

Abstract
In patients with human immunodeficiency virus (HIV) infection, neutrophil and monocyte functions, including phagocytosis, are impaired. The purpose of this study was to investigate changes of phagocytic function and respiratory burst occurring over the course of patients infected by the HIV-1 virus. Treatment-naive patients (group B), patients receiving highly active anti-retroviral treatment (HAART) (group C) and patients in which HAART has failed (group D) were studied and compared with healthy volunteers (group A). Phagocytosis and oxidative burst were evaluated using commercially available kits. Results clearly denote a significant decrease of the phagocytic function of both cell types of groups B and C compared with group A. Among group C patients, those in the upper quartile of CD4 increase had higher oxidative burst compared with patients of the other quartiles. In addition, comparisons clearly showed a lower degree of phagocytic function and of oxidative burst of both monocytes and neutrophils of group D compared with group B. Finally, it was found that monocyte and neutrophil function was correlated inversely to the change in viral load, i.e. the greater the decrease of viral load, the better the phagocytic and oxidative activity. Innate immunity defects appear to be present in HIV-positive patients, regarding phagocytic activity and oxidative burst of monocytes and neutrophils. These defects are greatly influenced by the level of treatment efficacy, with emphasis on CD4 cell counts and viral load.
Keywords: human immunodeficiency virus, innate immunity, oxidative burst, phagocytosis
Introduction
Polymorphonucleocytes (PMNs) and monocytes are important effectors of innate immunity. They can recognize and phagocytose bacteria and other microorganisms, and additionally they can present antigen and mediate prime adaptive immune responses. In patients with human immunodeficiency virus (HIV) infection, neutrophil and monocyte functions, such as chemotaxis, respiratory burst activity, bacterial killing and antibody-dependent cell-mediated cytotoxicity, are impaired. It has thus been suggested that these are, at least in part, responsible for the increased frequency and severity of bacterial and opportunistic infections in these patients [1]. Whether some of these defects result directly from the infection of the mononuclear phagocytes by HIV [2] or are secondary to other events remains to be determined.
Although there is general agreement that phagocytosis is impaired in HIV-infected patients, there are contradictory reports regarding individual indices of phagocytic function. For example, superoxide production by PMNs has been reported as increased, decreased or normal in different studies [3]–[6]. It is questionable whether function of monocytes and neutrophils for phagocytosis and oxidative burst is improved by efficient highly active anti-retroviral treatment (HAART) or impaired when HAART has failed. The purpose of this study was to investigate changes of phagocytic function and respiratory burst occurring over the course of patients infected by the HIV-1 virus. To this end, treatment-naive patients, patients receiving HAART and patients in which HAART has failed were studied and compared with healthy volunteers.
Patients and methods
The study protocol was approved by the competent institutional Ethics Review Board (‘G. Gennimatas’ Athens General Hospital). All patients provided written informed consent and the study was conducted in accordance with the provisions of the Declaration of Helsinki. HIV-1 seropositive patients, regularly followed-up at the HIV out-patient clinic, were eligible to participate.
Exclusion criteria were: (a) any active bacterial infection or current anti-microbial treatment; (b) long-term therapy for tuberculosis; (c) any acquired immunodeficiency syndrome-related opportunistic infection, as defined by the Centers for Disease Control (CDC) criteria [7]; (d) neutropenia defined as fewer than 1000 neutrophils/mm3; (e) chronic infection by hepatitis B or C viruses; and (f) chronic intake of corticosteroids defined as any daily oral intake of equal to or more than 1 mg/kg of equivalent prednisone for more than 1 month.
Gender- and age-matched healthy volunteers were enrolled.
The study was cross-sectional and every patient was studied on one single day. More precisely, enrolled individuals were assigned into four groups, as follows:
Group A: healthy volunteers;
Group B: HIV-1-seropositive patients who were treatment-naive; these patients remained treatment-naive because they did not present any criteria for start of HAART;
Group C: HIV-1 seropositive patients who were receiving HAART; and
Group D: HIV-1 seropositive patients who had discontinued HAART because of treatment failure. HAART failure was defined as the inability to maintain suppression of viral replication to viral load of fewer than 200 copies/ml.
