

Abstract
The immune receptor expressed on myeloid cells 1 (IREM-1/CD300F) has been shown to inhibit various inflammatory processes in myeloid cells, such as macrophages and mast cells. IREM-1 exerts its inhibitory effect through its intracellular immunoreceptor tyrosine-based inhibition motifs (ITIMs). In order to generate immunomodulatory molecules that can regulate the inflammatory activation of macrophages, decapeptides representing each of the five ITIM-like sequences in the cytoplasmic tail of IREM-1 were synthesized in conjugation with human immunodeficiency virus-transactivator of transcription (HIV-TAT48–57), which was added to promote internalization of the peptides. Interestingly, all these TAT–ITIM fusion peptides inhibited Toll-like receptor (TLR)-mediated production of proinflammatory molecules, including matrix metalloproteinase (MMP)-9, tumour necrosis factor (TNF)-α, monocyte chemotactic protein-1 (MCP-1) and interleukin (IL)-8. When various TLR ligands were used to stimulate the human macrophage-like cell line human acute monocytic leukaemia cell line (THP)-1, the TAT–ITIM peptides blocked both myeloid differentiation factor 88 (MyD88) and Toll-interleukin 1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF)-mediated TLR signalling pathways. Utilization of specific inhibitors and detection of the active form of signalling adaptors by Western blot analysis further demonstrated that the inhibitory effects of these TAT–ITIM peptides require activation of Src homology 2 (SH2)-containing tyrosine phosphatase (SHP) and/or phosphoinositide 3-kinase (PI3K). These data indicate that these synthetic peptides may be used to regulate immune responses that involve TLR-mediated inflammatory activation of macrophages.
Keywords: inflammation, IREM-1, ITIM, macrophage, TLR signalling
Introduction
The immune receptor expressed on myeloid cells-1 (IREM-1/CD300F/IgSF13/CMRF-35A5 in humans and CLM-1/LMIR3/MAIR-V/CD300LF in mice) belongs to the CD300 family of receptors and is expressed on a subset of B cells and myeloid cells, including macrophages, dendritic cells, granulocytes and mast cells [1]–[4]. Previous reports on IREM-1 have demonstrated its inhibitory action both in vivo and in vitro. IREM-1 expression was detected in inflammatory myeloid cells present in demyelinated areas in an animal model of experimental autoimmune encephalomyelitis, and suppression of IREM-1 expression resulted in the aggravation of clinical symptoms as well as increased production of proinflammatory mediators [5]. Over-expression of IREM-1 inhibited osteoclastogenesis induced by the receptor activator for nuclear factor κB ligand [RANKL, a member of the tumour necrosis factor (TNF) superfamily] in RAW264·7 cells [3]. Due to the lack of a known ligand for IREM-1, in-vitro analyses of its function have been performed using an agonistic monoclonal antibody (mAb). Antibody-mediated cross-linking of IREM-1 inhibited mast cells activation induced by FcεR stimulation [1,6]. Triggering of IREM-1 also inhibited macrophage inflammatory activation that was induced by the stimulation of B cell-activating factor (BAFF, another member of the TNF superfamily) [7], FasL [8] or Toll-like receptors [9]. The inhibitory activities of IREM-1 appear to be mediated by the intracellular immunoreceptor tyrosine-based inhibition motifs (ITIMs), which can interact with Src homology 2 (SH2)-containing tyrosine phosphatase (SHP)-1 [1]–[3].
Macrophages express most members of Toll-like receptors (TLRs) which recognize various pathogen-associated molecular patterns (PAMPs) [10]. A component of the Gram-negative bacterial cell wall, lipopolysaccharide (LPS), can stimulate TLR-4, and signalling from TLR-4 is transmitted through two main pathways: one via a complex containing myeloid differentiation factor 88 (MyD88) and Toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP) and the other via a complex containing TIR domain containing adapter-inducing interferon (IFN)-β (TRIF) and TRIF-related adaptor molecule (TRAM) (reviewed in [11]). Unmethylated deoxytidyl–phosphate–deoxyguanosine (CpG) motifs found commonly in viral and bacterial DNA stimulates TLR-9, which initiates activation signals through MyD88 and then TNF receptor-associated factor 6 (TRAF6). Conversely, double-stranded RNA from viral origins activate TLR-3, which initiates activation signals through TRIF [12]–[14]. Activation of these signalling pathways activate various transcription factors including nuclear factor (NF)-κB, the major transcription factor involved in the transcriptional activation of proinflammatory mediators [15]–[17].
Previously, IREM-1 was reported to inhibit both MyD88- and TRIF-mediated TLR signalling pathways through its association with SHP-1 in cells of the monocyte/macrophage lineage [9]. In addition, a synthetic peptide (TAT–YADL) that contains an ITIM-like sequence encompassing Y205 of IREM-1 cytoplasmic tail has been shown to inhibit inflammatory activation of macrophages that were activated through various signalling pathways initiated from BAFF, FasL and TLR [7]–[9]. However, the potential inhibitory effects of other ITIM-like sequences have not been evaluated. In order to compare all five ITIM-like sequences of IREM-1, synthetic peptides each representing one of five ITIM-like sequences were synthesized and compared with respect to their inhibitory action as well as downstream mediators that may be involved in these inhibitory effects. The results from this analysis led to two unexpected observations: all five TAT–ITIM peptides inhibited both MyD88 and TRIF-mediated TLR signalling pathways and the inhibitory action of these peptides were mediated not only by SHPs, but also PI3K.
