phosphatase (TRAP) accumulation of phosphorylated osteopontin | [102,103] |
| JMP, NNS, JASL, CANDLE | PSMB8 and others | Proteasome dysfunction and accumulation of polyubiquitilated proteins | [24]–[27] |
| Diseases with unknown genetics | |||
| Dermatomyositis (some forms) | nk | NA | [106]–[109] |
nk: not known; JMP: joint contractures, muscle atrophy, panniculitis induced lipodystrophy syndrome; NNS: Nakajo–Nishimura syndrome; JASL: Japanese autoinflammatory syndrome with lipodystrophy; CANDLE: chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperatures.
Concluding remarks
The study of rare monogenic autoinflammatory diseases has led to the discovery of the prominent role of IL-1 in mediating systemic and organ specific inflammation. In particular, the discovery that missense mutations in NLRP3 are the cause of the clinical spectrum of the CAPS has fuelled research that unravelled the role of the NLRP3 inflammasome as a sensor of cellular stress and danger that co-ordinates the activation and secretion of IL-1, an early response cytokine that can co-ordinate an immune response. The characterization of stimuli that lead to NLRP3 inflammasome activation have led to the further discoveries that the NLRP3 inflammasome activation and IL-1 release may be a common pathway in a number of apparently disparate disorders such as gout, type 2 diabetes and atherosclerosis, whereby the organ specific accumulation of inflammasome triggers confers tissue specificity of the immune response in these disorders. The use of IL-1 blocking agents in a wider spectrum of autoinflammatory diseases has revealed patient subsets who do not respond to IL-1 blockade, and together with clinical phenotyping led to the discovery of other autoinflammatory diseases, including a group of monogenic disorders that are associated with a prominent type 1 IFN pattern in peripheral blood. Whether IFN blocking strategies will result in effective therapies in these disorders needs to be examined further.
Disclosure
Dr Goldbach-Mansky has received financial support for clinical studies from Regeneron, Novartis and Biovitrum Inc.
References
- 1.International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. doi: 10.1016/s0092-8674(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 2.French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. doi: 10.1038/ng0997-25. [DOI] [PubMed] [Google Scholar]
- 3.McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97:133–44. doi: 10.1016/s0092-8674(00)80721-7. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat Genet. 2001;29:301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldmann J, Prieur AM, Quartier P, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46:3340–8. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Manji GA, Grenier JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002;277:29874–80. doi: 10.1074/jbc.M203915200. [DOI] [PubMed] [Google Scholar]
- 8.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 9.Dinarello CA. Mutations in cryopyrin: bypassing roadblocks in the caspase 1 inflammasome for interleukin-1beta secretion and disease activity. Arthritis Rheum. 2007;56:2817–22. doi: 10.1002/art.22841. [DOI] [PubMed] [Google Scholar]
- 10.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–5. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 11.Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–37. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360:2438–44. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA. 2008;105:1614–19. doi: 10.1073/pnas.0708616105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houten SM, Kuis W, Duran M, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22:175–77. doi: 10.1038/9691. [DOI] [PubMed] [Google Scholar]
- 15.Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999;22:178–81. doi: 10.1038/9696. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome) J Med Genet. 2005;42:551–57. doi: 10.1136/jmg.2005.030759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002;11:961–69. doi: 10.1093/hmg/11.8.961. [DOI] [PubMed] [Google Scholar]
- 18.Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet. 2001;29:19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 19.Kanazawa N, Okafuji I, Kambe N, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105:1195–97. doi: 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- 20.Ueki Y, Tiziani V, Santanna C, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28:125–26. doi: 10.1038/88832. [DOI] [PubMed] [Google Scholar]
