

Abstract
Diacylglycerol (DAG)/protein kinase C (PKC) signaling plays an integral role in the regulation of neuronal function. This is certainly true in Caenorhabditis elegans and in particular for thermosensory signaling and behavior. Downstream molecular targets for transduction of this signaling cascade remain, however, virtually uncharacterized. We investigated whether PKC phosphorylation of Munc18-1, an essential protein in vesicle trafficking and exocytosis, was the downstream effector for DAG regulation of thermosensory behavior. We demonstrate here that the C. elegans ortholog of Munc18-1, UNC-18, was phosphorylated in vitro at Ser322. Transgenic rescue of unc-18-null worms with Ser322 phosphomutants displayed altered thermosensitivity. C. elegans expresses three DAG-regulated PKCs, and blocking UNC-18 Ser322 phosphorylation was phenocopied only by deletion of calcium-activated PKC-2. Expression of nonphosphorylatable UNC-18 S322A, either pan-neuronally or specifically in AFD thermosensory neurons, converted wild-type worms to a pkc-2-null phenotype. These data demonstrate that an individual DAG-dependent thermosensory behavior of an organism is effected specifically by the downstream PKC-2 phosphorylation of UNC-18 on Ser322 in AFD neurons.
Introduction
Diacylglycerol (DAG) is a well characterized intracellular signaling messenger important in regulating neuronal function (Brose et al., 2004; Haucke and Di Paolo, 2007; de Jong and Verhage, 2009). Production of DAG can be stimulated by extracellular signals, resulting in the activation of downstream effectors, whereas DAG can be removed from cells by conversion into phosphatidic acid by diacylglycerol kinases (DGKs). Within neurons, the best characterized targets for DAG are the synaptic vesicle priming factor Munc13 (Lackner et al., 1999; Nurrish et al., 1999; Rhee et al., 2002) and protein kinase C (PKC), itself a potent intracellular signaling protein within the nervous system (Morgan et al., 2005). In Caenorhabditis elegans, DAG signaling regulates many intrinsic neural-dependent behaviors, in particular thermosensation. Loss-of-function mutations in pkc-1 display thermophilic thermotaxis, whereas dgk-1 diacylglycerol kinase-null worms are cryophilic (Okochi et al., 2005). In contrast, dgk-3 is specifically essential in the resetting of temperature memory in an alternate assay for isothermal tracking (Biron et al., 2006). Despite a defined role for DAG and PKC activation in regulating these thermosensory phenotypes, the downstream targets for the transduction of the regulatory signal within thermosensory neurons remain uncharacterized.
Munc18-1 is an essential synaptic protein required for synaptic vesicle fusion through its biochemical interactions with SNARE proteins. Importantly Munc18-1 is phosphorylated by PKC in vitro on Ser306 and Ser313 (Fujita et al., 1996; Barclay et al., 2003), the latter residue being rapidly phosphorylated upon in vivo depolarization (Craig et al., 2003). Although phosphorylation of Munc18-1 increases vesicle pool replenishment (Nili et al., 2006), alters fusion pore dynamics (Barclay et al., 2003) and is essential for DAG-induced synaptic potentiation (Wierda et al., 2007) a direct role for Munc18-1 phosphorylation in any defined DAG-dependent regulation of behavior is unknown. We aimed to identify a specific downstream effector for DAG-dependent regulation of behavior. We demonstrate that the C. elegans homolog UNC-18 is phosphorylated by PKC in vitro on Ser322, causing a reduction in closed-conformation binding to C. elegans syntaxin. Crucially, this single phosphorylation event on an individual protein underlies PKC-2 regulation of the thermosensitivity of C. elegans locomotion in a specific pair of thermosensory neurons.
Materials and Methods
Plasmid construction and recombinant protein production
UNC-18 was subcloned into pGEX-6p-1 for recombinant glutathione S-transferase (GST) fusion protein production. Recombinant proteins (GST, GST-UNC-18) were produced as described previously (Graham et al., 2009; Johnson et al., 2009). Removal of the GST tag from GST-UNC-18 was achieved by first binding GST-UNC-18 to glutathione-Sepharose beads, washing with PBS, and then incubating with 960 μl of chilled PreScission cleavage buffer and 40 μl of GST-PreScission protease (GE Healthcare) for 16 h at 4°C with rotation. Glutathione-Sepharose beads were then centrifuged to a pellet, and the supernatant containing cleaved UNC-18 protein was removed. Point mutations (R39C, S311A, S311E, S322A, S322E, E465K) were introduced using the GeneTailor mutagenesis kit (Invitrogen).
In vitro phosphorylation and mass spectroscopy
For phosphorylation, 2 μg of substrate protein (GST, GST-UNC-18, or cleaved UNC-18) were incubated in MES buffer (50 mm MES, 10 mm MgCl2, 1 mm DTT, 0.5 mm EDTA, pH 6.9) with 100 μm ATP and 2 μCi [γ-32P]ATP (GE Healthcare). The final reaction volume was 50 μl, containing 700 mU of an active PKC catalytic subunit purified from rat brain (Calbiochem). For mock phosphorylation, the PKC catalytic subunit was omitted. Reactions were incubated at 30°C for 3 h before termination. To assess the specificity of protein phosphorylation, 20 μl of arrested kinase reaction was separated on a 12.5% SDS-PAGE gel, stained with Coomassie Blue dye, and then destained overnight in destainer [35% ethanol, 2% glycerol (v/v)]. Gels were air-dried in Hoeffer Easy Breeze plastic frames (Thermo Fisher Scientific), exposed to a phosphor screen for 2–4 h, and scanned by a PhosphorImager 425 (Molecular Dynamics). To determine the in vitro phosphorylation site, both mock and PKC-phosphorylated samples were separated on NuPAGE 4–12% Bis-Tris precast gels (Invitrogen) and stained with Coomassie Blue dye before excising protein bands. Gel plugs were thoroughly destained in 50% acetonitrile (v/v)/50 mm ammonium bicarbonate, dried, and then incubated in trypsin solution (5 ng/μl in 50 mm ammonium bicarbonate) for 16 h at 37°C. Peptides were extracted from gel plugs by sonication in 60% (v/v) acetonitrile/1% (v/v) trifluoroacetic acid. Extracts were then dried completely and ZipTipped (Millipore) before electrospray ionization mass spectrometry (MS). Residual peptides were resuspended in 5% (v/v) acetonitrile/0.05% trifluoroacetic acid, and 5 μl of each suspension were delivered into a QStar Pulsar I hybrid quadrupole time-of-flight MS (AB Sciex by automated in-line liquid chromatography (integrated LCPackings System, 5 mm C18 nano-precolumn, and 75 μm × 15 cm C18 PepMap column (Dionex). A gradient from 5 to 48% acetonitrile/0.05% trifluoroacetic acid (v/v) in 60 min was applied at a flow rate of 300 nl/min, and survey scans of 1 s were acquired for m/z 400–2000. The three most intense ions were selected for tandem mass spectrometry (MS/MS) with accumulation times of 2 s and a dynamic exclusion of 30 s. Identification and analysis of peptide mass spectra was facilitated using MASCOT software (Matrix Science).