Materials and methods
Antibodies and reagents
IREM-1 specific mAbs were either generated in our laboratory (clones 2–3·6D and 2-1·7B), as described previously [7]. Polyclonal antibodies against SHP-2, phospho-SHP-2, protein kinase B (AKT), phospho-AKT (Ser473) and phospho-tyrosine (clone P-Tyr-100) were obtained from Cell Signaling (Danvers, MA, USA); SHP-1-specific mAb (clone PTY13) was purchased from Abcam (Cambridge, MA, USA); phospho-SHP-2-specific mAb (clone 82.TYR536) was from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA); and mouse IgG2a was from BD Pharmingen (San Jose, CA, USA). LY294002 was obtained from Calbiochem International Inc. (La Jolla, CA, USA). Protein tyrosine phosphatase inhibitor (PTP) inhibitor III and PTP inhibitor IV were purchased from Santa Cruz Biotechnology Inc. Bacterial LPS was purchased from Sigma (St Louis, MO, USA); palmitoyl-Cys((R,S)-2,3-di(palmitoyloxy)-propyl)-Ala-Gly-OH (PAM3-CSK4) was from Bachem AG (Budendorf, Switzerland); poly(I)-poly(C) double-strand RNA from GE Healthcare (Little Chalfont, Bucks, UK) and CpG 1826 (TLR-9 ligand) from Invivogen (San Diego, CA, USA). Fusion proteins containing ITIMs and HIV-TAT48–57 and its negative control containing only HIV-TAT48–57 were custom-designed and synthesized by Peptron Inc. (Daejeon, Korea). The human monocytic leukaemia cell line human acute monocytic leukaemia cell line (THP)-1 and the human embryonic kidney cell line 293T were obtained from the American Type Culture Collection (Rockville, MD, USA).
Immunoprecipitation and Western blot analysis
Immunoprecipitation of SHP-1 was conducted as described previously [7]. Briefly, cells (5 × 106/well in six-well plates) were incubated with 1 mM sodium pervanadate for 15 min and then 5 µM of synthetic peptides for 3 to 30 min. Cells were then lysed with NP-40 buffer (150 mM NaCl, 50 mM Tris-Cl (pH 7·5), 5 mM ethylenediamine tetraacetic acid (EDTA), 1% Nonidet P-40, 0·5% sodium deoxycholate and 1% of a protease inhibitor cocktail (Calbiochem). After removal of cellular debris by centrifugation, the supernatants were pre-cleared using protein G-Sepharose beads. SHP-1 was immunoprecipitated with 1 µg/ml of anti-SHP-1 mAb overnight at 4°C and 50 µl of protein G-Sepharose beads were added and incubated for 1 h at 4°C. The immunoprecipitates were mixed with 50 µl of sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer. Western blot analysis was performed as described previously [7,8].
Gelatin zymogram and enzyme-linked immunosorbent assay (ELISA)
The cells were activated by TLR ligands that were added to medium containing 1 × 106/ml THP-1 cells in RPMI-1640 supplemented with 0·1% fetal bovine serum (FBS). Matrix metalloproteinase (MMP) activity in the culture supernatant was determined via substrate gel electrophoresis, as described previously [18]. A sandwich ELISA (R&D Systems, Inc., MN, USA) was used to measure the concentrations of interleukin (IL)-8 in the supernatants. The detection limit was <10 pg/ml.
Statistical analysis
Statistical analysis of the data was performed using analysis of variance (anova) or Student's t-test, as appropriate. All data are presented as mean values ± standard deviation (s.d.), with the number of independent experiments indicated in the figure legends. Differences between experimental groups were considered significant for P < 0·05.
Results
All the synthetic peptides representing the ITIM-like sequences of IREM-1 exhibited inhibitory effect on TLR-mediated induction of IL-8 and MMP-9 expression
In order to test whether synthetic peptides representing each of the ITIM-like sequences exhibit a similar inhibitory effect, decapeptides encompassing ITIM-like sequences were synthesized in conjugation with HIV-TAT48–57 (Table 1). As a control, a peptide containing only the TAT sequence was also synthesized. Treatment of THP-1 cells with these peptides resulted in internalization of the peptides into the cells within 10 min [7]–[9]. When THP-1 cells were stimulated with LPS in the presence of these peptides, LPS-induced expression of IL-8 was blocked in all cases (Fig. 1a). The inhibitory effect of these peptides against other proinflammatory mediators was then tested. LPS-induced expression of MMP-9, which was tested using gelatin zymogram, was also inhibited by all of the synthetic peptides but not by TAT (Fig. 1b). The TAT–FADL peptide, which has a tyrosine-to-phenylalanine substitution in TAT–YADL, was synthesized as another control. TAT–FADL failed to inhibit LPS-induced expression of MMP-9. The expression levels of MMP-2, whose expression is independent of the activation status in macrophages, were neither induced by LPS nor affected by synthetic peptides. The expression of other LPS-induced proinflammatory cytokines such as TNF-α and monocyte chemotactic protein-1 (MCP-1) was also affected by all the synthetic peptides (Fig. 1b).
Table 1.
Synthetic peptides used in the experiments
| Name | Sequence | Source |
|---|---|---|
| TAT–YADL | GRKKRRQRRR-GDLCYADLTL | IREM-1 (AA 201 −210) |
| TAT–FADL | GRKKRRQRRR-GDLCFADLTL | IREM-1 (AA 201 −210, Y205F) |
| TAT–YVTM | GRKKRRQRRR-VEVEYVTMAS | IREM-1 (AA 232 −241) |
| TAT–YASL | GRKKRRQRRR-EDISYASLTL | IREM-1 (AA 245 −254) |
| TAT–YCNM | GRKKRRQRRR-QEPTYCNMGH | IREM-1 (AA 259 −268) |
| TAT–YSTI | GRKKRRQRRR-EPTEYSTISR | IREM-1 (AA 280 −289) |
| TAT–YMNM | GRKKRRQRRR-LHSDYMNMTP | CD28 (AA 187–196) |
| TAT | GRKKRRQRRR | – |
IREM: immune receptor expressed on myeloid cells; TAT: transactivator of transcription.
Fig. 1.
