

Abstract
Patients with hereditary angioedema (HAE) tend to produce autoantibodies and have a propensity to develop immunoregulatory disorders. We characterize the profile of autoantibodies in a group of HAE patients and investigate their memory B cells' phenotype and activation status. We studied the activity status phenotype, Toll-like receptor (TLR)-9 expression and total phosphotyrosine in B cells isolated from HAE patients. Additionally, the following autoantibodies were assessed in the serum of 61 HAE patients: anti-nuclear, rheumatoid factor, anti-cardiolipin, anti-tissue transglutaminase, anti-endomysial, anti-Saccharomyces cerevisiae, anti-thyroid and anti-neutrophil cytoplasmic antibodies. In 47·5% of HAE patients we detected at least one of the tested autoantibodies. Expression of CD69, CD5 and CD21 was found to be significantly higher on memory B cells from HAE patients compared to healthy controls (4·59 ± 4·41 versus 2·06 ± 1·81, P = 0·04, 8·22 ± 7·17 versus 3·65 ± 3·78, P = 0·05, 2·43 ± 0·54 versus 1·92 ± 0·41, P = 0·01, respectively). Total phosphotyrosine in B cells from HAE patients was significantly higher compared to healthy controls (4·8 ± 1·1 versus 2·7 ± 1·3, P = 0·0003). Memory B cells isolated from the HAE group contained higher amounts of TLR-9 compared to healthy controls (8·17 ± 4·1 versus 4·56 ± 1·6, P = 0·0027). Furthermore, the expression of TLR-9 in memory B cells from HAE patients with autoantibodies was significantly higher than the control group (10 ± 4·7 versus 4·56 ± 1·6, P = 0·0002) and from that in HAE patients without autoantibodies (10 ± 4·7 versus 5·8 ± 0·9, P = 0·036). HAE patients have enhanced production of autoantibodies due most probably to the increased activation of B cells, which was found to be in association with a high expression of TLR-9.
Keywords: autoantibodies, B cells, hereditary angioedema, TLR-9, total phosphotyrosine
Introduction
Hereditary angioedema (HAE) is a rare autosomal dominant inherited disease characterized by recurrent attacks of subcutaneous or submucosal oedema typically involving the arms, legs, hands, feet, bowels, genitalia, trunk, face or upper airway. In most patients, this is the result of a quantitative (type I) or qualitative (type II) deficiency of the active C1-esterase inhibitor (C1-INH) [1]. C1-INH has an important regulatory role in the complement, kallikrein-kinin, fibrinolytic and coagulation systems. Its deficiency leads to a release of excessive vasoactive peptides, among which bradykinin is considered to be most important in causing the development of angioedema [2,3].
Various immunoregulatory disorders have been described in patients suffering from HAE [4]–[10]. In an early study, 12% of the 157 HAE patients examined by Brickman et al. were found to have clinical immunoregulatory disorders, namely: glomerulonephritis (five patients), Sjögren's syndrome (three patients), inflammatory bowel disease (three patients), thyroiditis (three patients), systemic lupus erythematosus (one patient), drug-induced lupus (one patient), rheumatoid arthritis (one patient), juvenile rheumatoid arthritis with immunoglobulin (Ig)A deficiency (one patient), incipient pernicious anaemia (one patient) and sicca syndrome (one patient) [11]. The same group found a spectrum of cellular immunoregulatory abnormalities associated with C1-INH deficiency. These included a T cell subpopulation shift and an evidence for polyclonal B cell activation and high levels of circulating immune complexes [12]. Recently, Farkas et al. assessed the clinical data and immunoserological parameters of 130 Hungarian HAE patients. In agreement with the above early study, 12% were found to suffer from immunoregulatory disorders and in addition the authors revealed the presence of autoantibodies in 47·7% of their HAE patients. Interestingly, increased production of autoantibodies, especially anti-nuclear antibodies, was also found in a control group of patients with non-C1 INH-deficient angioedema [13]. The aim of this study was to characterize the autoantibody profile in a large group of HAE patients. Furthermore, we analysed the phenotype, including Toll-like receptor (TLR)-9 expression and activation status of memory B cells isolated from patients with HAE, aiming to propose a possible mechanism for this B cell autoreactivity.