The study visit for group C was scheduled at least 3 months after initiation of HAART; for group D, the study visit was scheduled at least 1 month and no more than 3 months after treatment discontinuation.
Absolute CD4 cell count and viral load were recorded for every enrolled patient at baseline of start of HAART and on the day of the study visit. Absolute CD4 cell count was determined after staining of lymphocytes with the monoclonal antibodies anti-CD45 at the fluorochrome PC5, anti-CD3 at the fluorochrome fluorescein isothiocyanate (FITC) and anti-CD4 at the fluorochrome phycoerythrin (PE) using appropriate isotypic antibody controls and magnetic beads after passage through a flow cytometer. Viral load was determined by real-time polymerase chain reaction (PCR) of whole blood.
On the study visit, 10 ml of whole blood were collected after venipuncture of one peripheral vein under aseptic conditions into heparin-coated, pyrogen-free tubes. Tubes were transported immediately for processing.
Phacogytosis parameters were measured with the Phagotest® kit (Orpegen Pharma, Heidelberg, Germany), according to the manufacturer's instructions [8]. Fresh heparinized blood was pre-incubated in a water bath for 15 min at 0°C. In this test, 2 × 107 FITC-labelled Escherichia coli, precooled at 0°C, was added to 100 µl of whole blood. All samples were then incubated for 10 min at 37°C. The bacteria were opsonized with immunoglobulin and complement from pooled sera. A negative control was kept on ice. At the end of the incubation time, phagocytosis was stopped by placing the samples on ice. Quenching solution (100 µl of Brilliant blue) was added in order to suppress the fluorescence of bacteria connected to leucocyte surfaces. After two consecutive washing steps, erythrocytes were lysed using lysis solution for 20 min at room temperature. Then, 200 µl propidium iodide was added to stain leucocytes and bacterial DNA. Subsequently, neutrophils and monocytes were analysed by flow cytometry, and phagocytic activity was quantified as the individual cellular phagocytic activity, expressed as the degree of fluorescence per cell (mean fluorescent intensity – MFI) after analysis by a flow cytometer.
Oxidative burst was determined quantitatively with the Bursttest® kit (Orpegen Pharma) according to the manufacturer's instructions [9]. Fresh heparinized blood was pre-incubated in a water bath for 15 min at 0°C. Then, four testing tubes were filled with 100 µl of blood each, and either 2 × 107 unlabelled opsonized bacteria E. coli (test sample), 20 µl of substrate solution (negative control), 20 µl of 5 µM peptide N-formyl-MetLeuPhe (fMLP) as chemotactic low physiological stimulus (low control) or 20 µl of 8·1 µM phorbol 12-myristate 13-acetate (PMA), a strong non-receptor activator (high control). All samples were incubated for 10 min at 37°C in a water bath. Dihydrorhodamine (DHR)-123 as a fluorogenic substrate was added and incubated again in the same conditions. The oxidative burst occurred with the production of reactive oxygen intermediates (ROIs) (superoxide anion, hydrogen peroxide) in granulocytes and monocytes stimulated in vitro. In ROI-stimulated granulocytes and monocytes, non-fluorescent DHR-123 underwent conversion to fluorescent rhodamine (R) 123. Erythrocytes were removed using lysing solution for 20 min at room temperature. Samples were washed and the supernatant was decanted. An amount of 200 µl of DNA staining solution (centrifuged and incubated for 10 min at 0°C in a dark place) was added to discriminate and exclude aggregation artefacts of bacteria and/or cells in cytometric flow analysis. Finally, neutrophils and monocytes were analysed by flow cytometry and oxidative burst was quantified as the enzymatic activity (the amount of released ROIs) per cell, expressed as the mean fluorescence intensity (MFI) after analysis by a flow cytometer [10].