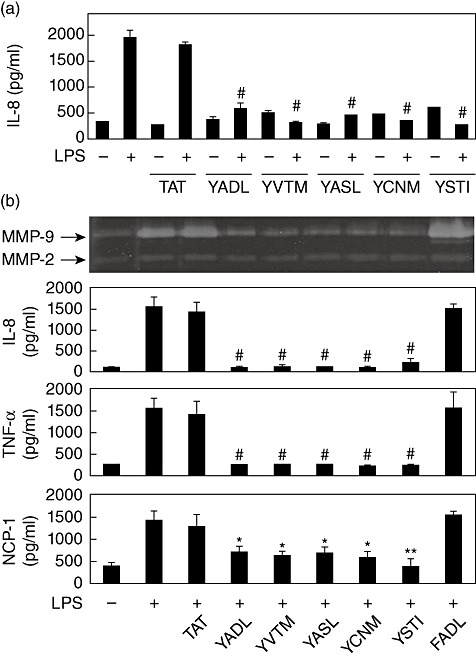
All five transactivator of transcription–immunoreceptor tyrosine-based inhibition motif (TAT–ITIM) peptides contain inhibitory potential against LPS stimulation in THP-1 cells. (a) Cells were pre-treated with 5 µM of TAT, TAT–YADL, TAT–YVTM, TAT–YASL, TAT–YCNM or TAT–YSTI for 30 min before stimulation with 1 µg/ml of lipopolysaccharide (LPS) for 24 h. Culture supernatants were then collected and interleukin (IL)-8 concentrations were measured using double-sandwich enzyme-linked immunosorbent assay (ELISA). (b) Cells were pretreated with 5 µM of synthetic peptides for 30 min before stimulation with 1 µg/ml of LPS for 24 h. Culture supernatants were then collected and matrix metalloproteinase (MMP) activity was analysed using gelatin zymogram and cytokine concentrations were measured using double-sandwich ELISA (n = 3, *P < 0·05, ** <0·01 and # <0·001 when compared with LPS-treated positive control).
The inhibitory action of the ITIM-peptides was then evaluated in cells treated with different TLR ligands. When poly(I:C) was used for the ligation of TLR-3, the expression of IL-8 was blocked by all the ITIM-representing peptides, but not by TAT or TAT–FADL (Fig. 2). This demonstrates the importance of the tyrosine residue on the inhibitory action of the peptides. When the cells were stimulated with CpG oligodeoxynucleotide (ODN), a TLR-9 ligand, in the presence of TAT–ITIM peptides, induction of IL-8 expression was also blocked while TAT and TAT–FADL were not inhibitory (Fig. 2). In all cases, the inhibitory action of TAT–YADL tended to be the strongest of all peptides.
Fig. 2.
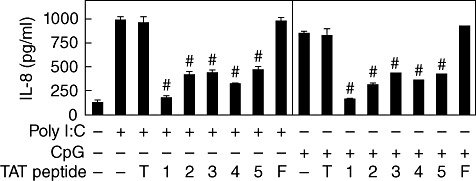
All five transactivator of transcription–immunoreceptor tyrosine-based inhibition motif (TAT–ITIM) peptides contain inhibitory potential against stimulation with deoxytidyl–phosphate–deoxyguanosine (CpG) and poly(I:C) in THP-1 cells. Cells were pre-treated with 5 µM of TAT (T), TAT–YADL (1), TAT–YVTM (2), TAT–YASL (3), TAT–YCNM (4), TAT–YSTI (5) or TAT–FADL (F) for 30 min before stimulation with 1 µg/ml of CpG oligodeoxynucleotide (ODN) or poly(I:C) for 24 h. Culture supernatants were then collected and interleukin (IL)-8 concentrations were measured using enzyme-linked immunosorbent assay (ELISA) (n = 3, #P < 0·001 when compared with corresponding positive control samples).
In order to determine whether the inhibitory action of these peptides required the presence of specific amino acid sequences other than tyrosine, another tyrosine-containing peptide (TAT–YMNM) was synthesized. YMNM represents a sequence motif associated with the co-stimulatory action in the cytoplasmic tail of CD28 [19]. When TAT–YCNM was compared with TAT–YMNM, only TAT–YCNM exhibited an inhibitory effect, while TAT–YMNM did not. Furthermore, the addition of TAT–YMNM alone to the culture medium resulted in the stimulation of MMP-9 and IL-8 expression (Fig. 3). These data indicate that the inhibitory action of the ITIM-representing peptides is a specific process requiring not only the presence of tyrosine but also the presence of certain sequences that accompany the tyrosine residue.
Fig. 3.
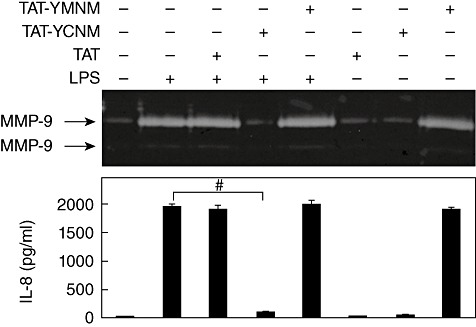
Transactivator of transcription (TAT)–YCNM, but not TAT–YMNM, has inhibitory potential against lipopolysaccharide (LPS) stimulation in human acute monocytic leukaemia cell line (THP)-1cells. Cells were pre-treated with 5 µM of TAT, TAT–YCNM or TAT–YMNM for 30 min before stimulation with 1 µg/ml of LPS for 24 h. Culture supernatants were then collected and matrix metalloproteinase (MMP)-9 and MMP-2 activity was analysed using gelatin zymogram and interleukin (IL-8) concentrations were measured using enzyme-linked immunosorbent assay (ELISA) (n = 3, #P < 0·001).
Synthetic peptides exert their inhibitory effects through activation of SHP-1, SHP-2 and/or PI3K
Inhibitors of signalling adaptors were used to identify the signalling adaptors that are responsible for the inhibitory actions of the synthetic peptides. Because SHP-1 and PI3K have been reported to interact with IREM-1 [20], PTP inhibitor III (a SHP-1-specific inhibitor) and LY294002 (a well known PI3K-specific inhibitor) were used. PTP inhibitor IV, which is a SHP-2-specific inhibitor, was also included, as SHP-2 is known to have inhibitory activity similar to SHP-1 [21,22]. PTP inhibitors III and IV have been shown to interact with the catalytic domain of SHP-1 and SHP-2 and suppress their phosphatase activities, respectively [23]. When THP-1 cells were stimulated with TLR ligands in the presence of anti-IREM-1 mAb, the expression of IL-8 was inhibited. As reported previously [9], the addition of PTP inhibitor III blocked the inhibitory action of anti-IREM-1 mAb in cells stimulated with LPS, CpG ODN or poly(I:C). The addition of PTP inhibitor IV or LY294002 also blocked the IREM-1 inhibitory activity (Fig. 4). The addition of inhibitor alone did not affect the induction of IL-8 by TLR ligands. These data indicate that multiple signalling adapters are involved in the inhibitory action of IREM-1.
Fig. 4.