- 21.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marrakchi S, Guigue P, Renshaw BR, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med. 2011;365:620–8. doi: 10.1056/NEJMoa1013068. [DOI] [PubMed] [Google Scholar]
- 23.Onoufriadis A, Simpson MA, Pink AE, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet. 2011;89:432–7. doi: 10.1016/j.ajhg.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal AK, Xing C, DeMartino GN, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87:866–72. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arima K, Kinoshita A, Mishima H, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo–Nishimura syndrome. Proc Natl Acad Sci USA. 2011;108:14914–19. doi: 10.1073/pnas.1106015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitamura A, Maekawa Y, Uehara H, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011;121:4150–60. doi: 10.1172/JCI58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Ramot Y, Torrelo A, et al. Mutations in PSMB8 cause CANDLE syndrome with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2011 doi: 10.1002/art.33368. Sep 27. doi: 10.1002/art.33368. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 29.Jin MS, Lee JO. Structures of the Toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–91. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Ausubel FM. Are innate immune signaling pathways in plants and animals conserved? Nat Immunol. 2005;6:973–79. doi: 10.1038/ni1253. [DOI] [PubMed] [Google Scholar]
- 31.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity. 2004;20:319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 32.Mankan AK, Kubarenko A, Hornung V. Immunology in the clinic review series; focus on autoinflammatory diseases: inflammasomes: mechanisms of activation. Clin Exp Immun. 2012;167:369–81. doi: 10.1111/j.1365-2249.2011.04534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–90. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–79. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USA. 2011;108:15324–29. doi: 10.1073/pnas.1100255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffman HM, Rosengren S, Boyle DL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–85. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–25. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 41.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 42.Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by NALP3 inflammasome. Nat Immunol. 2011;12:222–30. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tschopp J. Mitochondria: sovereign of inflammation? Eur J Immunol. 2011;41:1196–202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 44.Tassi S, Carta S, Delfino L, et al. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc Natl Acad Sci USA. 2010;107:9789–94. doi: 10.1073/pnas.1000779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saito M, Fujisawa A, Nishikomori R, et al. Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2005;52:3579–85. doi: 10.1002/art.21404. [DOI] [PubMed] [Google Scholar]
- 46.Arostegui JI, Lopez S, Pascal M, et al. A somatic NLRP3 mutation as a cause of a sporadic case of chronic infantile neurologic, cutaneous, articular syndrome/neonatal-onset multisystem inflammatory disease: novel evidence of the role of low-level mosaicism as the pathophysiologic mechanism underlying mendelian inherited diseases. Arthritis Rheum. 2010;62:1158–66. doi: 10.1002/art.27342. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in chronic infantile neurological cutaneous and articular syndrome patients: the results of an international multicenter collaborative study. Arthritis Rheum. 2011;63:3625–32. doi: 10.1002/art.30512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ozkurede VU, Franchi L. Immunology in the clinic review series; focus on autoinflammatory diseases: role of inflammasomes in autoinflammatory syndromes. Clin Exp Immun. 2012;167:382–90. doi: 10.1111/j.1365-2249.2011.04535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lachmann HJ, Lowe P, Felix SD, et al. In vivo regulation of interleukin 1beta in patients with cryopyrin-associated periodic syndromes. J Exp Med. 2009;206:1029–36. doi: 10.1084/jem.20082481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl. 1987;66:57–68. doi: 10.3109/03009748709102523. [DOI] [PubMed] [Google Scholar]