On each experiment day, two patient and one control samples were processed. Laboratory personnel were blinded as to clinical data and samples were labelled by serial numbers, without any reference to the identity of subjects.
Flow cytometry
At least 20 000 leucocytes were collected from each sample and measured on a fluorescence activated cell sorter (FACS)Calibur flow cytometer. The instrument was calibrated and standardized between each analysis by using Calibrite beads [Becton Dickinson (BD), Franklin Lakes, NJ, USA]. All samples analysis was performed with CellQuest software (BD). The granulocyte populations were gated using their forward- and side-scatter dot-plots.
Statistical analysis
Results were expressed as means ± standard error of the mean. Comparisons between groups were performed using one-way analysis of variance (anova), with Bonferroni post-hoc correction for multiple comparisons. For group C, patients were divided into quartiles of change of the absolute CD4 cell count (on the study visit from baseline). Patients belonging to the upper quartile were considered as subgroup C1 and the remaining of patients as subgroup C2. Comparisons between subgroups were performed using Student's t-test for unpaired data. Time from start of HAART in patients of group C and measurements of phagocytic function and of oxidative burst were correlated according to Spearman's rank order. Any value of P below 0·05 was considered significant.
Results
A total of 100 patients were enrolled in the study; 40 were assigned into group B; 40 into group C and 20 into group D. Thirty healthy volunteers were enrolled in group A. Demographic characteristics of the study population are summarized in Table 1. No patient was administered corticosteroids within the previous 6 weeks before the study visit.
Table 1.
Demographic characteristics of patients enrolled into the study
| Group A | Group B | Group C | Group D | P | |
|---|---|---|---|---|---|
| Male/female | 15/15 | 20/20 | 21/19 | 10/10 | 0·995 |
| Age (years, mean ± s.d.) | 36·6 ± 8·9 | 36·6 ± 8·9 | 36·6 ± 9·5 | 36·3 ± 8·4 | 0·999 |
| CD4 cells (/mm3, median–range) | 920 (328–1882) | 244 (147–550) | 262·5 (132–604) | 139 (73–447) | < 0·0001 |
| Viral load (log10 copies/ml, median–range) | – | 4·51* (2·65–5·66) | 1·69 (1·70–5·62) | 4·85 (2·67–5·47) | < 0·0001 |
Statistical comparison group B versus group D. P: non-significant (n.s.); s.d.: standard deviation.
Phagocytic activity and oxidative burst of monocytes and neutrophils of groups are shown in Fig. 1. These results clearly denote a significant decrease of the phagocytic function of both cell types of groups B and C compared with group A. No differences were found between the treatment-naive group B and group C, with the exception of the neutrophil oxidative burst being significantly lower in group C compared with group B. Comparisons clearly showed a lower degree of phagocytic function and of oxidative burst of both monocytes and neutrophils in group D compared to group B. A positive correlation was found between the phagocytic function of monocytes of patients of group C and the time from start of HAART (rs: +0·418, P = 0·007); between the oxidative burst of monocytes of patients of group C and the time from start of HAART (rs: +0·365, P = 0·021); and between the oxidative burst of neutrophils of patients of group C and the time from start of HAART (rs: +0·314, P = 0·049).
Fig. 1.