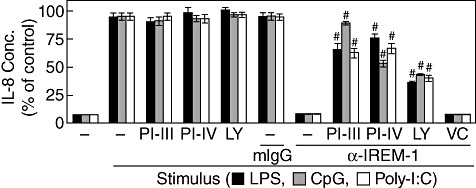
Inhibitory action of immune receptor expressed on myeloid cells 1 (IREM-1) requires Src homology 2 (SH2)-containing tyrosine phosphatase (SHP) and/or phosphoinositide 3-kinase (PI3K) activity. (a) Human acute monocytic leukaemia cell line (THP)-1cells were pre-treated with 1 mM of protein tyrosine phosphatase (PTP) inhibitor III (PI-III) and PTP inhibitor IV (PI-IV) for 30 min or 20 µM of LY294002 (LY) for 60 min. Cells were then treated with 1 µg/ml of anti-IREM-1 monoclonal antibody (mAb) or isotype-matching mouse immunoglobulin (Ig)G for 30 min, after which the cells were stimulated with 1 µg/ml of lipopolysaccharide (LPS), deoxytidyl–phosphate–deoxyguanosine (CpG) oligodeoxynucleotide (ODN) or poly(I:C). Dimethylsulphoxide (DMSO) (0·1%) was used as a vehicle control (VC). Culture supernatants were collected after 24 h for the measurement of interleukin (IL)-8 concentrations which were represented in percentages of the corresponding positive control samples. For LPS-, CpG- and poly(I:C)-treated positive controls, the measured concentrations were 1850 ± 35, 1024 ± 25 and 950 ± 18 pg/ml, respectively (mean ± standard deviation) (n = 3, #P < 0·001 when compared with corresponding positive control samples).
The involvement of SHP or PI3K in the inhibitory action of the synthetic peptides was then tested. As shown in Fig. 5, PTP inhibitor III blocked the inhibitory action of TAT–YADL, TAT–YVTM and TAT–YASL, but not that of TAT–YCNM and TAT–YSTI. PTP inhibitor IV blocked inhibitory action of all the peptides with statistical significance. The blocking effect was more prominent against TAT–YADL, TAT–YASL and TAT–YSTI than other peptides. LY294002 exhibited strong blocking activity against TAT–TVTM, TAT–YCNM and TAT–YSTI. These data indicate that each synthetic peptide uses different combination of signalling adapters to exert its inhibitory activity.
Fig. 5.
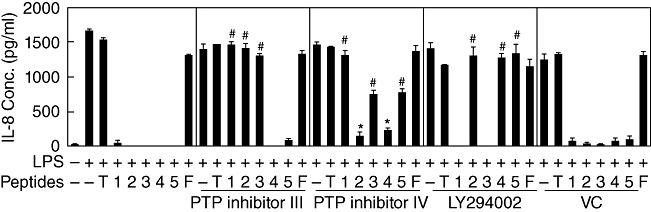
Inhibitory activities of synthetic peptides require Src homology 2 (SH2)-containing tyrosine phosphatase (SHP) and/or phosphoinositide 3-kinase (PI3K) activity. (a) Human acute monocytic leukaemia cell line (THP)-1cells were pre-treated with 1 mM of protein tyrosine phosphatase (PTP) inhibitor III or PTP inhibitor IV for 30 min or 20 µM of LY294002 for 60 min. Five µM of transactivator of transcription (TAT) (T), TAT–YADL (1), TAT–YVTM (2), TAT–YASL (3), TAT–YCNM (4), TAT–YSTI (5) or TAT–FADL (F) were then added for 30 min, after which cells were stimulated with 1 µg/ml of LPS. Dimethylsulphoxide (DMSO) (0·1%) was used as a vehicle control (VC). Culture supernatants were collected after 24 h and IL-8 concentrations were measured (n = 3, *P < 0·05 and # <0·001 when compared with lipopolysaccharide (LPS)-treated samples in the presence of corresponding synthetic peptides).
The catalytic activity of SHP-1 is regulated by the phosphorylation of its tyrosine residues (Y536 and Y564) by kinases such as Lyn, and phosphorylation of SHP-1 causes an increase in its activity [24,25]. Similarly, SHP-2 is phosphorylated at Y542 residues by src kinases [21,22]. Conversely, the activation of PI3K leads to the phosphorylation of its main substrate AKT at its S473 residue [26]. Blockage of the inhibitory action of the peptides by PTP inhibitor III, PTP inhibitor IV and LY294002 indicates that treatment with these peptides induces activation of SHPs and PI3K. Phosphorylation levels of SHP-1 and AKT were then tested. Phosphorylation of SHP-1 was tested by precipitating the cell lysates with anti-SHP-1 mAb and then analysing the phosphotyrosine levels of the SHP-1 in the precipitates using Western blot analysis. Phosphorylation levels of AKT were analysed using Western blot analysis of the cell lysates. As shown in Fig. 6, treatment of THP-1 cells with TAT–YASL induced phosphorylation of SHP-1 and did not affect the phosphorylation levels of AKT. In contrast, TAT–YCNM induced phosphorylation of AKT without affecting the phosphorylation levels of SHP-1.
Fig. 6.
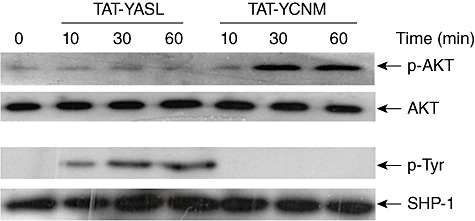
Src homology 2 (SH2)-containing tyrosine phosphatase (SHP) and/or phosphoinositide 3-kinase (PI3K) are activated differentially by different transactivator of transcription–immunoreceptor tyrosine-based inhibition motif (TAT–ITIM) peptides. Human acute monocytic leukaemia cell line (THP)-1 cells were treated with 5 µM of TAT–YASL or TAT–YCNM for the indicated times. Cell lysates were then collected and the levels of protein kinase B (AKT) and phospho-AKT were measured using Western blot analysis. To measure SHP-1 activation, cell lysates were immunoprecipitated with anti-SHP-1 monoclonal antibody (mAb) and the resulting precipitates were analysed with antibodies against phosphotyrosine or SHP-1. The data are representatives of three independent experiments.