- 51.Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep. 2011;13:123–31. doi: 10.1007/s11926-011-0165-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muckle TJ, Wells M. Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q J Med. 1962;31:235–48. [PubMed] [Google Scholar]
- 53.Dollfus H, Hafner R, Hofmann HM, et al. Chronic infantile neurological cutaneous and articular/neonatal onset multisystem inflammatory disease syndrome: ocular manifestations in a recently recognized chronic inflammatory disease of childhood. Arch Ophthalmol. 2000;118:1386–92. doi: 10.1001/archopht.118.10.1386. [DOI] [PubMed] [Google Scholar]
- 54.Ahmadi N, Brewer CC, Zalewski C, et al. Cryopyrin-associated periodic syndromes: otolaryngologic and audiologic manifestations. Otolaryngol Head Neck Surg. 2011;145:295–302. doi: 10.1177/0194599811402296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kummerle-Deschner JB, Tyrrell PN, Reess F, et al. Risk factors for severe Muckle–Wells syndrome. Arthritis Rheum. 2010;62:3783–91. doi: 10.1002/art.27696. [DOI] [PubMed] [Google Scholar]
- 56.Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355:581–92. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neven B, Marvillet I, Terrada C, et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62:258–67. doi: 10.1002/art.25057. [DOI] [PubMed] [Google Scholar]
- 58.Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology. 2010;74:1267–70. doi: 10.1212/WNL.0b013e3181d9ed69. [DOI] [PubMed] [Google Scholar]
- 59.Hill SC, Namde M, Dwyer A, Poznanski A, Canna S, Goldbach-Mansky R. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA) Pediatr Radiol. 2007;37:145–52. doi: 10.1007/s00247-006-0358-0. [DOI] [PubMed] [Google Scholar]
- 60.Lepore L, Paloni G, Caorsi R, et al. Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J Pediatr. 2010;157:310–5. doi: 10.1016/j.jpeds.2010.02.040. [DOI] [PubMed] [Google Scholar]
- 61.Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle–Wells syndrome. Arthritis Rheum. 2011;63:840–9. doi: 10.1002/art.30149. [DOI] [PubMed] [Google Scholar]
- 62.Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 2008;58:2443–52. doi: 10.1002/art.23687. [DOI] [PubMed] [Google Scholar]
- 63.Goldbach-Mansky R, Shroff SD, Wilson M, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58:2432–42. doi: 10.1002/art.23620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–25. doi: 10.1056/NEJMoa0810787. [DOI] [PubMed] [Google Scholar]
- 65.Kuemmerle-Deschner JB, Hachulla E, Cartwright R, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. 2011;70:2095–102. doi: 10.1136/ard.2011.152728. [DOI] [PubMed] [Google Scholar]
- 66. Anakinra package insert. Available at: http://www.kineretrx.com/professional/pi.jsp (accessed 16 December 2011)
- 67. Rilonacept package insert. Available at: http://www.regeneron.com/ARCALYST-fpi.pdf (accessed 16 December 2011)
- 68. Canakinumab package insert. Available at: http://www.pharma.us.novartis.com/jsp/search/info/search.jsp (accessed 16 December 2011)
- 69.Jesus AA, Osman M, Silva CA, et al. A novel mutation of IL1RN in the deficiency of interleukin-1 receptor antagonist syndrome. Description of two unrelated cases from Brazil. Arthritis Rheum. 2011;63:4007–17. doi: 10.1002/art.30588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stenerson M, Dufendach K, Aksentijevich I, Brady J, Austin J, Reed AM. The first case of compound heterozygous IL1RN mutations causing deficiency of the interleukin-1-receptor antagonist. Arthritis Rheum. 2011;63:4018–22. doi: 10.1002/art.30565. [DOI] [PubMed] [Google Scholar]
- 71.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial) Ann Rheum Dis. 2011;70:747–54. doi: 10.1136/ard.2010.134254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci USA. 2011;108:7148–53. doi: 10.1073/pnas.1103681108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de Koning HD, Bodar EJ, Simon A, van der Hilst JC, Netea MG, van der Meer JW. Beneficial response to anakinra and thalidomide in Schnitzler's syndrome. Ann Rheum Dis. 2006;65:542–44. doi: 10.1136/ard.2005.045245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryan JG, de Koning HD, Beck LA, Booty MG, Kastner DL, Simon A. IL-1 blockade in Schnitzler syndrome: ex vivo findings correlate with clinical remission. J Allergy Clin Immunol. 2008;121:260–2. doi: 10.1016/j.jaci.2007.09.021. [DOI] [PubMed] [Google Scholar]