Patients and methods
Patients
We studied 61 patients with C1-INH deficiency {36 women and 25 men aged 43·3 ± 14 [mean ± standard deviation (s.d.) years, range 19–70 years]}. Fifty-six had type 1 HAE and five had type 2 HAE. The diagnosis of HAE was based on the patient's family history, clinical presentation and laboratory results of levels of functional or antigenic C1 esterase inhibitor of less than half the normal levels. The patients were recruited from Israel (30 patients, 15 women, 15 men) and Italy (31 patients, 21 women, 10 men).
Thirty-seven of 61 (60%) patients were treated with danazol. Seventy healthy age- and sex-matched volunteers from the medical staff of our medical centre served as controls. Twenty controls were used for the B cell phenotype and activation profiles and 50 controls were used for the analysis of serum autoantibodies. The controls were healthy by self-report, with no clinical symptoms of autoimmune or infectious diseases. The local Committee on Human Experimentation approved the study.
Detection of autoantibodies
Blood samples were drawn from HAE patients during their visits in the out-patient clinic and the serum was stored at –20°C until assayed.
The detection of anti-nuclear antibodies (ANA) in the patients' serum was assayed by indirect immunofluorescence using slides covered with HEp-2 cells (Zeus Scientific, Inc., Branchburg, NJ, USA). Anti- extractable nuclear antigen (ENA) antibodies were analysed using a commercial enzyme-linked immunosorbent assay (ELISA) kit (Orgentec Diagnostika GmbH, Mainz, Germany). Rheumatoid factor was assayed by the 2-min latex slide test (Biokit, SA, Barcelona, Spain). Anti-cardiolipin antibodies were analysed using a commercial ELISA kit (Genesis Diagnostics, Cambridgeshire, UK).
Antibodies to tissue transglutaminase (ttG) were analysed using a commercial ELISA kit (Inova Diagnostics, Inc., San Diego, CA, USA) Anti-endomysial antibodies were analysed using a commercial ELISA kit (Inova Diagnostics, Inc.) Anti-Saccharomyces cerevisiae antibody (ASCA) levels were determined by ELISA commercial kits for IgG-ASCA (Inova Diagnostics, Inc.). Anti-thyroid antibodies (thyroglobulin and thyroid peroxidase) were analysed using a commercial ELISA kit (Orgentec Diagnostika GmbH). Anti-neutrophil cytoplasmatic antibodies were detected by indirect immunofluorescence using ethanol/(formalin)-fixed human neutrophil slides (Inova Diagnostics, Inc.).
Complement measurements
Complement 4 (C4) levels were analysed using BN Prospec System (Dade Behring Marburg GmbH, Marburg, Germany). Human C1 inactivator levels were analysed using radial immunodiffusion (Binding Site Group Ltd, Birmingham, UK).
Purification of B cells
Peripheral blood mononuclear cells (PBMCs) were isolated on Lymphoprep (Axis-Shield, Oslo, Norway). B lymphocytes were isolated by negative selection using the B cell isolation kit II for magnetic affinity cell sorting (MACS) system (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer's instructions, achieving >95% purity determined by flow cytometry.
Flow cytometric analysis
B cell activation phenotype was performed using three-colour flow cytometry. Freshly isolated B cells were incubated in the dark for 20 min with saturating concentrations of fluorochrome-labelled monoclonal antibodies. The cells were labelled with directly conjugated mouse monoclonal IgG antibody to CD19 FITC and CD27 phycoerythrin (PE)-cyanin 5 (Cy5) and directly conjugated mouse monoclonal IgG antibody to either CD21, CD40, CD86, CD69, CD5 or major histocompatibility complex class II (MHC-II) antibodies (PE, Immunotech, Beckman Coulter Co., Marseille, France).
For detection of intracellular TLR-9 expression in memory B cells, isolated B cells were stained with anti- CD19-FITC and anti-CD27-PC5 (Immunotech). In addition, these cells were fixed and permeabilized with a cell permeabilization kit (Caltag Laboratories, An Der Grub, Austria) and stained for the detection of intracellular TLR-9 using PE-conjugated anti-TLR-9 monoclonal antibodies (R&D Systems, Minneapolis, MN, USA).