Signalling adapter usage may change depending upon the stimuli
Previous experiments (Fig. 5) indicated that the inhibitory effects of the synthetic peptide were mediated through a combination of SHP-1, SHP-2 and PI3K. In order to dissect the inhibitory action of TAT–ITIM peptides with regard to MyD88 and TRIF-mediated pathways, the blocking effects of PTP inhibitor III, PTP inhibitor IV or LY294002 against TAT–ITIMs were tested in cells stimulated with LPS, CpG ODN or poly(I:C). As shown in Fig. 7, PTP inhibitor III blocked the inhibitory activity of TAT–YADL, TAT–YVTM and TAT–YASL after LPS stimulation, which is in agreement with the results obtained in Fig. 5. Interestingly, PTP inhibitor III blocked the inhibitory activity of a different set of peptides (TAT–YADL, TAT–YASL and TAT–YSTI) after stimulation with CpG ODN or poly(I:C). This indicates that the usage of SHP-1 by TAT–IRIM peptides changes according to the stimuli.
Fig. 7.
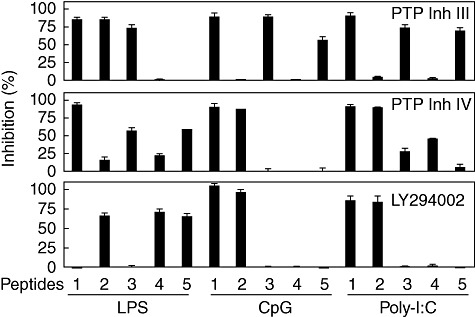
The usage of signalling adapter may change depending upon the stimuli. Human acute monocytic leukaemia cell line (THP)-1 cells were pre-treated with 1 mM of the protein tyrosine phosphatase (PTP) inhibitor III or PTP inhibitor IV for 30 min or 20 µM of LY294002 for 60 min. Five µM of transactivator of transcription (TAT)–YADL (1), TAT–YVTM (2), TAT–YASL (3), TAT–YCNM (4) and TAT–YSTI (5) were then added for 30 min, after which cells were stimulated with 1 µg/ml of LPS, deoxytidyl–phosphate–deoxyguanosine (CpG) oligodeoxynucleotide (ODN) or poly(I:C). Culture supernatants were collected after 24 h for the measurement of interleukin (IL)-8 concentrations. The inhibitory activities of the inhibitors were calculated as a percentage of complete reversion. The data are representative of three independent experiments.
When the PTP inhibitor IV was used to test its blocking effect against TAT–ITIMs in the context of LPS, CpG ODN or poly(I:C)-mediated cellular activation, similar results were obtained. TAT–YADL-, TAT–YASL- or TAT–YSTI-mediated suppression of LPS-induced expression of IL-8 was severely blocked by PTP inhibitor IV. TAT–YVTM and TAT–YCNM-mediated blockage was only slightly affected, which is in agreement with results described in Fig. 5. Conversely, the PTP-inhibitor IV blocked the inhibitory effects of TAT–YADL and TAT–YVTM against CpG ODN and poly(I:C)-induced IL-8 expression (Fig. 7). Low levels of suppressive effect were also observed against TAT–YASL and TAT–YCNM in cells stimulated with poly(I:C). This indicates that the involvement of SHP-2 in peptide-mediated inhibition also changes according to the TLR ligands.
The utilization of PI3K was then tested using LY294002. When the cells were stimulated with LPS, LY294002 blocked the inhibitory effects of TAT–YVTM, TAT–YCNM and TAT–YSTI. When cells were stimulated with CpG-ODN or poly(I:C), only TAT–YADL and TAT–YVTM-mediated inhibition was blocked by LY294002 (Fig. 7). This also indicates that usage of PI3K by synthetic peptide changes depending upon the stimuli. All inhibitors failed to affect LPS-, CpG ODN- or poly(I:C)-induced expression of IL-8 when treated in the absence of synthetic peptides (data not shown). In addition, LPS-, CpG ODN- or poly(I:C)-induced expression of IL-8 was not suppressed by 0·1% dimethylsulphoxide (DMSO), which was used as a vehicle control (data not shown).
Another interesting observation was that the inhibitory effect of TAT–YCNM against CpG ODN-induced IL-8 expression was not blocked by any of the three inhibitors used in the experiments. This raises the possibility that TAT–YCNM may use another yet unidentified signalling adapter for the suppression of CpG ODN-induced IL-8 expression.
The experimental results shown in Fig. 7 indicates that TAT–YADL uses SHP-1 to suppress LPS-, CpG ODN- and poly poly(I:C)-induced expression of IL-8, while TAT–YVTM uses SHP-1 to suppress LPS- but not CpG ODN- or poly(I:C)-induced expression of IL-8. To verify this hypothesis, Western blot analysis was performed for the detection of phospho-SHP-1 in cells treated with TAT–YADL (or TAT–YVTM) and stimulated with LPS, CpG ODN or poly(I:C). As shown in Fig. 8, phosphorylation of SHP-1 was detected in cells treated with TAT–YADL and stimulated with any of the three stimuli. In contrast, phosphorylation of SHP-1 was detected in cells treated with TAT–YVTM and stimulated with LPS but not in cells treated with TAT–YVTM and stimulated with CpG ODN or poly(I:C). The results of the Western blot supports those reported in Fig. 7.
Fig. 8.
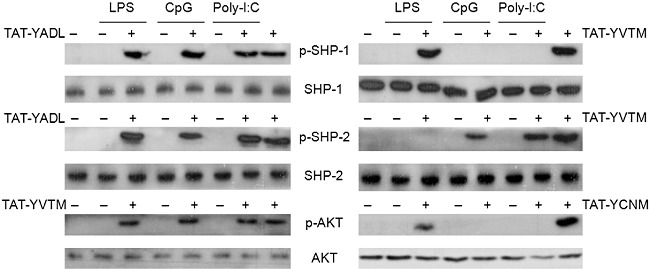
The usage of signalling adapter may change depending upon the stimuli. Human acute monocytic leukaemia cell line (THP)-1 cells were treated with 5 µM of transactivator of transcription–immunoreceptor tyrosine-based inhibition motif (TAT–ITIM) peptides for 30 min and then stimulated with 1 µg/ml of lipopolysaccharide (LPS), deoxytidyl–phosphate–deoxyguanosine (CpG) oligodeoxynucleotide (ODN) or poly(I:C). Cell lysates were then collected after 30 min and the levels of Src homology 2 (SH2)-containing tyrosine phosphatase (SHP), phospho-SHP-1, SHP-2, phospho-SHP-2, protein kinase B (AKT) and phospho-AKT were measured using Western blot analysis. These data are representative of three independent experiments.