Binding of 35S-labeled in vitro transcription-translation derived proteins to immobilized GST fusion proteins
The cytoplasmic domain of UNC-64 was produced using the TNT T7 Quick Coupled Transcription/Translation system (Promega) in the presence of ∼0.6 MBq of [35S]methionine (GE Healthcare) per reaction, as per the manufacturer's protocol. Glutathione-Sepharose beads were washed three times in binding buffer [150 mm potassium acetate, 1 mm MgCl2, 0.05% (v/v) Tween 20 and 20 mm HEPES, pH 7.4] and then incubated with either 2 μM GST or GST-UNC-18 (wild-type or mutant) in a total volume of 200 μl of binding buffer for 30 min. Fifty microliters of the bead/protein mix was then incubated with 5 μl of 35S-labeled in vitro transcription/translation product in a final volume of 100 μl for 2 h at 4°C. Beads were then washed three times with binding buffer before eluting with Laemmli buffer. Proteins were separated by SDS-PAGE and transferred to nitrocellulose paper. Radiolabeled bands were visualized by exposure to autoradiography film for 3–4 h.
Protein three-dimensional structure production
Predicted model of UNC-18 (C. elegans ortholog of Munc18-1) was generated by the I-TASSER (Iterative-Threading/Assembly/Refinement) online server (Roy et al., 2010), which predicts unknown structures based upon known ortholog information, protein-protein alignment, ab initio folding, and modeling. The three-dimensional representation of the structure was produced in PyMol (Delano Scientific).
Nematode culture and strains
C. elegans were grown under standard conditions on nematode growth medium (NGM) agar plates at 20°C with Escherichia coli OP50 as a food source as described previously (Graham et al., 2009; Johnson et al., 2009). The wild-type reference strain was Bristol N2. Mutant strains used in this study were unc-18 (e81), pkc-2 (ok328), pkc-1 (nu448), pkc-1 (nj3), tpa-1 (k501), hsf-1 (sy441), dgk-1 (ok1462), dgk-2 (gk124), and dgk-3 (gk110).
Nematode constructs and transgenes
Transgenic strains were generated by germline injection of expression constructs (5–20 ng/μl) along with the co-injection marker sur-5::GFP (20 ng/μl) into unc-18 (e81), dgk-2 (gk214), pkc-2 (ok328), or Bristol N2 worms. Rescue experiments involved expression constructs carrying unc-18 cDNA (wild-type or with individual point mutations) under the control of the unc-18 genomic flanking regions. We and others have demonstrated previously that this construct rescues unc-18 (e81) worms phenotypically (Graham et al., 2009; Johnson et al., 2009). For pkc-2 (ok328) and dgk-2 (gk124) rescue experiments, cDNAs encoding for pkc-2 (E01H11.1a) or dgk-2 (F46H6.2a.1) were first produced from a reverse-transcribed total RNA preparation isolated by standard protocols from Bristol N2 worms. Rescuing expression constructs were then generated by placing the pkc-2 and the dgk-2 cDNAs under the control of the pan-neuronal rab-3 promoter (gift from M. Nonet, Washington University School of Medicine, St. Louis, MO) (Mahoney et al., 2006). AFD-specific expression was achieved using a gcy-8 promoter, a DNA fragment cloned from 2 kb upstream of the gcy-8 start codon (Mori et al., 2007). AFD-specific expression was verified by co-injection with a GFP marker under the same promoter. Total injected DNA concentration was made up to 130 ng/μl with empty pcDNA3.1(−) vector for all injections. Successful transgenic animals were indicated by GFP fluorescence or rescue of paralyzed phenotype where indicated. For each construct, 3–5 individual independently derived transgenic lines were isolated and analyzed; results presented here were consistent for all generated lines.
Protein extraction and Western blotting
Animals were harvested from 10 × 60 mm diameter seeded NGM plates in chilled M9 buffer (42.3 mm Na2HPO4, 22 mm KH2PO4, 85.6 mm NaCl, 1 mm MgSO4). After the settling into a pellet, worms were washed three times in fresh M9 buffer and finally resuspended in 1 ml of worm lysis buffer [50 mm Tris/HCl, 150 mm NaCl, pH 7.6, 1.5% (w/v) n-octyl glucoside, and Protease Inhibitor Cocktail (Sigma)]. Worm suspension was lysed at 4000 psi in a One-Shot cell disrupter (Constant Systems) and incubated with rotation at 4°C for 1 h. Insoluble material was removed by centrifugation at 15,000 × g for 30 min at 4°C. Soluble proteins were dissolved in Laemmli buffer (Sigma) and boiled. Protein lysates were separated by SDS-PAGE (12.5% gels) and transferred to nitrocellulose paper for immunoblotting. Antibodies used for immunoblotting: 1:1500 anti-β-actin (Sigma) and 1:500 anti-PKCα/β/γ (Calbiochem).
Reverse-transcription PCR
Animals were grown on two 60 mm NGM plates and harvested with chilled M9 buffer. Worms were centrifuged at 5000 × g for 5 min, and the worm pellet was lysed in 400 μl of TRI Reagent (Sigma). RNA was purified and first-strand cDNA was produced from 1 μg of total RNA template using avian myeloblastosis virus reverse transcriptase and random primers (Promega) following the manufacturer's instructions. End-point PCR was performed with specific primers for pkc-1, pkc-2, tpa-1, or act-1 using GoTaq DNA polymerase (Promega).
Behavioral assays
All experiments were performed on young hermaphrodite animals from sparsely populated plates grown at 20°C. All C. elegans strains were cultured and assays performed in a temperature controlled room at 20°C.
Locomotion assays.
Locomotion rate was measured as described previously (Graham et al., 2009). Briefly, locomotion was quantified in solution as thrashes per minute (one thrash defined as one complete sinusoidal movement from maximum to minimum amplitude and back again). Single young adult hermaphrodites were removed from NGM plates and placed in a Petri dish containing 200 μl of freshly made Dent's solution (140 mm NaCl, 6 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.4 with bovine serum albumin at 0.1 mg/ml). Assessment of locomotion was performed 10 min following immersion in solution. At least 20 animals were recorded per strain tested per experiment. All data are expressed as mean ± SE. Significance was assessed by Student's t test.
Temperature assays.
To assess the temperature of locomotion failure, single adult hermaphrodites were placed in a Petri dish containing 200 μl of freshly made Dent's solution. The Petri dish was then placed onto the center of a small Peltier effect thermoelectric device (Melcor) controlled by a DC power supply. A thermocouple was placed in the Dent's solution to record the temperature of the solution accurately and in real time. By gradually increasing the current into the Peltier device, the temperature of the Dent's solution was then raised ∼1°C/min until the animal was deemed paralyzed. Paralysis was defined here as a total lack of movement in response to mechanical stimulation (gentle prod with a 0.125 mm tungsten probe). To assess the temperature dependence of locomotion rate, thrashing was recorded at both 20°C and 28°C following 10 min of acclimatization to that temperature. Single adult hermaphrodites were removed from NGM plates and placed in a Petri dish containing 200 μl of freshly made Dent's solution. The Petri dish was placed onto the center of a small Peltier effect thermoelectric device and a thermocouple was placed in the Dent's solution. By increasing the current into the Peltier device, the temperature of the Dent's solution was raised to 28°C in ∼1 min. Locomotion was then quantified 10 min after a stable temperature (±0.5°C) was attained. Thrashing was defined as described previously. Initial experiments on Bristol N2 wild types had determined that 28°C was an optimal temperature to determine differences in thermosensitivity of locomotion between strains. In these initial experiments, the experimental setup was identical to that described above; however, temperature was raised in 2°C increments from 20°C until animals were paralyzed. At each increment, the new temperature was reached in ∼1 min followed by 1 min acclimatization and a 1 min locomotion recording time. When determining temperature dependence of locomotion, measurements were made as a percentage of mean locomotion rate for each strain at 20°C measured each day (at least 10 control animals per day). At least 20 animals were tested per strain. All data are expressed as mean ± SE. Significance was tested by Kruskal-Wallis one-way ANOVA and post hoc comparison of means using the Mann–Whitney U test.