Expression of these markers was carried out with a fluorescence activated cell sorter (FACS) using FC-500 software (Beckman Coulter).
All markers were expressed with mean flow cytometry intensity (MFI). Results were shown as mean ± s.d.
Total phosphotyrosine Western blot analysis
Protein phosphorylation in lymphocytes is a mechanism that controls signal transduction and protein activity and can modulate cellular proliferation, survival, differentiation, function and motility. Therefore, in order to further analyse the activation status of B cells by total phosphotyrosine, we performed Western blotting. Briefly, isolated B cells were lysed and proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose extra blotting membrane (Sartorius AG, Göttingen, Germany). Blots were probed with anti-phosphotyrosine, mouse anti-human (Cell Signaling Technology, Danvers, MA, USA) and goat anti-human for detecting actin (from Santa Cruz Laboratories, Santa Cruz, CA, USA). To reveal bound antibodies we used horseradish peroxidase (HRP)-conjugated secondary antibodies. Blots were developed with enhanced chemiluminescence (ECL) reagent (Pierce; Thermo Scientific, Rockford, IL, USA). To obtain semi-quantitative estimates for the total tyrosine phosphorylation, it was quantified and densitometry analysis was performed using Tina 2·0 software (Raytest, Straubenhardt, Germany). Values were normalized to the intensity of actin bands.
Statistical analysis
For comparisons of quantitative values we used the unpaired Student's t-test. The frequency of autoantibodies in HAE patients and control group was compared using Fisher's exact test. Two-tailed P-values of 0·05 or less were considered statistically significant. Data are expressed as mean values of MFI ± s.d.
Results
Autoantibodies
In 29 of the 61 (47·5%) patients, at least one of the tested autoantibodies was found in the serum, as detailed in Table 1. We did not find any difference in gender ratio when HAE patients with autoantibodies were compared with those without autoantibodies [male (12 of 25), female (17 of 36)]. Additionally, we did not find a difference in the average mean of the complement 4 (C4) levels between these two groups of HAE patients [0·095 ± 0·05 versus 0·088 ± 0·05, P = not significant (n.s.)]. In the healthy control group, five of 50 (10%) had serum autoantibodies. This frequency is statistically lower compared to HAE patients [five of 50 (10%) versus 29 of 61 (47·5%), P = 0·0001]. Two had positive anti-nuclear antibodies (4%), two of 50 (4%) had anti-cardiolipin antibodies and in one serum we found positive anti-S. cerevisiae antibodies. Seven of 61 HAE patients (11·4%) suffered from the following immunoregulatory disorders; one patient had systemic lupus erythematosus (SLE), two patients had coeliac disease, one patient had mixed connective tissue disease, one patient had systemic sclerosis, one patient had Crohn's disease and one patient multiple sclerosis-like syndrome.
Table 1.
Autoantibodies and immunoregulatory disorders in patients with hereditary angioedema
| Gender | HEp-21 | a.Cal2 | ENA3 | ANCA4 | RF5 | a.thy6 | a.tTG7 | ASCA8 | IR Dis. | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | + | + | + | ||||||
| 2 | F | Centriole ++ | ||||||||
| 3 | F | c-ANCA | ||||||||
| 4 | M | Fine speckled +++ | RNP | p-ANCA | MCTD | |||||
| 5 | M | + | ||||||||
| 6 | F | IgM:24 | + | + | ||||||
| 7 | F | Nucleolar + | ||||||||
| 8 | F | + | ||||||||
| 9 | M | Fine speckled + | anti-SSA; Scl-70 | Systemic sclerosis | ||||||
| 10 | M | c-ANCA | ||||||||
| 11 | M | Nucleolar + | ||||||||
| 12 | F | Coarse speckled +++ | anti-SSA; anti-SSB | SLE | ||||||
| 13 | F | Nucleolar + | ||||||||
| 14 | F | + | ||||||||
| 15 | F | + | ||||||||
| 16 | M | + | ||||||||
| 17 | F | IgM:45 | ||||||||
| 18 | F | Homogeneous ++ | IgG:19; IgM:20 | + | ||||||
| 19 | M | c-ANCA | ||||||||
| 20 | M | IgG:21; IgM:97 | Crohn | |||||||
| 21 | M | + | MS-like | |||||||
| 22 | F | Fine speckled + | ||||||||
| 23 | F | Speckled +++ | IgG:24 | anti-SSA; anti-SSB | + | |||||
| 24 | F | + | Coeliac | |||||||
| 25 | F | + | Coeliac | |||||||
| 26 | M | + | ||||||||
| 27 | F | IgG:23 | + | |||||||
| 28 | M | IgG:19 | ||||||||
| 29 | M | IgG:19 |
HEp2: anti-nuclear antibodies were detected using indirect immunofluorescence. The serum was diluted 1 : 40. The positivity strength was determined according to the fluorescence intensity (concentration levels of antibodies: + low, ++ moderate, +++ high).