SHP-2 is used by TAT–YADL in cells stimulated with any of the three stimuli. In contrast, SHP-2 is used by TAT–YVTM in cells stimulated with CpG ODN or poly(I:C), but not with LPS (Fig. 7). Western blot analysis was also performed to evaluate the phosphorylation of SHP-2 in cells treated with TAT–YADL (or TAT–YVTM) and stimulated with LPS, CpG ODN or poly(I:C). As expected, phosphorylation pattern of SHP-2 (Fig. 7) correlated with the inhibition by the PTP inhibitor IV (Fig. 8).
PI3K appeared to be used by TAT–YVTM in cells stimulated with any of the three stimuli. In contrast, PI3K is used for TAT–YCNM in cells stimulated with LPS but not with CpG ODN or poly(I:C). Western blot analysis was also performed to evealuate the phosphorylation of AKT in cells treated with TAT–YYTM (or TAT–YCNM) and stimulated with LPS, CpG ODN or poly(I:C). The phosphorylation pattern of AKT was also correlated with inhibition by LY294002 (Figs 7 and 8). These results further support the notion that utilization of signalling adapters by the synthetic peptides can somehow change depending upon the TLR ligand that has been used for the stimulation of cells.
Discussion
Current data indicate that synthetic peptides containing the ITIM-like sequences of IREM-1 can block the MyD88 or TRIF-mediated TLR signalling through activation of SHP-1, SHP-2 and/or PI3K. Similar to SHP-1/2, the p85 subunit of PI3K also contains an SH2 domain [27,28]. Thus, it is highly likely that these molecules interact directly with the synthetic peptides.
Five putative ITIMs encompassing Y205, Y236, Y249, Y284 and Y263 have been identified, and substitution (tyrosine into phenylalanine) analysis revealed that two of them (Y205 and Y249 corresponding to YADL and YASL, respectively) were required for the IREM-1-mediated inhibitory effects, while the other two (Y236 and Y263 corresponding to YVTM and YCNM, respectively) were responsible for the interaction with PI3K and subsequent induction of degranulation in rat basophilic leukaemia cells [20]. In this regard, it was expected that the inhibitory action of the some of the peptides will be mediated by SHP-1. SHP-1 is a protein tyrosine phosphatase (PTP) that is expressed mainly in lymphocytes and myeloid cells. SHP-1 is composed of two SH2 N-terminal domains and a C-terminal protein-tyrosine phosphatase domain. SHP-1 has been shown to modulate various cellular signals that involve PI3K, Janus kinase 2, signal transducers and activators of transcription (STATs), mitogen-activated protein kinases (MAPKs), extracellular regulated kinase (ERK) and NF-κB [21,22]. Moreover, LPS-induced expression of proinflammatory mediators was inhibited by the over-expression of SHP-1 in murine macrophages [29] and depletion of SHP-1 expression resulted in enhanced macrophage activities in clinical cases and experimental models [30]–[32]. Furthermore, studies using SHP-1-deficient mice demonstrated that SHP-1 inhibits the production of TLR-mediated proinflammatory cytokines through binding and subsequent inactivation of IL-1 receptor-associated kinase 1 (IRAK1), which is a key signalling molecule in TLR signalling [33].
The relationship between SHP-2 and IREM-1 has not yet been reported. SHP-2 is believed to interact with IREM-1 or ITIM-containing synthetic peptides through its SH2 domain and exert inhibitory activity in a manner similar to SHP-1.
PI3K is another signalling adapter molecule that mediates the inhibitory activities of IREM-1 and ITIM-containing peptides. Although PI3K is involved in cellular activation in most cases, PI3K has a dual role and it may also function as an inhibitory molecule in certain cases. Signalling involving PI3K/AKT has been shown to inhibit certain signalling events that mediate inflammation. Activation of the PI3K/AKT pathway limited TLR-4 or TLR-2-mediated inflammatory activation of human monocytes and neutrophils through modulation of MAPK activity [34,35]. PI3K activity has been shown to be associated with inhibition of LPS- or cytokine-induced expression of inducible nitric oxide synthase (iNOS) through suppression of NF-κB activity in macrophages, glial cells and endothelial cells [36]–[38]. Co-immunoprecipitation analysis demonstrated the physical interaction between TRIF and PI3K and inhibition of PI3K activity enhanced TRIF-dependent NF-κB activity in dendritic cells [39]. These previous observations are in agreement with the findings of this study, where ITIMs encompassing Y236, Y263, and Y284 of IREM-1 were shown to inhibit TLR-mediated signalling through activation of PI3K activity.
It is interesting that the usage of SHP-1, SHP-2 and PI3K by the synthetic peptides differed depending upon the TLR ligands used for cell stimulation. Overall, the pattern of adaptor usage by synthetic peptide for the inhibition of LPS-induced IL-8 expression differed from those for the inhibition of CpG ODN (or poly-I:C)-induced IL-8 expression (Fig. 7). TLR-4, which is the receptor for LPS, is expressed mainly on the cell surface while TLR-3 (the receptor for poly-I:C) and TLR-9 (the receptor for CpG ODN) interacts with their ligand in internal vesicles such as endosomes. It appears likely that the interaction of TLR ligands with cell surface TLR may alter the arrangement or distribution of signalling adapters or signalling regulators which is different from that induced by interaction of TLR ligands with internal receptors. In that case, the signalling adapters interacting with TAT–ITIM could be affected, resulting in different adapter usage patterns between cells stimulated with LPS and CpG ODN (or poly-I:C). In agreement with this interpretation, Wu et al. have demonstrated recently that CLM-3 (a novel murine molecule belonging to CD300/IREM/CLM superfamily) affects TLR-9-induced/MyD88-mediated expression of TNF-α without affecting TLR-4-induced expression of TNF-α[40]. Further analysis is required to identify the underlining mechanism responsible for the differential usage of signalling adapters by synthetic peptides under the influence of different TLRs.