Aldicarb assays.
Acute sensitivity to aldicarb (1 mm; Sigma) was assessed by measuring time to onset of paralysis following exposure to the drug as described previously (Lackner et al., 1999; Graham et al., 2009). For each experiment 20–30 animals were placed onto the center of a 60 mm unseeded NGM plate containing aldicarb. To assess paralysis worms were lightly mechanically stimulated every 5–10 min following drug exposure. Worms were deemed to be paralyzed when they failed to respond to stimulation. Where indicated worms were pretreated with 2 μg/ml PMA for 2 h followed by exposure to aldicarb in combination with PMA. Experiments were performed at least three times.
Results
C. elegans UNC-18 is phosphorylated by PKC on Ser322
A prime candidate for a DAG/PKC signaling effector within the nervous system is the essential exocytotic protein Munc18-1. The ortholog of Munc18-1 in C. elegans, UNC-18, is also phosphorylated by PKC (Sassa et al., 1996); however, the precise phosphorylation site(s) and the physiological consequences within the nervous system are unknown. We first verified that UNC-18 was indeed phosphorylated in vitro. In the presence of 32P-labeled ATP and the catalytic subunit of PKC, both GST-UNC-18 and proteolytically cleaved, tag-free UNC-18 were readily phosphorylated by PKC, whereas GST alone was not (Fig. 1A). Under mock phosphorylation conditions where the kinase was omitted, none of the recombinant proteins were phosphorylated. Previous work speculated on Ser2, Ser322, Thr462, and Ser515 as potential PKC phosphorylation sites of UNC-18 (Sassa et al., 1996). None of these residues are orthologous to the evolutionarily conserved Munc18-1 phosphorylation site (Ser313); therefore, an unbiased approach was employed to determine in vitro phosphorylation sites by mass spectrometry. Protein sequence coverage for phosphorylated UNC-18 (66% compared to 72% for mock-phosphorylation) unfortunately did not cover Ser311, the orthologous residue to Munc18-1 Ser313. In addition, Ser2, Thr462, and Ser515 were not covered, thus leaving their status as potential phosphorylation sites unanswered. Mass spectrometry, however, did positively identify Ser322 as an in vitro site for PKC phosphorylation (Fig. 1B).
Figure 1.

Phosphorylation of UNC-18 at Ser322 reduces closed conformation binding to syntaxin. A, Recombinant protein (GST, GST-UNC-18, or tag-free UNC-18) was incubated with 32P-labeled ATP in the presence (+PKC) or absence (MOCK) of the catalytic subunit of PKC. Proteins were separated by SDS-PAGE and specific phosphorylation of GST-UNC-18 and tag-free UNC-18 was determined by PhosphorImager. Coomassie staining demonstrated equal protein loading of all samples. B, Phosphorylated protein was excised from gels, thoroughly destained, and incubated with trypsin. Peptides were then analyzed by liquid chromatography-MS/MS, resulting in positive identification of phosphorylated Ser322 of UNC-18. C, Location of the phosphorylation target of PKC in the UNC-18 protein structure. Both Ser322 and the orthologous residue to the phosphorylation site in Munc18-1 (Ser311) are indicated by arrows. D, Sequence alignment of C. elegans UNC-18 and mammalian Munc18-1 proteins. Identical amino acids are highlighted. Demonstrated in vitro phosphorylation sites are indicated by arrows (Ser306 and Ser313 in Munc18-1; Ser322 in UNC-18). E, Radiolabeled wild-type UNC-64 (C. elegans syntaxin) was incubated with recombinant wild-type GST-UNC-18 or various point mutations (S311A, S311E, S322A, S322E). Samples of bound protein were detected by autoradiography. Quantification of binding as a percentage of wild type demonstrated a specific reduction for S322E (*p < 0.05; N = 4).
With no existing structural data for the C. elegans protein, we generated a predicted model of UNC-18 using the I-TASSER online server (Roy et al., 2010). As diverse Sec1/Munc18-1 (SM) proteins have very similar structures (Misura et al., 2000; Bracher and Weissenhorn, 2002; Hu et al., 2007), we reasoned that an UNC-18 structure would likely be comparable. Our generated structure is arch shaped, similar to other SM proteins (Fig. 1C). Ser322 is located within domain 3a in an adjacent region of the protein orthologous to Ser313 of Munc18-1 and partially matches the consensus recognition site of PKC (Fig. 1D). Critically, Ser322 is structurally accessible to a kinase contained within a disordered region of the protein and thus would act as a feasible in vivo phosphorylation site. That Ser322 is not the orthologous residue to the phosphorylation target of the mammalian protein is perhaps not surprising, as a recent evolutionary study of Cdk1 substrates indicates that precise phosphorylation site positioning is not conserved, with flexible movement within disordered regions being sufficient to preserve context-dependent regulation of protein interactions (Holt et al., 2009).
Phosphorylation of Munc18-1 reduces closed conformation binding to the SNARE protein syntaxin (Fujita et al., 1996; Barclay et al., 2003). Attempts to replicate this result with UNC-18 were unsuccessful, an outcome likely the consequence of a relatively low in vitro stoichiometry of phosphorylation. To circumvent this problem, we constructed recombinant proteins where Ser322 was mutated to either a phosphomimetic glutamate (UNC-18 S322E) or a phospho-null alanine (UNC-18 S322A), a technique previously used successfully for Munc18-1 (Barclay et al., 2003). In a binding assay with the full cytoplasmic domain of C. elegans syntaxin (UNC-64), we found that UNC-18 S322E indeed had a specific reduction in binding as predicted (Fig. 1E). A reduction was not evident for the S322A mutation, nor were there any effects of mutations to Ser311 (the orthologous residue to Munc18-1 Ser313). These results indicate that UNC-18 is phosphorylated in vitro on Ser322 and support a role for PKC phosphorylation in reducing closed-conformation syntaxin binding, a similar outcome to that seen for the mammalian protein.