- 76.Schuster C, Kranke B, Aberer E, Arbab E, Sturm G, Aberer W. Schnitzler syndrome: response to anakinra in two cases and a review of the literature. Int J Dermatol. 2009;48:1190–4. doi: 10.1111/j.1365-4632.2009.04151.x. [DOI] [PubMed] [Google Scholar]
- 77.Botsios C, Sfriso P, Furlan A, Punzi L, Dinarello CA. Resistant Behçet disease responsive to anakinra. Ann Intern Med. 2008;149:284–86. doi: 10.7326/0003-4819-149-4-200808190-00018. [DOI] [PubMed] [Google Scholar]
- 78.Bilginer Y, Ayaz NA, Ozen S. Anti-IL-1 treatment for secondary amyloidosis in an adolescent with FMF and Behçet's disease. Clin Rheumatol. 2010;29:209–10. doi: 10.1007/s10067-009-1279-8. [DOI] [PubMed] [Google Scholar]
- 79.Ferguson PJ, Bing X, Vasef MA, et al. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone. 2006;38:41–7. doi: 10.1016/j.bone.2005.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ferguson PJ, El Shanti HI. Autoinflammatory bone disorders. Curr Opin Rheumatol. 2007;19:492–98. doi: 10.1097/BOR.0b013e32825f5492. [DOI] [PubMed] [Google Scholar]
- 81.McGonagle D, Tan AL, Shankaranarayana S, Madden J, Emery P, McDermott MF. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann Rheum Dis. 2007;66:1683–84. doi: 10.1136/ard.2007.073759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Terkeltaub R, Sundy JS, Schumacher HR, et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis. 2009;68:1613–17. doi: 10.1136/ard.2009.108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.So A, De Meulemeester M, Pikhlak A, et al. Canakinumab for the treatment of acute flares in difficult-to-treat gouty arthritis: results of a multicenter, phase II, dose-ranging study. Arthritis Rheum. 2010;62:3064–76. doi: 10.1002/art.27600. [DOI] [PubMed] [Google Scholar]
- 84.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 85.Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–77. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blumberg H, Dinh H, Trueblood ES, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med. 2007;204:2603–14. doi: 10.1084/jem.20070157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Almeida MQ, Tsang KM, Cheadle C, et al. Protein kinase A regulates caspase-1 via Ets-1 in bone stromal cell-derived lesions: a link between cyclic AMP and pro-inflammatory pathways in osteoblast progenitors. Hum Mol Genet. 2011;20:165–75. doi: 10.1093/hmg/ddq455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blumberg H, Dinh H, Dean C, et al. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J Immunol. 2010;185:4354–62. doi: 10.4049/jimmunol.1000313. [DOI] [PubMed] [Google Scholar]
- 90.Vigne S, Palmer G, Lamacchia C, et al. IL-36R ligands are potent regulators of dendritic and T cells. Blood. 2011;118:5813–23. doi: 10.1182/blood-2011-05-356873. [DOI] [PubMed] [Google Scholar]
- 91.Spencer SD, Di Marco F, Hooley J, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–78. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–23. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 93.Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wallace GR, Kondeatis E, Vaughan RW, et al. IL-10 genotype analysis in patients with Behçet's disease. Hum Immunol. 2007;68:122–27. doi: 10.1016/j.humimm.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 95.Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behçet's disease. Nat Genet. 2010;42:698–702. doi: 10.1038/ng.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kitano Y, Matsunaga E, Morimoto T, Okada N, Sano S. A syndrome with nodular erythema, elongated and thickened fingers, and emaciation. Arch Dermatol. 1985;121:1053–56. [PubMed] [Google Scholar]
- 97.Tanaka M, Miyatani N, Yamada S, et al. Hereditary lipo-muscular atrophy with joint contracture, skin eruptions and hyper-gamma-globulinemia: a new syndrome. Intern Med. 1993;32:42–5. doi: 10.2169/internalmedicine.32.42. [DOI] [PubMed] [Google Scholar]
- 98.Kasagi S, Kawano S, Nakazawa T, et al. A case of periodic-fever-syndrome-like disorder with lipodystrophy, myositis, and autoimmune abnormalities. Mod Rheumatol. 2008;18:203–7. doi: 10.1007/s10165-008-0033-4. [DOI] [PubMed] [Google Scholar]
- 99.Garg A, Hernandez MD, Sousa AB, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab. 2010;95:E58–E63. doi: 10.1210/jc.2010-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Megarbane A, Sanders A, Chouery E, Delague V, Medlej-Hashim M, Torbey PH. An unknown autoinflammatory syndrome associated with short stature and dysmorphic features in a young boy. J Rheumatol. 2002;29:1084–87. [PubMed] [Google Scholar]