a.Cal: anti-cardiolipin immunoglobulin (Ig)G and anti-cardiolipin IgM were assayed using enzyme-linked immunosorbent assay (ELISA). The positive value of cardiolipin IgG is higher than 18 IgG phospholipid units (GPLU)/ml and of cardiolipin IgM is higher than 15 U/ml according to the kit.
ENA: extractable nuclear antigens (ENA) were detected using ELISA. If the index value of a sample was higher than 1.00 then positivity was determined.
Anti-neutrophil cytoplasmic antibodies (ANCA): autoantibodies were detected using indirect immunofluorescence. p-ANCA or c-ANCA were determined based on the staining pattern.
RF: rheumotoid factor was assayed by the 2-min latex slide test and their positivity was determined according to the presence of agglutination on the slide.
a.thy: Anti-thyroid antibodies [anti-thyroid peroxidase antibodies (TPO) and anti-thyroglobulin (TG)] were detected by ELISA. If antibody concentration was higher than 150 IU/ml then positivity was determined.
a.tTG: anti-tissue transglutaminase antibody levels were detected using ELISA. If tTG antibody level was higher than 20 units, the result was considered positive.
ASCA: anti-Saccharomyces cerevisiae antibodies; IgG antibodies to S. cerevisiae were determined using ELISA. When the antibody level was higher than 25 units then sample was classified as positive. F, female; M, male; MCTD, mixed connective tissue disease; MS, multiple sclerosis; RNP, ribo2nucleoprotein; SSA, Sjogren's syndrome A; SSB, Sjogren's syndrome B; SLE, systemic lupus erythematosus.
Surface B cell markers
Expression of CD69 and CD5 was found to be statistically higher on memory B cells (CD19+CD27+) from HAE patients compared to healthy controls (4·59 ± 4·41 versus 2·06 ± 1·81, P = 0·04, 8·22 ± 7·17 versus 3·65 ± 3·78, P = 0·05, respectively). Expression of CD21 on memory B cells was also significantly higher when compared to that on memory B cells from healthy controls (2·43 ± 0·54 versus 1·92 ± 0·41, P = 0·01). In contrast, we did not find any statistical difference in the expression of MHC-II, CD40 and CD86 on the memory B cells of the two groups. Results are summarized in Table 2.
Table 2.
Phenotype of memory B cells (CD19+CD27+) in hereditary angioedema (HAE) patients versus healthy controls
| HAE patients | Healthy controls | P-value | |
|---|---|---|---|
| TLR-9 | 8·17 ± 4·1 | 4·76 ± 1·55 | 0·0027 |
| MHC-II | 5·75 ± 1·82 | 5·05 ± 1·32 | n.s. |
| CD5 | 8·22 ± 7·17 | 3·65 ± 3·78 | 0·05 |
| CD21 | 2·43 ± 0·54 | 1·92 ± 0·41 | 0·01 |
| CD40 | 8·89 ± 2·48 | 8·56 ± 2·6 | n.s. |
| CD69 | 4·59 ± 4·41 | 2·06 ± 1·81 | 0·04 |
| CD86 | 0·79 ± 0·71 | 0·81 ± 0·74 | n.s. |
MHC, major histocompatibility complex; TLR, Toll-like receptor.