Phenotype of C. elegans UNC-18 Ser322 phosphomutants
The functional relevance of UNC-18 Ser322 phosphorylation was characterized in vivo by transgenic rescue of C. elegans unc-18 (e81)-null worms with plasmids expressing either wild-type (Wt), S322A, or S322E unc-18 under the control of its endogenous genomic promoter. The e81 allele carries a premature stop codon resulting in a null phenotype, reduced neuromuscular transmission, and paralysis (Sassa et al., 1999; Weimer et al., 2003). Transgenic rescue with unc-18 S322A or S322E created phenotypically normal worms, qualitatively indistinguishable from transgenic rescues with Wt unc-18 (Fig. 2A; Table 1). Physiological effects of the phosphomutants on locomotion were quantified by measuring the rate of thrashing and found to be statistically equal for transgenic rescue with any of the unc-18 constructs (Fig. 2B). Results were consistent for multiple independently derived transgenic lines as demonstrated previously (Graham et al., 2009). We also measured acute aldicarb sensitivity, which directly assesses the strength of neuromuscular signaling. Once again there was no difference between the Wt transgenic rescue and either Ser322 phosphomutant (Fig. 2C). It remained possible, however, that effects of the mutations would not be evident unless under conditions of high phosphorylation. Worms pretreated with the DAG analog PMA are hypersensitive to aldicarb caused by the activation of UNC-13 (Lackner et al., 1999) and PKC-1-dependent phosphorylation of an as yet unknown target (Sieburth et al., 2007). We tested the hypothesis that phosphorylation of UNC-18 Ser322 was involved in PMA-induced aldicarb hypersensitivity. Although an effect of PMA pretreatment on aldicarb sensitivity was evident, the effect was identical for all transgenic worm rescues (Fig. 2C). These data indicate that phosphorylation of UNC-18 at Ser322 does not affect the strength of neuromuscular signaling in C. elegans and that the phosphorylation target for PKC-1 involved in PMA-induced hypersensitivity is not Ser322 of UNC-18.
Figure 2.
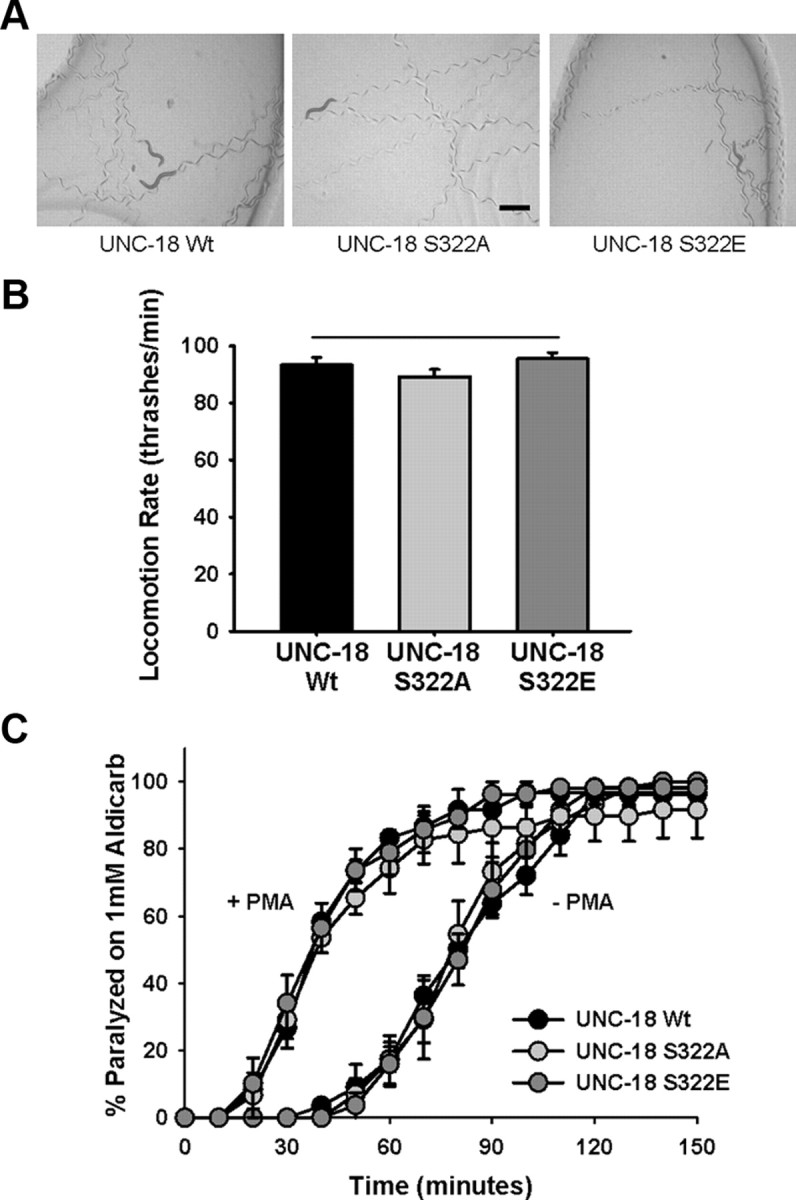
Transgenic C. elegans expressing Ser322 phosphomutations are phenotypically wild type. A, The unc-18 (e81)-null allele was rescued transgenically by constructs expressing wild type (UNC-18 Wt) or mutant (UNC-18 S322A or UNC-18 S322E) UNC-18. Photographs depict trails left by five worms after 5 min moving through a bacterial lawn. Scale bar, 0.5 mm. B, Rate of locomotion of individual e81 transgenic rescues was quantified by counting thrashes per minute in solution. No statistical difference was evident for either phosphomutant versus transgenic rescue with wild-type UNC-18. C, Neuromuscular cholinergic release was quantified by exposing populations of individual transgenic rescues to 1 mm aldicarb (−PMA) and determining onset of paralysis following drug exposure by mechanical stimulation until paralysis of the total population of worms was attained. Concurrently, aldicarb experiments were performed following a 2 h pre-exposure to plates containing 2 μg/ml PMA (+PMA) to hypersensitize worms to aldicarb. Data were analyzed from at least 20–25 worms per transgenic line per experiment (N = 3).
Table 1.
Basal locomotion rates of C. elegans strains
| C. elegans strain | Locomotion rate (thrashes/min) |
|---|---|
| Bristol N2 | 83.9 ± 1.5 |
| Bristol N2+ unc-18 S322A | 69.0 ± 2.8 |
| Bristol N2+ unc-18 S322E | 74.5 ± 3.0 |
| Bristol N2+ Pafd::unc-18 S322A | 77.5 ± 2.6 |
| pkc-2 (ok328) | 80.1 ± 2.2 |
| pkc-2+ Prab-3::pkc-2 | 65.6 ± 3.4 |
| pkc-2+ unc-18 S322A | 82.8 ± 3.5 |
| pkc-2+ unc-18 S322E | 78.8 ± 1.6 |
| unc-18 (e81) | 1.6 ± 0.8 |
| unc-18+ unc-18 Wt | 93.3 ± 2.6 |
| unc-18+ unc-18 S322A | 89.1 ± 2.6 |
| unc-18+ unc-18 S322E | 95.5 ± 2.2 |
| unc-18+ unc-18 R39C | 92.5 ± 1.9 |
| unc-18+ unc-18 E465K | 67.8 ± 2.9 |
| pkc-1 (nu448) | 49.0 ± 5.5 |
| pkc-1 (nj3) | 79.9 ± 2.5 |
| dgk-1 (ok1462) | 38.4 ± 5.4 |
| dgk-3 (gk110) | 85.8 ± 2.2 |
| dgk-2 (gk124) | 75.4 ± 3.5 |
| dgk-2+ Prab-3::dgk-2 | 62.3 ± 2.1 |
| dgk-2+ unc-18 S322A | 83.6 ± 2.7 |
| dgk-2+ unc-18 S322E | 81.3 ± 3.1 |
Locomotion was quantified as thrashing per minute in 200 μl of Dent's solution at room temperature (20°C).