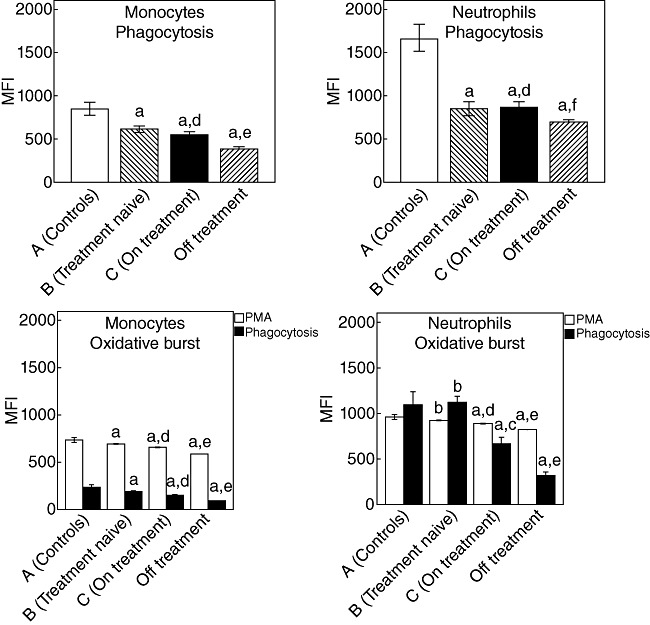
Phagocytosis and oxidative burst of monocytes and neutrophils of 30 healthy volunteers (group A), 40 treatment-naive human immunodeficiency virus (HIV-1) patients (group B), 40 patients on highly active anti-retroviral treatment (HAART) (group C) and 20 patients who discontinued HAART due to treatment failure (group D). For the oxidative burst, values after stimulation with a phagocytosis stimulus and with phorbol myristate acetate (PMA) are added. P for comparisons: a < 0·0001 group A versus the other groups; bnon-significant versus the other groups; c < 0·0001 group B versus group C; dnon-significant group B versus group C; e < 0·0001 group B versus group D; f0·049 group B versus group D.
We hypothesized that the impaired activity of monocytes and of neutrophils of group C (patients on HAART) may not be similar in all treated patients, being comparatively less impaired in patients of group C with better immunological response compared to those with a less satisfactory response to HAART. We assumed that immunological response to HAART may be indexed by the level of increase of the absolute CD4 cell count on the time of sampling compared to the respective count at baseline (before start of HAART). To this end, quartiles of changes of the absolute CD4 cell count from baseline of start of HAART were determined. The upper quartile of patients (subgroup C1) showed increases of CD4 cells between 273 and 373/mm3 from baseline. These patients had greater oxidative burst of monocytes and neutrophils, compared to patients belonging to subgroup C2 (CD4 cell increase in the range of 9–272/mm3) (Fig. 2).
Fig. 2.
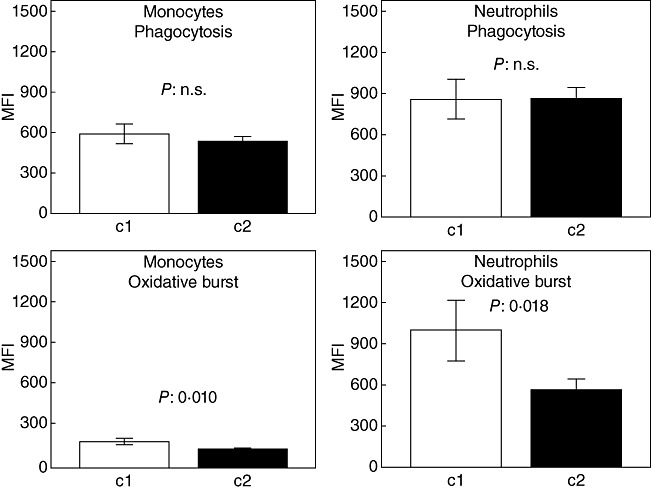
Phagocytosis and oxidative burst of monocytes and of neutrophils of patients infected by human immunodeficiency virus (HIV-1) after start of highly active anti-retroviral treatment (HAART). Patients are divided into group C1 (n = 10) with change of CD4 cells from 273 to 373/mm3 from baseline of start of HAART and C2 (n = 30) with change of CD4 cells from 9 to 272/mm3 from baseline of start of HAART. P-values refer to comparisons between groups C1 and C2; n.s.: non-significant.
Another question was whether changes of viral load from baseline may be correlated with monocyte and neutrophil function. Indeed, in patients of groups C and D, monocyte and neutrophil function was correlated inversely with the change in viral load (Fig. 3), i.e. the greater the decrease of viral load, the better the phagocytic and oxidative activity.