TLR-9 expression in B cells
Memory B cells isolated from the HAE group expressed a significantly higher amount of TLR-9 (8·17 ± 4·1 versus 4·56 ± 1·6, P = 0·0027). Furthermore, the expression of TLR-9 in B cells from HAE patients who had autoantibodies was much higher than that of memory B cells from both the control group (10 ± 4·7 versus 4·56 ± 1·6, P = 0·0002) and from HAE patients without autoantibodies (10 ± 4·7 versus 5·8 ± 0·9, P = 0·036). Expression of TLR-9 in B cells from HAE patients without autoantibodies compared to the control group was only slightly higher, with borderline significance (5·8 ± 0·9 versus 4·56 ± 1·6, P = 0·07). All results are shown in Fig. 1.
Fig. 1.
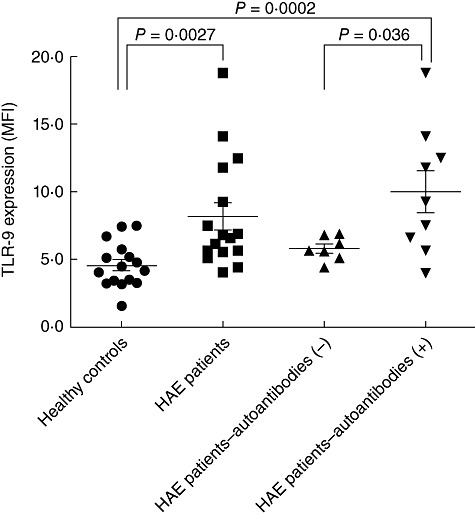
Toll-like receptor (TLR)-9 expression in memory B cells from hereditary angioedema (HAE) patients and healthy controls. TLR-9 expression in memory B cells from the HAE group expressed a significantly higher amount of TLR-9 compared to a group of age- and sex-matched healthy volunteers without any symptoms of autoimmune disease (P = 0·0027). Moreover, the expression of TLR-9 in HAE patients who had autoantibodies was much higher than in the control group HAE (P = 0·0002) or from HAE patients without autoantibodies (P = 0·036). TLR-9 expression in HAE patients without autoantibodies compared to the control group was slightly higher, with borderline significance (P = 0·07).
Total phosphotyrosine Western blot analysis
B cells isolated from HAE patients contained higher amounts of total phosphotyrosine in comparison to B cells isolated from healthy controls (4·8 ± 1·1 versus 2·7 ± 1·3, P = 0·0003; see Fig. 2).
Fig. 2.
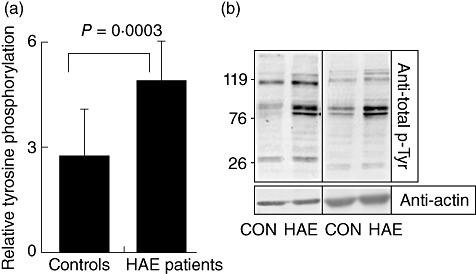
Purified B cells from hereditary angioedema (HAE) patients represent increased total tyrosine phosphorylation compared to healthy controls. Purified B cells were collected, washed and lysed for total cell lysates. After lysis, electrophoresis and transfer, membranes were probed with anti-phosphotyrosine antibody stripped and reprobed with anti-actin antibody. (a) Histogram representing relative tyrosine phosphorylation after quantification using Tina 2·0 software. The results shown are representative of 12 HAE patients (eight patients with autoantibodies and four patients without autoantibodies) versus 12 age- and sex-matched healthy volunteers without any symptoms of autoimmune disease, P < 0·05. (b) Two representative examples of Western blot analysis from HAE patients and healthy controls.
Discussion
The deficiency of functioning C1-INH in patients with hereditary angioedema has already been reported to occur in association with various immunoregulatory disorders and enhanced autoantibodies production, as detailed in the Introduction. However, the mechanisms underlining this phenomenon are still obscure. In this study, we further established the recent finding by Farkas et al., where almost half of our patients with hereditary angioedema had autoantibodies in their serum [13]. We found that memory B cells isolated from these patients expressed high levels of TLR-9 compared to B cells isolated from healthy controls. Furthermore, these cells were over-activated compared to B cells isolated from healthy controls, as demonstrated by the high level of total phosphorylated tyrosine and high expression of CD69 and CD5.