Phenotype of C. elegans PKC mutants
The PKCs are a family of serine/threonine protein kinases implicated in a host of cellular functions (Steinberg, 2008). The family is subdivided into cPKCs (conventional isoforms activated by both calcium and DAG), nPKCs (novel isoforms activated by DAG alone), and aPKCs (atypical isoform activity regulated by protein–protein interactions and PDK-1 phosphorylation). C. elegans only express three PKCs regulated by DAG (Tabuse, 2002). Of these, both pkc-1 and tpa-1 are nPKCs, leaving pkc-2 as the only calcium-activated PKC (Fig. 3A). Mutants lacking PKC-1 are resistant to the paralytic effect of aldicarb and are partially resistant to PMA-induced aldicarb hypersensitivity (Sieburth et al., 2007). Initially isolated in a mutational screen for resistance to growth on phorbol esters (Tabuse and Miwa, 1983), tpa-1 has also been implicated in a number of DAG-regulated phenotypes (Waggoner et al., 2000; van der Linden et al., 2003; Van Buskirk and Sternberg, 2007). Despite a clear link between the two nPKCs and DAG-signaling, virtually nothing is known about the role of PKC-2 in C. elegans. We obtained loss-of-function alleles for pkc-1 and pkc-2 and characterized the worms for locomotion and aldicarb defects. As reported previously (Sieburth et al., 2007), pkc-1 (nu448) worms moved slower in solution than Bristol N2 wild-type worms (Fig. 3B; Table 1). This reduction in locomotion rate was also evident, although to a lesser degree, for pkc-2 worms. We verified that pkc-2 worms did not express PKC-2 by blotting worm extracts with an antibody for mammalian cPKCs. This determined that pkc-2-null animals did not express a protein of ∼75 kDa that was expressed in Bristol N2, pkc-1, and tpa-1 worms (Fig. 3C). This would equal a predicted size of two of the pkc-2 isoforms. We also confirmed that pkc-2 worms expressed transcripts for pkc-1 and tpa-1, indicating a specific lack of pkc-2 (Fig. 3D). Finally, aldicarb sensitivity was measured for pkc-1 and pkc-2-null worms. Both PKC mutants showed a measurable resistance to aldicarb phenotype in comparison to wild type (Fig. 3E). In addition, both mutants partially blocked PMA-induced aldicarb hypersensitivity (Fig. 3F). Finally, we tested whether the aldicarb phenotypes of pkc-2 were a direct result of pkc-2 loss-of-function by transgenically expressing wild-type pkc-2 under the control of the pan-neuronal rab-3 promoter. Rescue of pkc-2-null worms indeed reverted the aldicarb phenotype similar to wild-type Bristol N2 sensitivity (Fig. 3E,F). The involvement of PKC-1 in phorbol ester-mediated regulation of aldicarb responses has been demonstrated previously (Sieburth et al., 2007); however, these data indicate a novel role for PKC-2.
Figure 3.

PKC-2 partially mediates the effects of phorbol esters to potentiate neuromuscular signaling. A, Comparison of the three diacylglycerol-regulated PKC proteins in C. elegans. Approximate positions of the pseudosubstrate (PS), DAG-binding domain (C1), calcium-binding domain (C2), and kinase domain (kinase) are depicted. Note that only pkc-2 has an active C2 domain. B, Rate of locomotion of individual PKC mutant worms (pkc-1, pkc-2, tpa-1) was quantified by counting thrashes per minute in solution and comparing to those of wild-type worms (N2). Loss-of-function worms of pkc-1 and pkc-2 were significantly slower in comparison to wild types (*p < 0.05). C, Loss-of-function pkc-2 (ok328) worms do not express PKC-2 protein. Worm lysates were separated by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblotted with an antibody to anti-PKCα/β/γ and to β-actin. Protein bands indicated by PKC-2 arrow were ∼75 kDa and were absent in the pkc-2 (ok328) worm lysates. D, Loss-of-function pkc-2 worms do express other PKC isoforms. Total RNA was purified from individual strains and used to generate cDNA. Primers specific to pkc-1, pkc-2, and tpa-1 were used to determine expression of mRNA by endpoint RT-PCR. E, Neuromuscular cholinergic release for pkc-1 and pkc-2 worms were quantified by exposing populations of individual transgenic rescues to 1 mm aldicarb. Both pkc-1 and pkc-2-null worms showed a resistance to the paralyzing effects of aldicarb in comparison to wild-type (N2) or pkc-2 rescue worms (pkc-2+Prab-3::pkc-2). F, Release was also quantified in response to 1 mm aldicarb following pre-exposure to 2 μg/ml PMA. Both pkc-1 and pkc-2-null worms showed a reduction in PMA-induced hypersensitization to aldicarb in comparison to wild-type (N2) or pkc-2 rescue worms (pkc-2+Prab-3::pkc-2). Data for both aldicarb experiments were analyzed from at least 20–25 worms per transgenic line per experiment (N = 3).
Temperature sensitivity of locomotion in PKC-null worms and UNC-18 Ser322 phosphomutants
DAG signaling and PKC activation also underlie thermosensation in C. elegans (Okochi et al., 2005; Biron et al., 2006); however, nothing is known regarding pkc-2 in temperature-dependent phenotypes. Both pkc-1 and pkc-2 are expressed in the nervous system and specifically in sensory neurons (Tabuse, 2002); thus, each could have potential roles in thermosensory signaling. We screened PKC-null mutants using a simplified acute thermal behavioral assay. Raising the temperature by 1°C per minute until paralysis determined that, in comparison to Bristol N2 controls, pkc-2 worms had a significantly higher temperature limit for locomotion (Fig. 4A). Transgenic rescue of pkc-2-null worms with neuronally expressed pkc-2 reduced the temperature of paralysis to wild-type levels. As an additional control, we used a loss-of-function hsf-1 (sy441) strain (Hajdu-Cronin et al., 2004). In Drosophila, a link between heat shock protein expression and temperature sensitivity of locomotion has previously been established (Xiao et al., 2007). In our experiments, hsf-1 worms were paralyzed at a lower temperature than wild types (Fig. 4A). Of two loss-of-function pkc-1-null alleles, nu448 had a lower temperature of paralysis, whereas nj3 was not different from wild type. These results indicated pkc-2-null worms specifically had a reduced temperature-dependency of locomotion.
Figure 4.

Loss-of-function of PKC-2 or blocking Ser322 phosphorylation of UNC-18 results in decreased temperature sensitivity of locomotion. A, Indicated worm strains were placed in solution and temperature was increased 1°C/min until paralysis (verified by mechanical stimulation). In comparison to wild type (N2), both hsf-1 and pkc-1 (nu448) worms were paralyzed at a lower temperature (*p < 0.01), whereas pkc-2 worms were paralyzed at a higher temperature (*p < 0.01). Both pkc-1 (nj3) and the pkc-2 rescue worms (pkc-2+Prab-3::pkc-2) were not significantly different from wild type. B, Locomotion rates of indicated strains at 28°C (open bars) were quantified as a percentage of locomotion at room temperature (20°C; filled bars). In comparison to wild-type (N2), there was a substantial reduction in locomotion at elevated temperature for hsf-1 worms and for both pkc-1 alleles (*p < 0.01). Loss-of-function pkc-2 worms moved significantly faster at elevated temperatures in comparison to wild-type and to pkc-2 worms transgenically rescued with pkc-2 (pkc-2+Prab-3::pkc-2) (*p < 0.01). C, Temperature of paralysis of unc-18 (e81) worms rescued with the indicated unc-18 transgene was assessed as described above. In comparison to rescue with wild-type unc-18 (Wt) worms expressing either S322E or R39C, unc-18 point mutations were paralyzed at a lower temperature (*p < 0.01) whereas those expressing S322A unc-18 were paralyzed at a higher temperature (*p < 0.01). There was difference in paralysis temperature between wild-type and E465K unc-18 worms. D, Locomotion rates at 28°C (open bars) were quantified as a percentage of locomotion at room temperature (20°C; filled bars) as described above. In comparison to unc-18 (e81) worms transgenically expressing wild-type (Wt) unc-18, worms expressing unc-18 with S322E or R39C point mutations moved significantly slower at elevated temperatures (*p < 0.01). Transgenic rescues expressing unc-18 with the S322A point mutation moved significantly faster at elevated temperatures (*p < 0.01). There was no difference in paralysis temperature between wild-type and E465K unc-18 worms.