Fig. 3.
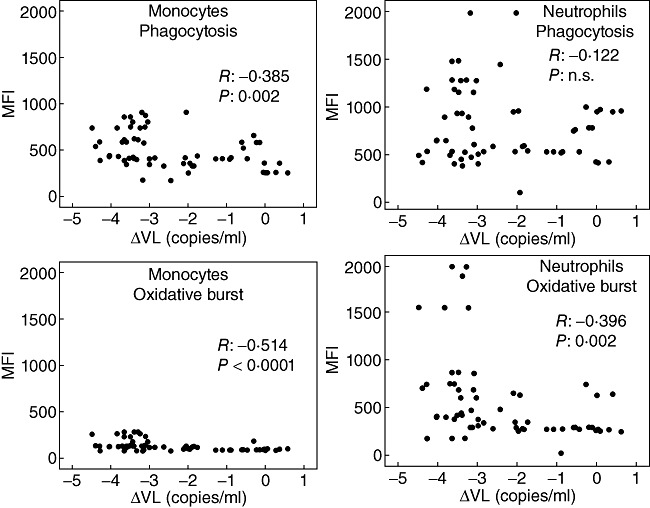
Correlation between logarithmic changes of the viral load (VL) from baseline and phagocytic and oxidative burst activity of monocytes and neutrophils. Patients assigned to group C [n = 40, on highly active anti-retroviral treatment (HAART)] and to group D (n = 20, HAART failure) are included in this analysis; n.s.: non-significant.
Discussion
In the present report we show that the functions of phagocytic activity and oxidative burst of neutrophils and of monocytes are impaired among HIV-1 infected patients compared with healthy controls. Previous data exist on the influence of HIV infection on phagocytosis and intracellular killing of microorganisms [11], as well as on neutrophil function [12] which, however, refer to the ability for killing of intracellular microorganisms, such as mycobacteria, Salmonella spp., Pneumocystis jirovecii, Cryptococcus neoformans and Candida albicans[13]–[16]. Available evidence on phagocytosis of extracellular bacteria in HIV infection leads to contradictory results; some authors report increased phagocytosis and intracellular bactericidal activity, whereas others report normal or decreased function [3]–[6,17]. These contradictory findings can be explained on the basis of several factors, such as patient age, stage of HIV infection, concurrent infections and concomitant medication. Methodological differences, such as the choice of microorganism for the phagocytosis and intracellular killing assays, could have also played a role. Finally, the assays used are often difficult to standardize; therefore, comparison of the results of different studies is problematic.
To control for some of these problems, we have compared three groups of HIV patients who are representative of different stages of the physical course of HIV infection: treatment-naïve, on HAART and after HAART failure. Although all three study populations had impaired functions compared with controls, it was obvious that the better the immunological status of the patients and the greater the decrease of viral load, the better were the functions of monocytes and of neutrophils. This conclusion was based on (a) the better oxidative burst of monocytes and of neutrophils of patients with HAART achieving higher increases of CD4 cell counts; (b) the better functions of both monocytes and of neutrophils among patients who were treatment-naive compared with those who failed HAART; and (c) the negative correlation between decreases of viral load and the function of monocytes and neutrophils. The impact of effective management of HIV infection, reflected by the absolute CD4 cell count and the viral load, on the function of phagocytosis and oxidative burst is in agreement with a previous report by Mastroianni et al. [18], suggesting functional improvement of neutrophils and monocytes against C. albicans, in HIV-1 patients treated with HAART.