- 101.Torrelo A, Patel S, Colmenero I, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. 2010;62:489–95. doi: 10.1016/j.jaad.2009.04.046. [DOI] [PubMed] [Google Scholar]
- 102.Ramot Y, Czarnowicki T, Maly A, Navon-Elkan P, Zlotogorski A. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: a case report. Pediatr Dermatol. 2011;28:538–41. doi: 10.1111/j.1525-1470.2010.01163.x. [DOI] [PubMed] [Google Scholar]
- 103.Oyanagi K, Sasaki K, Ohama E, et al. An autopsy case of a syndrome with muscular atrophy, decreased subcutaneous fat, skin eruption and hyper gamma-globulinemia: peculiar vascular changes and muscle fiber degeneration. Acta Neuropathol. 1987;73:313–19. doi: 10.1007/BF00688252. [DOI] [PubMed] [Google Scholar]
- 104.Moebius J, van den Broek M, Groettrup M, Basler M. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur J Immunol. 2010;40:3439–4. doi: 10.1002/eji.201040620. [DOI] [PubMed] [Google Scholar]
- 105.Seifert U, Bialy LP, Ebstein F, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142:613–24. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 106.Baechler EC, Bauer JW, Slattery CA, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. 2007;13:59–68. doi: 10.2119/2006-00085.Baechler. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. 2009;60:1815–24. doi: 10.1002/art.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rider LG, Miller FW. Mast cells and type I interferon responses in the skin of patients with juvenile dermatomyositis: are current therapies just scratching the surface? Arthritis Rheum. 2010;62:2619–22. doi: 10.1002/art.27525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum. 2010;62:2813–22. doi: 10.1002/art.27529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hannema AJ, Kluin-Nelemans JC, Hack CE, Eerenberg-Belmer AJ, Mallee C, van Helden HP. SLE like syndrome and functional deficiency of C1q in members of a large family. Clin Exp Immunol. 1984;55:106–14. [PMC free article] [PubMed] [Google Scholar]
- 111.McAdam RA, Goundis D, Reid KB. A homozygous point mutation results in a stop codon in the C1q B-chain of a C1q-deficient individual. Immunogenetics. 1988;27:259–64. doi: 10.1007/BF00376120. [DOI] [PubMed] [Google Scholar]
- 112.Botto M, Dell'Agnola C, Bygrave AE, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 113.Crow YJ, Leitch A, Hayward BE, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi–Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–6. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 114.Crow YJ, Hayward BE, Parmar R, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi–Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–20. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 115.Lausch E, Janecke A, Bros M, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet. 2011;43:132–37. doi: 10.1038/ng.749. [DOI] [PubMed] [Google Scholar]
- 116.Briggs TA, Rice GI, Daly S, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet. 2011;43:127–31. doi: 10.1038/ng.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Elkon KB, Stone VV. Type I interferon and systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31:803–12. doi: 10.1089/jir.2011.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ozen S, Bilginer Y, Aktay AN, Calguneri M. Anti-interleukin 1 treatment for patients with familial Mediterranean fever resistant to colchicine. J Rheumatol. 2011;38:516–18. doi: 10.3899/jrheum.100718. [DOI] [PubMed] [Google Scholar]
- 120.Meinzer U, Quartier P, Alexandra JF, Hentgen V, Retornaz F, Kone-Paut I. Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum. 2011;41:265–71. doi: 10.1016/j.semarthrit.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 121.Moser C, Pohl G, Haslinger I, et al. Successful treatment of familial Mediterranean fever with Anakinra and outcome after renal transplantation. Nephrol Dial Transplant. 2009;24:676–78. doi: 10.1093/ndt/gfn646. [DOI] [PubMed] [Google Scholar]