The measured differences in paralysis temperature were significant, but limited. Therefore, we revised our protocol to assay locomotion rates at 28°C in an acutely acclimatized animal as a normalized percentage of room temperature (20°C) locomotion. The change in protocol served two purposes. First, we reasoned that the small differences seen for paralysis temperature during a rapid temperature upstep might be amplified for animals acutely acclimatized to the higher temperature. Second, we wanted to normalize the effects of temperature to endogenous locomotion rates, as wild-type Bristol N2 animals moved intrinsically faster than all three PKC mutants. We selected 28°C, as preliminary experiments determined it as an optimal temperature for demonstrating departure from room temperature locomotion rate in acclimatized animals. In agreement with earlier results, pkc-2 worms had a significantly enhanced rate of locomotion at 28°C in comparison to wild type (Fig. 4B). We then verified that this enhancement in temperature-sensitivity of locomotion was specifically the result of pkc-2 loss-of-function by testing the transgenic pkc-2 rescue worms and found that this completely eliminated the enhancement in temperature sensitivity of locomotion for pkc-2-null worms, reverting the temperature phenotype to a level statistically indistinguishable from that of wild type (Fig. 4B). Our control, hsf-1, was completely paralyzed at 28°C, as were the pkc-1 (nu448) worms. Using this revised protocol revealed that the alternative loss-of-function pkc-1 allele (nj3) also had an increased temperature-sensitivity, confirming that pkc-1 mutant worms demonstrate the opposite temperature phenotype to pkc-2 worms.
As UNC-18 could conceivably be phosphorylated by either PKC, we next examined whether our Ser322 phosphomutants would phenocopy either the pkc-2 or pkc-1 thermosensory effect. In the thermal paralysis assay, blocking PKC phosphorylation with the UNC-18 S322A transgenic mutant increased the temperature threshold for locomotion in comparison to rescues with wild type UNC-18 (Fig. 4C). The phosphomimetic UNC-18 S322E had the opposite phenotype, with paralysis occurring at a significantly decreased temperature. A further reduction from the wild-type worm phenotype would be expected if endogenous phosphorylation was not 100%, a result compatible with dynamics of Munc18-1 phosphorylation (de Vries et al., 2000; Craig et al., 2003). Phenocopy of pkc-2 was also evident when assessing the rate of locomotion at 28°C as a percentage of room temperature locomotion. UNC-18 S322A mutants were barely affected by the temperature increase, whereas S322E mutants were completely paralyzed at 28°C (Fig. 4D). Biochemically, the S322E mutation decreased binding to closed conformation syntaxin, similar to that seen with the R39C mutation in either Munc18-1 or UNC-18 (Fisher et al., 2001; Johnson et al., 2009). Transgenic rescue of unc-18 (e81)-null worms with a plasmid expressing unc-18 R39C produced an identical temperature-dependent locomotor phenotype as the S322E worms (Fig. 4C,D). R39C worms were paralyzed at a lower temperature than wild types, and locomotion at 28°C was completely abolished. The effects of transgenic UNC-18 expression were specific to the mutations introduced, as an E465K mutation had no effect on thermosensitivity of C. elegans locomotion (Fig. 4C,D) despite its having an aldicarb resistance phenotype (Graham et al., 2011). Taken together, these results indicate that the nonphosphorylatable UNC-18 S322A mutant indeed phenocopies PKC-2 loss-of-function to reduce the temperature sensitivity of locomotion, and that the effect is likely the result of a phosphorylation-dependent decrease in binding to closed-conformation syntaxin.
Temperature sensitivity of locomotion is dependent on diacylglycerol levels
The increased temperature sensitivity of UNC-18 S322E phosphomimetic mutants implies that endogenous levels of wild-type phosphorylation is not absolute. Therefore, mutations that increase upstream PKC activation should have increased temperature sensitivity in comparison to wild-type worms. As cPKCs require coactivation by both calcium and DAG, we analyzed mutations that block the upstream intracellular removal of DAG for changes in temperature sensitivity. DAG can be removed from cells by diacylglycerol kinases through its conversion into phosphatidic acid. There are five dgk genes in C. elegans (dgk-1–5), of which at least two have previously been associated with temperature sensation (Okochi et al., 2005; Biron et al., 2006). Testing specific loss-of-function mutants for dgk-1, 2, and 3 determined that only dgk-2 increased temperature sensitivity in comparison to wild type and phenocopied the gain-of-function mutation seen for S322E UNC-18 (Fig. 5). The enhanced temperature sensitivity was specific for the dgk-2 loss-of-function allele, as transgenic dgk-2 worms expressing dgk-2 pan-neuronally had their temperature sensitivity enhanced to a level similar to that of pkc-2-null worms (Fig. 5). As a reduction in neuronal DAG levels should occur in response to transgenic overexpression of dgk-2, this decrease in temperature sensitivity is consistent with the interpretation that endogenous phosphorylation of UNC-18 is not absolute. We then tested whether this dgk-2-specific phenotype was responsible for the increased sensitivity of UNC-18 S322E worms by creating transgenic worms expressing the Ser322 phosphomutants in the dgk-2 genetic background. There was no additive increase in dgk-2 temperature sensitivity upon expression of UNC-18 S322E; however, UNC-18 S322A increased temperature sensitivity to a level comparable with pkc-2-null mutants (Fig. 5). Taken together these data support the conclusion that control of temperature dependency of locomotion via PKC-2 phosphorylation of UNC-18 is downstream of DAG signaling.
Figure 5.

Changes in diacylglycerol levels affect temperature sensitivity of locomotion. Locomotion rates at 28°C (open bars) were quantified as a percentage of locomotion at room temperature (20°C; filled bars) as described in previous figures. Wild-type N2 and loss-of-function pkc-2 worms were compared to loss-of-function mutations in individual diacylglycerol kinase mutant worms. There was a specific increase in temperature-sensitivity of dgk-2 worms in comparison to wild-type, dgk-1, or dgk-3 (*p < 0.01). Transgenic overexpression of dgk-2 pan-neuronally (dgk-2 + Prab-3::dgk-2) or expression of the UNC-18 S322A phosphomutant in the dgk-2 background (dgk-2 + unc-18 S322A) decreased temperature sensitivity of the dgk-2 allele to a level similar to that of the pkc-2-null phenotype (*p < 0.01). Transgenic expression of the UNC-18 S322E phosphomutant (dgk-2 + unc-18 S322E) had an increased temperature sensitivity in comparison to wild-type (*p < 0.01) but were statistically equivalent to the dgk-2-null phenotype. Significance was assessed by Kruskal–Wallis one-way ANOVA and post hoc comparison of means by Mann–Whitney U test.