The low degree of phagocytosis by neutrophils and monocytes suggests a defect in the initial phase of phagocytosis, i.e. opsonization of microorganism, binding of opsonized and non-opsonized microorganisms to phagocyte receptors and formation of the phagosome via F-actin-dependent cytoskeletal changes [19]. Abnormalities in the recognition of the microorganism by the phagocyte receptors and the intracellular signalling by those receptors might be responsible for our findings. Kedzierska et al. suggested that HIV causes defects in phagocytosis by interfering with intracellular signalling of Fc-gamma receptors rather than interfering with the expression of phagocyte receptors or the binding of these receptors to their ligands [20,21]. Two studies also suggest that formation of the phagosome is defective among HIV-positive patients [22,23]; this may be mediated through impaired recycling of endosomal compartment membranes [22].
The following limitations of the present study should be acknowledged: (a) the lack of report on the mechanisms of failure of oxidative burst and of phagocytosis potential of HIV-1-infected patients; (b) the lack of follow-up of these patients over time; and (c) the lack of study of the ability of neutrophils to contain infections by extracellular pathogens.
In conclusion, innate immunity defects appear to be present in HIV-positive patients with regard to phagocytic activity and oxidative burst of monocytes and of neutrophils. These defects are influenced greatly by the level of management of HIV-positive patients with emphasis on CD4 cell counts and viral load.
Disclosure
The authors have no conflicts of interest to declare.
References
- 1.Engelich G, Wright DG, Hartshorn KL. Acquired disorders of phagocyte function complicating medical and surgical illnesses. Clin Infect Dis. 2001;33:2040–8. doi: 10.1086/324502. [DOI] [PubMed] [Google Scholar]
- 2.Noursadeghi M, Katz DR, Miller RF. HIV-1 infection of mononuclear phagocytic cells: the case for bacterial innate immune deficiency in AIDS. Lancet Infect Dis. 2006;6:794–804. doi: 10.1016/S1473-3099(06)70656-9. [DOI] [PubMed] [Google Scholar]
- 3.Bandres JC, Trial J, Musher DM, et al. Increased phagocytosis and generation of reactive oxygen products by neutrophils and monocytes of men with stage 1 human immunodeficiency virus infection. J Infect Dis. 1993;168:75–83. doi: 10.1093/infdis/168.1.75. [DOI] [PubMed] [Google Scholar]
- 4.Pos O, Stevenhagen A, Meenhorst PL, et al. Impaired phagocytosis of Staphylococcus aureus by granulocytes and monocytes of AIDS patients. Clin Exp Immunol. 1992;88:23–8. doi: 10.1111/j.1365-2249.1992.tb03033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pitrak DL, Bak PM, DeMarais P, et al. Depressed neutrophil superoxide production in human immunodeficiency virus infection. J Infect Dis. 1993;167:1406–10. doi: 10.1093/infdis/167.6.1406. [DOI] [PubMed] [Google Scholar]
- 6.Nottet HS, de Graaf L, de Vos NM, et al. Phagocytic function of monocyte-derived macrophages is not affected by human immunodeficiency virus type 1 infection. J Infect Dis. 1993;168:84–91. doi: 10.1093/infdis/168.1.84. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan JE, Benson C, Holmes KH, et al. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm Rep. 2009;58:1–207. [PubMed] [Google Scholar]
- 8. Operator's Manual PHAGOTEST. Available at: http://www.icp.ucl.ac.be/mexp/phagotst.pdf (accessed 1 May 2011). Orpegen Pharma GmBH, Heidelberg, Germany.
- 9. Operator's Manual BURSTTEST. Available at: http://www.pvi.uni-bonn.de/bursttst.pdf (accessed 1 May 2011). Orpegen Pharma GmBH, Heidelberg, Germany.