- 122.Roldan R, Ruiz AM, Miranda MD, Collantes E. Anakinra: new therapeutic approach in children with familial Mediterranean fever resistant to colchicine. Joint Bone Spine. 2008;75:504–5. doi: 10.1016/j.jbspin.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 123.Calligaris L, Marchetti F, Tommasini A, Ventura A. The efficacy of anakinra in an adolescent with colchicine-resistant familial Mediterranean fever. Eur J Pediatr. 2008;167:695–96. doi: 10.1007/s00431-007-0547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gattringer R, Lagler H, Gattringer KB, et al. Anakinra in two adolescent female patients suffering from colchicine-resistant familial Mediterranean fever: effective but risky. Eur J Clin Invest. 2007;37:912–14. doi: 10.1111/j.1365-2362.2007.01868.x. [DOI] [PubMed] [Google Scholar]
- 125.Kuijk LM, Govers AM, Frenkel J, Hofhuis WJ. Effective treatment of a colchicine-resistant familial Mediterranean fever patient with anakinra. Ann Rheum Dis. 2007;66:1545–46. doi: 10.1136/ard.2007.071498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Simon A, Bodar EJ, van der Hilst JC, et al. Beneficial response to interleukin 1 receptor antagonist in traps. Am J Med. 2004;117:208–10. doi: 10.1016/j.amjmed.2004.02.039. [DOI] [PubMed] [Google Scholar]
- 127.Sacre K, Brihaye B, Lidove O, et al. Dramatic improvement following interleukin 1beta blockade in tumor necrosis factor receptor-1-associated syndrome (TRAPS) resistant to anti-TNF-alpha therapy. J Rheumatol. 2008;35:357–58. [PubMed] [Google Scholar]
- 128.Gattorno M, Pelagatti MA, Meini A, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:1516–20. doi: 10.1002/art.23475. [DOI] [PubMed] [Google Scholar]
- 129.Obici L, Meini A, Cattalini M, et al. Favourable and sustained response to anakinra in tumour necrosis factor receptor-associated periodic syndrome (TRAPS) with or without AA amyloidosis. Ann Rheum Dis. 2011;70:1511–12. doi: 10.1136/ard.2010.143438. [DOI] [PubMed] [Google Scholar]
- 130.Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. 2011;70:2155–8. doi: 10.1136/ard.2011.149922. [DOI] [PubMed] [Google Scholar]
- 131.Cailliez M, Garaix F, Rousset-Rouviere C, et al. Anakinra is safe and effective in controlling hyperimmunoglobulinaemia D syndrome-associated febrile crisis. J Inherit Metab Dis. 2006;29:763. doi: 10.1007/s10545-006-0408-7. [DOI] [PubMed] [Google Scholar]
- 132.Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ. Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology (Oxf) 2005;44:406–8. doi: 10.1093/rheumatology/keh479. [DOI] [PubMed] [Google Scholar]
- 133.Brenner M, Ruzicka T, Plewig G, Thomas P, Herzer P. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009;161:1199–201. doi: 10.1111/j.1365-2133.2009.09404.x. [DOI] [PubMed] [Google Scholar]
- 134.Gattorno M, Piccini A, Lasiglie D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:1505–15. doi: 10.1002/art.23437. [DOI] [PubMed] [Google Scholar]
- 135.Vasques Godinho FM, Parreira Santos MJ, Canas DS. Refractory adult onset Still's disease successfully treated with anakinra. Ann Rheum Dis. 2005;64:647–48. doi: 10.1136/ard.2004.026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fitzgerald AA, Leclercq SA, Yan A, Homik JE, Dinarello CA. Rapid responses to anakinra in patients with refractory adult-onset Still's disease. Arthritis Rheum. 2005;52:1794–803. doi: 10.1002/art.21061. [DOI] [PubMed] [Google Scholar]
- 137.Kotter I, Wacker A, Koch S, et al. Anakinra in patients with treatment-resistant adult-onset Still's disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum. 2007;37:189–97. doi: 10.1016/j.semarthrit.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 138.Maier J, Birkenfeld G, Pfirstinger J, Scholmerich J, Fleck M, Bruhl H. Effective treatment of steroid refractory adult-onset Still's disease with anakinra. J Rheumatol. 2008;35:939–41. [PubMed] [Google Scholar]
- 139.Tamaki H, Shimizu H, Hiraoka E, et al. Marked effect and steroid-sparing ability of anakinra on a patient with refractory adult-onset Still's disease. Mod Rheumatol. 2010;20:200–4. doi: 10.1007/s10165-009-0254-1. [DOI] [PubMed] [Google Scholar]
- 140.Naumann L, Feist E, Natusch A, et al. IL1-receptor antagonist anakinra provides long-lasting efficacy in the treatment of refractory adult-onset Still's disease. Ann Rheum Dis. 2010;69:466–67. doi: 10.1136/ard.2009.108068. [DOI] [PubMed] [Google Scholar]
- 141.Debiais S, Maillot F, Luca L, Buret J, Fautrel B, Renard JP. Efficacy of anakinra in a case of refractory Still disease. J Clin Rheumatol. 2008;14:357–58. doi: 10.1097/RHU.0b013e318188b8d8. [DOI] [PubMed] [Google Scholar]
- 142.Rice GI, Bond J, Asipu A, et al. Mutations involved in Aicardi–Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–32. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]