Phenotypic effects of blocking UNC-18 Ser322 phosphorylation is cell specific
Transgenic worms were next created overexpressing the UNC-18 Ser322 phosphomutants pan-neuronally in a Bristol N2 genetic background. Expression of UNC-18 S322A pan-neuronally increased locomotion rate at 28°C, making the temperature dependency of wild-type locomotion indistinguishable from that of pkc-2-null worms indicating that S322A overexpression could convert wild-type worms into the pkc-2-null phenotype (Fig. 6A). Conversely, UNC-18 S322E expression in wild-type worms caused an increase in temperature sensitivity consistent with the interpretation that endogenous phosphorylation of UNC-18 is not absolute (Fig. 6A). We also expressed the UNC-18 Ser322 phosphomutants pan-neuronally in the pkc-2 genetic background and confirmed that while UNC-18 S322A expression was not additive with the pkc-2 temperature phenotype, UNC-18 S322E expression was able to convert the pkc-2 worms to a phenotype similar to that seen with wild-type worms (Fig. 6A).
Figure 6.

AFD-specific block of UNC-18 Ser322 phosphorylation converts wild-type temperature-sensitivity into a pkc-2 loss-of-function phenotype. A, Locomotion rates at 28°C (open bars) were quantified as a percentage of locomotion at room temperature (20°C; filled bars) as described in previous figures. Wild-type (N2) and pkc-2-null worms were transgenically engineered to overexpress the UNC-18 phosphomutants. In comparison to untransformed wild types, worms expressing S322A either pan-neuronally (+unc-18 S322A) or specifically in AFD neurons (+Pafd unc-18 S322A) moved significantly faster at 28°C (*p < 0.01) and were statistically indistinguishable from either pkc-2-null worms or pkc-2 worms expressing S322A. The locomotion rate at 28°C of pkc-2 worms and pkc-2 worms expressing S322A (+unc = 18 S322A) were not statistically different from each other. Conversely, expression of S322E (+unc-18 S322E) pan neuronally increased temperature sensitivity (*p < 0.01) in either the Bristol N2 or pkc-2-null background. Temperature sensitivity of locomotion in N2 worms expressing S322E (+unc-18 S322E) was increased in comparison to pkc-2 worms expressing S322E (*p < 0.01). Significance was assessed by Kruskal–Wallis one-way ANOVA and post hoc comparison of means by Mann–Whitney U test. B, Photograph depicts wild-type worm expressing GFP specifically in the AFD neurons. Bristol N2 worms were transgenically engineered to express UNC-18 S322A in the AFD neurons using a gcy-8 promoter. Successful transformation was identified by co-injection of a GFP reporter plasmid under the control of the same promoter.
Finally, we determined whether the reduction in temperature dependence of locomotion caused by blocking Ser322 phosphorylation had a cell-specific point of origin. Temperature sensation in C. elegans involves pairs of parallel sensory neurons, the AFD neurons that respond specifically to increases in temperature (Kimura et al., 2004), and the olfactory AWC neurons whose intracellular calcium levels fluctuate in response to changes in temperature (Kuhara et al., 2008). Downstream of these neurons are a number of interneurons involved in sensory integration and processing, eventually synapsing onto the motor neurons (Mori et al., 2007). PKC-2 is expressed in many, but not all, neurons, including head sensory neurons anterior and posterior to the nerve ring (Islas-Trejo et al., 1997). We determined whether an AFD-specific block of UNC-18 phosphorylation could also convert wild-type N2 worms to a pkc-2-null phenotype by expressing UNC-18 S322A in the AFD thermosensory neurons using a gcy-8 promoter (Fig. 6B) and found that AFD-specific expression of the S322A mutant UNC-18 was as effective in conversion of wild-type phenotype as pan-neuronal expression (Fig. 6A). This result does not exclude phosphorylation of UNC-18 in other cells of the organism having phenotypic effects; however, it is consistent with the temperature-dependent effect on locomotion having its origin solely in the AFD thermosensory neuron.
Discussion
DAG signaling is intrinsically involved in the regulation of thermosensory signaling in C. elegans (Okochi et al., 2005; Biron et al., 2006); however, the downstream effectors for DAG remain uncharacterized. Our results suggest that DAG signaling occurs through PKC-2 activation, resulting in Ser322 phosphorylation of the essential exocytotic protein UNC-18. Mutational and binding experiments point to phosphorylation mechanistically reducing closed-conformation binding to syntaxin, as is the case for phosphorylation of the mammalian ortholog Munc18-1 (Fujita et al., 1996; Barclay et al., 2003). Ser322 is contained within domain 3a of UNC-18, a region of the protein implicated in both 1:1 binding to syntaxin (Misura et al., 2000), but also interaction with the assembled SNARE complex (Rathore et al., 2010; Hu et al., 2011). The R39C mutation of Munc18-1/UNC-18 however reduces binding to isolated syntaxin (Fisher et al., 2001; Johnson et al., 2009), but not the SNARE complex (Shen et al., 2007). Phenocopy of the temperature dependency of locomotion by the R39C mutation then supports the interpretation that the effects of the S322E mutation are due to changes in closed-conformation binding, although concomitant effects on SNARE binding cannot be excluded. We have previously shown that closed-conformation binding of UNC-18 to syntaxin is not the essential function of the protein in exocytosis (Johnson et al., 2009), in agreement with previous results (Weimer et al., 2003). Nevertheless, our data point to an important regulatory role for UNC-18 phosphorylation in response to DAG signaling, possibly limited to specific cells or under certain environmental conditions. Phosphorylation of Munc18-1 can increase vesicle pool replenishment (Nili et al., 2006) and alter fusion pore dynamics (Barclay et al., 2003). Either of these changes to exocytosis, via classical or peptidergic transmission, could alter the signaling dynamics from the AFD thermosensory neurons, ultimately regulating the downstream locomotor patterns and behavior of the organism.
Our data point to a specific signaling pathway involving DAG activation of the classical PKC, PKC-2, and phosphorylation of UNC-18 in the regulation of acute temperature sensitivity of locomotion. It remains possible however that these effects on locomotion rate may be different in response to increased duration or more severe thermal challenges. Any role of the DAG-PKC-2 signaling pathway in longer-term changes to temperature sensitivity then remains to be formally tested. In addition to pkc-2, however, a role for pkc-1 was also implicated. Analysis of two independent null alleles confirmed that pkc-1 mutants display the opposite phenotype to that seen for pkc-2, an increase in temperature sensitivity. Although the phenotype was indeed consistent for both pkc-1 alleles, there was a noticeable difference in the magnitude of the phenotype. Such a difference may be the result of secondary mutations, inherent strain differences, or even differences in the specific allele mutation. PKC-1 is expressed in the AFD neurons, and loss-of-function pkc-1 worms display thermophilic thermotaxis (Okochi et al., 2005). Therefore, a role for pkc-1 in temperature sensitivity is perhaps unsurprising. It is unexpected, however, that loss-of-function mutations in two similar DAG-activated PKC isoforms have contrary phenotypes. This suggests the possibility that distinct DAG pathways, possibly within the same cells, could regulate temperature-dependent phenotypes in different ways. Indeed, three separate thermal phenotypes in C. elegans have specifically implicated different dgk isoforms, dgk-1 (Okochi et al., 2005), dgk-2 (this paper), and dgk-3 (Biron et al., 2006).