Phosphotyrosine signalling plays a central role in many cell-to-cell communication pathways, including those that regulate proliferation, differentiation, adhesion and immune defence. Recent studies have demonstrated that TLR-9 plays an important role in the induction and maintenance of autoimmunity, especially in SLE patients [14]. In addition, the role of TLR-9 in promoting autoantibody production was established further by Christensen et al., who demonstrated a specific requirement for TLR-9 in autoantibody formation in a murine model of lupus, indicating a critical role for innate immune activation in autoimmunity [15]. Memory B cells isolated from SLE patients expressed high levels of TLR-9, and the stimulation of TLR-9 in B cells with a synthetic ligand, cytosine–guanine dinucleotide (CpG) oligodeoxynucleotide, induced further B cell proliferation, cytokine secretion such as interleukin (IL)-10, IL-6 and IL-12 and the up-regulation of co-stimulatory molecules such as CD40 and CD86 [16,17]. Similarly to SLE, we indeed found that B cells from our HAE patients expressed high levels of TLR-9. The most commonly produced autoantibody that we found in these patients (10 of 61 patients, 16·4%) was ANA. In agreement with our finding, Farkas et al. found a marked elevation in the ANA titres in 27·6% of HAE patients [13]. This incidence of ANA is of significance when compared to the less than 5% reported in the general population or to 4% in our control group [18]–[20]. Furthermore, we found that the group of HAE patients who had autoantibodies in their serum expressed higher levels of TLR-9 compared to HAE patients without autoantibodies. This finding supports the relationship between TLR-9 expression and the generation of autoantibodies.
We also found that memory B cells from our patients expressed higher levels of CD5 compared to healthy controls. These cells are known to produce low-affinity polyreactive antibodies (natural antibodies), which recognize autoantigens or conserved structures on self-antigens such as polysaccharide residues [21]. They have a reduced capacity to enter the cell cycle and have a longer lifespan. Although the precise role of these cells in autoimmunity is still obscure, the numbers of peripheral CD5+ B cells were found to be increased in several autoimmune diseases, such as rheumatoid arthritis, primary Sjögren's syndrome, autoimmune thyroid disease and multiple sclerosis [22]. Therefore, it seems that these cells might play a role in the pathogenesis of autoimmune diseases [23].
The finding of low C4 levels, along with low functional C1INH in HAE, remains the most important immunological finding in this disease. C4 is important for the immune complex solubilization and removal [24]. Therefore, inherited deficiencies of C1q and C4 are associated with the chronic activation of the classical complement pathway and the development of autoimmune disease such as lupus-like disease early in life [25].
Activation of the classical complement arm through immune complexes causes the production of C3 convertase, and the cleavage of C3 by C3 convertase leads to the production of C3b being an essential product for the immune complex removal. In addition, deficiencies of C4 render mice unable to clear apoptotic cells/debris [26]. Mevorach et al. demonstrated that apoptotic materials are immunogenic and accelerate the production of autoantibody in mice not prone to autoimmunity [27]. Apoptotic material, especially when associated with microbial products in the form of immune complexes (ICs), might activate autoreactive B lymphocytes and induce serum autoantibodies [28]. One can speculate that the persistence of ICs could possibly activate B cell receptors and up-regulate the expression of TLR-9, allowing HAE patients to overproduce autoantibodies.
Another possible explanation for the over-activation of B cells in HAE could be through increased signalling of the human complement receptor type 2 (CR2) on B cells. CR2 (CD21) plays a pivotal role in the activation and proliferation of B cells and is a prerequisite for T-dependent immune responses. Engagement of CR2 with the B cell receptor lowers the threshold required for B cell activation by an antigen, enhances cell activation, reduces inhibitory signals and prevents apoptosis [29]–[32].
Only seven of our 61 (11·4%) patients had a defined immunoregulatory disorder. This incidence of immunoregulatory disorders is similar to the 12% found by Brickman et al. and 11·5% that was found by Farkas et al. [11,13]. It is not yet clear if this finding represents increased incidence compared to that in the general population. However, we assume that HAE patients are predisposed to developing immunoregulatory disorders, but only after additional precipitating factors such as human leucocyte antigen (HLA) genotype, infection or environmental factors that are necessary for the development of particular autoimmune or immunoregulatory disorders. This finding indicates the need for periodical autoantibody analysis and inspection for the appearance of symptoms suggesting autoimmune disease. However, treatment of these patients remains the same.