All the ITIM-containing synthetic peptides have 10 AAs derived from the intracellular domain of IREM-1. ITIM sequences are characterized by the presence of S/I/V/LxYxxI/V/L [41]. The presence of tyrosine is essential for the ITIM activity, as this residue is phosphorylated and interacts with downstream mediators containing the SH2 domain [6]. It was also unexpected that all the sequences containing the ITIM-like sequence exerted inhibitory activity. It is likely that the inhibitory activity of individual ITIM-like sequence was masked by YADL, which appears to have strongest inhibitory activity both in vivo and in vitro, due to either steric hindrance or competition between ITIMs. When the ITIMs were used separateli and in isolation from each other, their hidden functions were manifested.
In order to assess the importance of the AA sequence around the essential tyrosine for the ITIM activity, another tyrosine-containing peptide (TAT–YMNM) derived from CD28 was used. YMNM represents a sequence motif associated with the co-stimulatory action in the cytoplasmic tail of CD28 [19]. Unlike TAT–YCNM, TAT–YMNM failed to show any inhibitory activity. This indicates that the flanking sequence, in addition to the essential tyrosine, is important for ITIM activity. It is likely that TAT–YMNM interacts with other SH2-containing proteins with activating potential. Preliminary data indicate that TAT–YMNM-induced activation was blocked by MAPK inhibitors (our unpublished observation), indicating that MAPK are the signalling mediators activated (probably through an indirect manner) by TAT–YMNM.
In conclusion, synthetic peptides representing the ITIM domains of IREM-1 blocked both MyD88- and TRIF-mediated TLR-signalling pathways through activation of SHP-1, SHP-2 and/or PI3K. Thus, synthetic peptides that mimic the inhibitory action of IREM-1 could be used to regulate inflammatory activation of macrophages, which are closely associated with the pathogenesis of chronic inflammatory diseases such as atherosclerosis, rheumatoid arthritis and cancer.
Acknowledgments
This work was supported by a grant from the Korean Ministry of Education, Science and Technology (the Regional Core Research Program ‘Anti-aging and Well-being Research Center’ School of Medicine, Kyungpook National University).
Disclosure
The authors have nothing to declare.
References
- 1.Alvarez-Errico D, Aguilar H, Kitzig F, Brckalo T, Sayos J, Lopez-Botet M. IREM-1 is a novel inhibitory receptor expressed by myeloid cells. Eur J Immunol. 2004;34:3690–701. doi: 10.1002/eji.200425433. [DOI] [PubMed] [Google Scholar]
- 2.Sui L, Li N, Liu Q, et al. IgSF13, a novel human inhibitory receptor of the immunoglobulin superfamily, is preferentially expressed in dendritic cells and monocytes. Biochem Biophys Res Commun. 2004;319:920–8. doi: 10.1016/j.bbrc.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 3.Chung DH, Humphrey MB, Nakamura MC, Ginzinger DG, Seaman WE, Daws MR. CMRF-35-like molecule-1, a novel mouse myeloid receptor, can inhibit osteoclast formation. J Immunol. 2003;171:6541–8. doi: 10.4049/jimmunol.171.12.6541. [DOI] [PubMed] [Google Scholar]
- 4.Shibuya A, Nakahashi-Oda C, Tahara-Hanaoka S. Regulation of immune responses by the activating and inhibitory myeloid-associate immunoglobuline-like receptors (MAIR) (CD300) Immune Netw. 2009;9:41–5. doi: 10.4110/in.2009.9.2.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xi H, Katschke KJ, Jr, Helmy KY, et al. Negative regulation of autoimmune demyelination by the inhibitory receptor CLM-1. J Exp Med. 2010;207:7–16. doi: 10.1084/jem.20091508. S1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izawa K, Kitaura J, Yamanishi Y, et al. An activating and inhibitory signal from an inhibitory receptor LMIR3/CLM-1: LMIR3 augments lipopolysaccharide response through association with FcRgamma in mast cells. J Immunol. 2009;183:925–36. doi: 10.4049/jimmunol.0900552. [DOI] [PubMed] [Google Scholar]
- 7.Lee SM, Nam YP, Suk K, Lee WH. IREM-1 inhibits BAFF-mediated inflammatory regulation of THP-1 cells through modulation of the activities of ERK. Clin Exp Immunol. 2010;161:504–11. doi: 10.1111/j.1365-2249.2010.04211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee SM, Kim EJ, Suk K, Lee WH. Stimulation of FasL induces production of pro-inflammatory mediators through activation of mitogen activated protein kinases and nuclear factor-kappaB in THP-1 cells. Inflammation. 2012 doi: 10.1007/s10753-010-9283-3. (in press) [DOI] [PubMed] [Google Scholar]
- 9.Lee SM, Kim EJ, Suk K, Lee WH. CD300F blocks both MyD88 and TRIF-mediated TLR signaling through activation of SHP-1. J Immunol. 2011;186:6296–303. doi: 10.4049/jimmunol.1002184. [DOI] [PubMed] [Google Scholar]
- 10.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 11.Nasu K, Narahara H. Pattern recognition via the Toll-like receptor system in the human female genital tract. Mediators Inflamm. 2010 doi: 10.1155/2010/976024. doi: 10.1155/2010/976024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 13.Verstak B, Nagpal K, Bottomley SP, Golenbock DT, Hertzog PJ, Mansell A. MyD88 adapter-like (Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem. 2009;284:24192–203. doi: 10.1074/jbc.M109.023044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang FX, Kirschning CJ, Mancinelli R, et al. Bacterial lipopolysaccharide activates nuclear factor-kappaB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J Biol Chem. 1999;274:7611–14. doi: 10.1074/jbc.274.12.7611. [DOI] [PubMed] [Google Scholar]
- 15.Wietek C, Miggin SM, Jefferies CA, O'Neill LA. Interferon regulatory factor-3-mediated activation of the interferon-sensitive response element by Toll-like receptor (TLR) 4 but not TLR3 requires the p65 subunit of NF-kappa. J Biol Chem. 2003;278:50923–31. doi: 10.1074/jbc.M308135200. [DOI] [PubMed] [Google Scholar]