- 10.Shalekoff S, Tiemessen CT, Gray CM, et al. Depressed phagocytosis and oxidative burst in polymorphonuclear leukocytes from individuals with pulmonary tuberculosis with or without human immunodeficiency virus type 1 infection. Clin Diagn Lab Immunol. 1998;5:41. doi: 10.1128/cdli.5.1.41-44.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Covelli V, Pece S, Giuliani G, et al. Pathogenetic role of phagocytic abnormalities in human virus immunodeficiency infection: possible therapeutical approaches. A review. Immunopharmacol Immunotoxicol. 1997;19:147–64. doi: 10.3109/08923979709007655. [DOI] [PubMed] [Google Scholar]
- 12.Pitrak DL. Neutrophil deficiency and dysfunction in HIV-infected patients. Am J Health Syst Pharm. 1999;56(Suppl. 5):S9–16. doi: 10.1093/ajhp/56.suppl_5.S9. [DOI] [PubMed] [Google Scholar]
- 13.Crowe SM, Vardaxis NJ, Kent SJ, et al. HIV infection of monocyte-derived macrophages in vitro reduces phagocytosis of Candida albicans. J Leukoc Biol. 1994;56:318–27. doi: 10.1002/jlb.56.3.318. [DOI] [PubMed] [Google Scholar]
- 14.Monari C, Baldelli F, Pietrella D, et al. Monocyte dysfunction in patients with acquired immunodeficiency syndrome (AIDS) versus Cryptococcus neoformans. J Infect. 1997;35:257–63. doi: 10.1016/s0163-4453(97)93042-5. [DOI] [PubMed] [Google Scholar]
- 15.Koziel H, Eichbaum Q, Kruskal BA, et al. Reduced binding and phagocytosis of Pneumocystis carinii by alveolar macrophages from persons infected with HIV-1 correlates with mannose receptor downregulation. J Clin Invest. 1998;102:1332–44. doi: 10.1172/JCI560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kedzierska K, Churchill M, Maslin CL, et al. Phagocytic efficiency of monocytes and macrophages obtained from Sydney blood bank cohort members infected with an attenuated strain of HIV-1. J Acquir Immune Defic Syndr. 2003;34:445–53. doi: 10.1097/00126334-200312150-00001. [DOI] [PubMed] [Google Scholar]
- 17.Schafer V, Kreuter J, Rubsamen-Waigmann H, et al. Influence of HIV-infection on the phagocytic activity of monocytes/macrophages and granulocytes. Clin Diagn Virol. 1994;1:279–87. doi: 10.1016/0928-0197(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 18.Mastroianni CM, Lichtner M, Mengoni F, et al. Improvement in neutrophil and monocyte function during highly active antiretroviral treatment of HIV-1-infected patients. AIDS. 1999;13:883–90. doi: 10.1097/00002030-199905280-00003. [DOI] [PubMed] [Google Scholar]
- 19.Brown EJ, Gresham HD. Phagocytosis. In: Paul WE, editor. Fundamental immunology. Philadelphia, PA: Lippincott Williams & Wilkins; 2003. pp. 1105–26. [Google Scholar]
- 20.Kedzierska K, Ellery P, Mak J, et al. HIV-1 down-modulates gamma signaling chain of Fc gamma R in human macrophages: a possible mechanism for inhibition of phagocytosis. J Immunol. 2002;168:2895–903. doi: 10.4049/jimmunol.168.6.2895. [DOI] [PubMed] [Google Scholar]
- 21.Kedzierska K, Azzam R, Ellery P, et al. Defective phagocytosis by human monocyte/macrophages following HIV-1 infection: underlying mechanisms and modulation by adjunctive cytokine therapy. J Clin Virol. 2003;26:247–63. doi: 10.1016/s1386-6532(02)00123-3. [DOI] [PubMed] [Google Scholar]
- 22.Mazzolini J, Herit F, Bouchet J, et al. Inhibition of phagocytosis in HIV-1 infected macrophages relies on Nef-dependent alteration of focal delivery of recycling compartments. Blood. 2010;115:4226–36. doi: 10.1182/blood-2009-12-259473. [DOI] [PubMed] [Google Scholar]
- 23.Ryder MI, Winkler JR, Weinreb RN. Elevated phagocytosis, oxidative burst, and F-actin formation in PMNs from individuals with intraoral manifestations of HIV infection. J Acquir Immune Defic Syndr. 1988;1:346–53. [PubMed] [Google Scholar]