Relevant to this, it was demonstrated that treatment with danazol in HAE patients significantly increases C4, haemolytic complement 50% levels and the disappearance of circulating immune complexes [33]. Therefore, it could be speculated that the promotion of C4 synthesis by danazol could possibly result in the decrease of B cell activation and autoantibody generation. However, we did not find any difference between treated and non-treated patients with regard to B cell activation and autoantibody generation. Nevertheless, further studies are needed to clarify this point.
In summary, we suggest that HAE patients have enhanced production of autoantibodies compared to the general healthy population, due most probably to activation of B cells which associate with high expression of TLR-9. B cells might be activated by immune complex and thereby have the potential, in certain genetic backgrounds, to break tolerance and trigger autoimmunity.
Disclosure
None.
References
- 1.Zuraw BL. Hereditary angioedema. N Engl J Med. 2008;359:1027–36. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
- 2.Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–7. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 3.Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE., III Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057–63. doi: 10.1172/JCI14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan S, Tarzi MD, Doré PC, Sewell WA, Longhurst HJ. Secondary systemic lupus erythematosus: an analysis of 4 cases of uncontrolled hereditary angioedema. Clin Immunol. 2007;123:14–17. doi: 10.1016/j.clim.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 5.Youinou P, Dorval JC, Cledes J, Leroy JP, Miossec P, Masse R. A study of familial lupus erythematosus-like disease and hereditary angio-oedema treated with danazol. Br J Dermatol. 1983;108:717–22. doi: 10.1111/j.1365-2133.1983.tb01085.x. [DOI] [PubMed] [Google Scholar]
- 6.Farkas H, Gyeney L, Nemesánszky E, et al. Coincidence of hereditary angioedema (HAE) with Crohn's disease. Immunol Invest. 1999;28:43–53. doi: 10.3109/08820139909022722. [DOI] [PubMed] [Google Scholar]
- 7.Massa MC, Connolly SM. An association between C1 esterase inhibitor deficiency and lupus erythematosus: report of two cases and review of the literature. J Am Acad Dermatol. 1982;7:255–64. doi: 10.1016/s0190-9622(82)70115-x. [DOI] [PubMed] [Google Scholar]
- 8.Muhlemann MF, Macrae KD, Smith AM, et al. Hereditary angioedema and thyroid autoimmunity. J Clin Pathol. 1987;40:518–23. doi: 10.1136/jcp.40.5.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duncan IJ, Tymms KE, Carney G. Rheumatoid arthritis and hereditary angioedema. J Rheumatol. 1988;15:700–2. [PubMed] [Google Scholar]
- 10.Hory B, Haultier JJ. Glomerulonephritis and hereditary angioedema: report of 2 cases. Clin Nephrol. 1989;31:259–63. [PubMed] [Google Scholar]
- 11.Brickman CM, Tsokos GC, Balow JE, et al. Immunoregulatory disorders associated with hereditary angioedema: I. Clinical manifestations of autoimmune disease. J Allergy Clin Immunol. 1986;77:749–57. doi: 10.1016/0091-6749(86)90424-0. [DOI] [PubMed] [Google Scholar]
- 12.Brickman CM, Tsokos GC, Chused TM, et al. Immunoregulatory disorders associated with hereditary angioedema. II. Serologic and cellular abnormalities. J Allergy Clin Immunol. 1986;77:758–67. doi: 10.1016/0091-6749(86)90425-2. [DOI] [PubMed] [Google Scholar]
- 13.Farkas H, Csuka D, Gács J, et al. Lack of increased prevalence of immunoregulatory disorders in hereditary angioedema due to C1-inhibitor deficiency. Clin Immunol. 2011;141:58–66. doi: 10.1016/j.clim.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 14.Papadimitraki ED, Choulaki C, Koutala E, et al. Expansion of toll-like receptor 9- expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum. 2006;54:3601–11. doi: 10.1002/art.22197. [DOI] [PubMed] [Google Scholar]