- 16.Chang M, Jin W, Sun SC. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol. 2009;10:1089–95. doi: 10.1038/ni.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meylan E, Burns K, Hofmann K, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5:503–7. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 18.Lee WH, Kim SH, Lee Y, et al. Tumor necrosis factor receptor superfamily 14 is involved in atherogenesis by inducing proinflammatory cytokines and matrix metalloproteinases. Arterioscler Thromb Vasc Biol. 2001;21:2004–10. doi: 10.1161/hq1201.098945. [DOI] [PubMed] [Google Scholar]
- 19.Harada Y, Tokushima M, Matsumoto Y, et al. Critical requirement for the membrane-proximal cytosolic tyrosine residue for CD28-mediated costimulation in vivo. J Immunol. 2001;166:3797–803. doi: 10.4049/jimmunol.166.6.3797. [DOI] [PubMed] [Google Scholar]
- 20.Alvarez-Errico D, Sayos J, Lopez-Botet M. The IREM-1 (CD300f) inhibitory receptor associates with the p85alpha subunit of phosphoinositide 3-kinase. J Immunol. 2007;178:808–16. doi: 10.4049/jimmunol.178.2.808. [DOI] [PubMed] [Google Scholar]
- 21.Chong ZZ, Maiese K. The Src homology 2 domain tyrosine phosphatases SHP-1 and SHP-2: diversified control of cell growth, inflammation, and injury. Histol Histopathol. 2007;22:1251–67. doi: 10.14670/hh-22.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz U. SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels. Immunol Rev. 2009;228:342–59. doi: 10.1111/j.1600-065X.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arabaci G, Guo XC, Beebe KD, Coggeshall KM, Pei D. α-Haloacetophenone derivatives as photoreversible covalent inhibitors of protein tyrosine phosphatases. J Am Chem Soc. 1999;121:5085–6. [Google Scholar]
- 24.Lorenz U, Ravichandran KS, Pei D, Walsh CT, Burakoff SJ, Neel BG. Lck-dependent tyrosyl phosphorylation of the phosphotyrosine phosphatase SH-PTP1 in murine T cells. Mol Cell Biol. 1994;14:1824–34. doi: 10.1128/mcb.14.3.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchida T, Matozaki T, Noguchi T, et al. Insulin stimulates the phosphorylation of Tyr538 and the catalytic activity of PTP1C, a protein tyrosine phosphatase with Src homology-2 domains. J Biol Chem. 1994;269:12220–8. [PubMed] [Google Scholar]
- 26.Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter CL, Auger KR, Chanudhuri M, et al. Phosphoinositide 3-kinase is activated by phosphopeptides that bind to the SH2 domains of the 85-kDa subunit. J Biol Chem. 1993;268:9478–83. [PubMed] [Google Scholar]
- 28.Street A, Macdonald A, Crowder K, Harris M. The hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J Biol Chem. 2004;279:12232–41. doi: 10.1074/jbc.M312245200. [DOI] [PubMed] [Google Scholar]
- 29.Hardin AO, Meals EA, Yi T, Knapp KM, English BK. SHP-1 inhibits LPS-mediated TNF and iNOS production in murine macrophages. Biochem Biophys Res Commun. 2006;342:547–55. doi: 10.1016/j.bbrc.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Christophi GP, Hudson CA, Panos M, Gruber RC, Massa PT. Modulation of macrophage infiltration and inflammatory activity by the phosphatase SHP-1 in virus-induced demyelinating disease. J Virol. 2009;83:522–39. doi: 10.1128/JVI.01210-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christophi GP, Massa PT. Central neuroinvasion and demyelination by inflammatory macrophages after peripheral virus infection is controlled by SHP-1. Viral Immunol. 2009;22:371–87. doi: 10.1089/vim.2009.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christophi GP, Panos M, Hudson CA, et al. Macrophages of multiple sclerosis patients display deficient SHP-1 expression and enhanced inflammatory phenotype. Lab Investig. 2009;89:742–59. doi: 10.1038/labinvest.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An H, Hou J, Zhou J, et al. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat Immunol. 2008;9:542–50. doi: 10.1038/ni.1604. [DOI] [PubMed] [Google Scholar]
- 34.Strassheim D, Asehnoune K, Park JS, et al. Phosphoinositide 3-kinase and Akt occupy central roles in inflammatory responses of Toll-like receptor 2-stimulated neutrophils. J Immunol. 2004;172:5727–33. doi: 10.4049/jimmunol.172.9.5727. [DOI] [PubMed] [Google Scholar]
- 35.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 36.Pahan K, Raymond JR, Singh I. Inhibition of phosphatidylinositol 3-kinase induces nitric-oxide synthase in lipopolysaccharide- or cytokine-stimulated C6 glial cells. J Biol Chem. 1999;274:7528–36. doi: 10.1074/jbc.274.11.7528. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Tupper JC, Bannerman DD, Winn RK, Rhodes CJ, Harlan JM. Phosphoinositide 3 kinase mediates Toll-like receptor 4-induced activation of NF-kappa B in endothelial cells. Infect Immun. 2003;71:4414–20. doi: 10.1128/IAI.71.8.4414-4420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diaz-Guerra MJ, Castrillo A, Martin-Sanz P, Bosca L. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J Immunol. 1999;162:6184–90. [PubMed] [Google Scholar]
- 39.Aksoy E, Vanden Berghe W, Detienne S, et al. Inhibition of phosphoinositide 3-kinase enhances TRIF-dependent NF-kappa B activation and IFN-beta synthesis downstream of Toll-like receptor 3 and 4. Eur J Immunol. 2005;35:2200–9. doi: 10.1002/eji.200425801. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Zhu X, Li N, et al. CMRF-35-like molecule 3 preferentially promotes TLR9-triggered proinflammatory cytokine production in macrophages by enhancing TNF receptor-associated Factor 6 ubiquitination. J Immunol. 2011;187:4881–9. doi: 10.4049/jimmunol.1003806. [DOI] [PubMed] [Google Scholar]
- 41.Barrow AD, Trowsdale J. You say ITAM and I say ITIM, let's call the whole thing off: the ambiguity of immunoreceptor signalling. Eur J Immunol. 2006;36:1646–53. doi: 10.1002/eji.200636195. [DOI] [PubMed] [Google Scholar]