- 15.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–31. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–9. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 17.Hartmann G, Krieg AM. Mechanism and function of a newly identified CpG DNA motif in human primary B cells. J Immunol. 2000;164:944–53. doi: 10.4049/jimmunol.164.2.944. [DOI] [PubMed] [Google Scholar]
- 18.Tan EM, Feltkamp TE, Smolen JS, et al. Range of antinuclear antibodies in ‘healthy’ individuals. Arthritis Rheum. 1997;40:1601–11. doi: 10.1002/art.1780400909. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh P, Dwivedi S, Naik S, et al. Antinuclear antibodies by indirect immunofluorescence: optimum screening dilution for diagnosis of systemic lupus erythematosus. Indian J Med Res. 2007;126:34–8. [PubMed] [Google Scholar]
- 20.Marin GG, Cardiel MH, Cornejo H, Viveros ME. Prevalence of antinuclear antibodies in 3 groups of healthy individuals: blood donors, hospital personnel, and relatives of patients with autoimmune diseases. J Clin Rheumatol. 2009;15:325–9. doi: 10.1097/RHU.0b013e3181bb971b. [DOI] [PubMed] [Google Scholar]
- 21.Hayakawa K, Asano M, Shinton SA, et al. Positive selection of natural autoreactive B cells. Science. 1999;285:13–16. doi: 10.1126/science.285.5424.113. [DOI] [PubMed] [Google Scholar]
- 22.Talal N, Dauphinee M, Ahmed SA. CD5 B cells in autoimmunity. Ann NY Acad Sci. 1992;651:551–6. doi: 10.1111/j.1749-6632.1992.tb24661.x. [DOI] [PubMed] [Google Scholar]
- 23.Pers JO, Jamin C, Predine-Hug F, Lydyard P, Youinou P. The role of CD5-expressing B cells in health and disease [review] Int J Mol Med. 1999;3:239–45. doi: 10.3892/ijmm.3.3.239. [DOI] [PubMed] [Google Scholar]
- 24.Walport MJ, Davies KA, Morley BJ, Botto M. Complement deficiency and autoimmunity. Ann NY Acad Sci. 1997;815:267–81. doi: 10.1111/j.1749-6632.1997.tb52069.x. [DOI] [PubMed] [Google Scholar]
- 25.Pickering MC, Botto M, Taylor PR, Lachmann PJ, Walport MJ. Systemic lupus erythematosus, complement deficiency and apoptosis. Adv Immunol. 2000;76:227–324. doi: 10.1016/s0065-2776(01)76021-x. [DOI] [PubMed] [Google Scholar]
- 26.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–92. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor PR, Carugati A, Fadok VA, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med. 2000;192:359–66. doi: 10.1084/jem.192.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology. 1998;199:265–85. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- 29.Tedder TF, Inaoki M, Sato S. The CD19–CD21 complex regulates signal transduction thresholds governing humoral immunity and autoimmunity. Immunity. 1997;6:107–18. doi: 10.1016/s1074-7613(00)80418-5. [DOI] [PubMed] [Google Scholar]
- 30.Mongini PK, Vilensky MA, Highet PF, Inman JK. The affinity threshold for human B cell activation via the antigen receptor complex is reduced upon co-ligation of the antigen receptor with CD21 (CR2) J Immunol. 1997;159:3782–91. [PubMed] [Google Scholar]
- 31.Lyubchenko T, dal Porto J, Cambier JC, Holers VM. Coligation of the B cell receptor with complement receptor type 2 (CR2/CD21) using its natural ligand C3dg: activation without engagement of an inhibitory signaling pathway. J Immunol. 2005;174:3264–72. doi: 10.4049/jimmunol.174.6.3264. [DOI] [PubMed] [Google Scholar]
- 32.Barrington RA, Zhang M, Zhong X, et al. CD21/CD19 coreceptor signaling promotes B cell survival during primary immune responses. J Immunol. 2005;175:2859–67. doi: 10.4049/jimmunol.175.5.2859. [DOI] [PubMed] [Google Scholar]
- 33.Fabiani JE, Paulin P, Simkin G, Leoni J, Palombarani S, Squiquera L. Hereditary angioedema: therapeutic effect of danazol on C4 and C1 esterase inhibitors. Ann Allergy. 1990;64:388–92. [PubMed] [Google Scholar]