We have characterized Ser322 of UNC-18 as an identified phosphorylation site sufficient to determine regulation of temperature-dependent phenotypes. Phosphorylation of UNC-18 Ser322, however, was not involved in DAG-regulated potentiation of signaling strength at the neuromuscular junction, although we have demonstrated a novel role for activation of PKC-2. Original work in this area pointed to DAG acting through the C1 domain-containing synaptic protein UNC-13 (Lackner et al., 1999; Nurrish et al., 1999). More recent work has also implicated PKC-1; however, effector targets for PKC-1 were left uncharacterized, although both UNC-18 and SNAP-25 were excluded (Sieburth et al., 2007). Our results support the conclusion that UNC-18 is not a part of the pathway underlying DAG stimulation of neuromuscular junction signaling. It remains possible, however, that synergistic phosphorylation of multiple sites within the same protein, or indeed among distinct proteins, could be either the PKC-1 or PKC-2 targets within motor neurons.
Sensory signaling and resultant behavior can be regulated via DAG signaling pathways within diverse sensory neurons. In C. elegans, for example, odor preference can be altered by DAG signaling in the AWC neurons through pkc-1 activation, resulting in altered postsynaptic calcium responses (Tsunozaki et al., 2008). Olfactory adaptation in AWC neurons also requires DAG signaling as dgk-3; dgk-1 double mutants have defective adaptation (Matsuki et al., 2006). Regulation of mechanoreception in C. elegans sensory neurons utilizes a DAG pathway, signaling through pkc-1, to alter evoked calcium transients (Kindt et al., 2007). In each instance, the targets of the DAG signaling pathway should affect a presynaptic alteration to exocytosis/synaptic transmission that results in altered postsynaptic activation; however, the involved downstream effectors have not been identified. We characterize here a specific downstream effector, identified as a prime component of the exocytotic machinery, in the regulation of C. elegans behavior.
Thermosensation in C. elegans involves the integration of bidirectional sensory information from both inhibitory AFD and stimulatory AWC thermosensory neurons (Ohnishi et al., 2011). AFD neurons can also regulate the heat shock response of somatic cells, likely through long-range neuroendocrine signaling (Prahlad et al., 2008). Synaptic transmission from the AFD neuron is both glutamatergic (Ohnishi et al., 2011) and peptidergic (Narayan et al., 2011) in nature. Regulation of thermosensation in C. elegans can originate from the AFD neurons via cell-specific DAG signaling (Okochi et al., 2005; Biron et al., 2006). Although the upstream molecular components of thermosensation within AFD have been extensively characterized, identification of regulatory downstream effectors has thus far been lacking. AFD neurons discriminate differences in temperature by raising calcium concentration transiently in response to warming (Kimura et al., 2004). This increase in calcium, in turn, would activate PKC-2, as it is the only calcium-dependent cPKC within the C. elegans genome, thus initiating reversible phosphorylation of downstream targets such as UNC-18. Using genetic approaches, we have demonstrated that AFD cell-specific, PKC-dependent phosphorylation of UNC-18 regulates the thermosensitivity of C. elegans locomotion. Interestingly, thermosensitivity of larval locomotion in Drosophila can be altered by increased activity of the serine/threonine protein phosphatase PP2A (Dawson-Scully et al., 2007), which would be an alternative route to regulate levels of PKC-dependent phosphorylation. We conclude that dynamic regulation of animal behavior in response to changing environmental temperature can occur via specific PKC-2-dependent phosphorylation of the exocytotic protein UNC-18 in a single thermosensory neuron pair.
Footnotes
This work was supported by grants from the Biotechnology and Biological Sciences Research Council (A.M. and R.D.B.) Wellcome Trust (J.W.B., A.M., and R.D.B.) and a Research Councils UK Academic Fellowship (J.W.B.). M.R.E. was supported by a Medical Research Council Studentship. C. elegans strains used in this work was provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources. We thank Prof. Kevin Park (University of Liverpool, Liverpool, UK) for access to mass spectrometry facilities.
References
- Barclay JW, Craig TJ, Fisher RJ, Ciufo LF, Evans GJ, Morgan A, Burgoyne RD. Phosphorylation of Munc18 by protein kinase C regulates the kinetics of exocytosis. J Biol Chem. 2003;278:10538–10545. doi: 10.1074/jbc.M211114200. [DOI] [PubMed] [Google Scholar]
- Biron D, Shibuya M, Gabel C, Wasserman SM, Clark DA, Brown A, Sengupta P, Samuel AD. A diacylglycerol kinase modulates long-term thermotactic behavioral plasticity in C. elegans. Nat Neurosci. 2006;9:1499–1505. doi: 10.1038/nn1796. [DOI] [PubMed] [Google Scholar]
- Bracher A, Weissenhorn W. Structural basis for the Golgi membrane recruitment of Sly1p by Sed5p. EMBO J. 2002;21:6114–6124. doi: 10.1093/emboj/cdf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Betz A, Wegmeyer H. Divergent and convergent signaling by the diacylglycerol second messenger pathway in mammals. Curr Opin Neurobiol. 2004;14:328–340. doi: 10.1016/j.conb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Craig TJ, Evans GJ, Morgan A. Physiological regulation of Munc18/nSec1 phosphorylation on serine-313. J Neurochem. 2003;86:1450–1457. doi: 10.1046/j.1471-4159.2003.01955.x. [DOI] [PubMed] [Google Scholar]
- Dawson-Scully K, Armstrong GA, Kent C, Robertson RM, Sokolowski MB. Natural variation in the thermotolerance of neural function and behavior due to a cGMP-dependent protein kinase. PLoS One. 2007;2:e773. doi: 10.1371/journal.pone.0000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong AP, Verhage M. Presynaptic signal transduction pathways that modulate synaptic transmission. Curr Opin Neurobiol. 2009;19:245–253. doi: 10.1016/j.conb.2009.06.005. [DOI] [PubMed] [Google Scholar]
- de Vries KJ, Geijtenbeek A, Brian EC, de Graan PN, Ghijsen WE, Verhage M. Dynamics of munc18-1 phosphorylation/dephosphorylation in rat brain nerve terminals. Eur J Neurosci. 2000;12:385–390. doi: 10.1046/j.1460-9568.2000.00931.x. [DOI] [PubMed] [Google Scholar]
- Fisher RJ, Pevsner J, Burgoyne RD. Control of fusion pore dynamics during exocytosis by Munc18. Science. 2001;291:875–878. doi: 10.1126/science.291.5505.875. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Sasaki T, Fukui K, Kotani H, Kimura T, Hata Y, Südhof TC, Scheller RH, Takai Y. Phosphorylation of Munc-18/n-Sec1/rbSec1 by protein kinase C. J Biol Chem. 1996;271:7265–7268. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- Graham ME, Edwards MR, Holden-Dye L, Morgan A, Burgoyne RD, Barclay JW. UNC-18 modulates ethanol sensitivity in Caenorhabditis elegans. Mol Biol Cell. 2009;20:43–55. doi: 10.1091/mbc.E08-07-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ME, Prescott GR, Johnson JR, Jones M, Walmesley A, Haynes LP, Morgan A, Burgoyne RD, Barclay JW. Structure–function study of mammalian Munc18-1 and C. elegans UNC-18 implicates domain 3b in the regulation of exocytosis. PLoS One. 2011;6:e17999. doi: 10.1371/journal.pone.0017999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdu-Cronin YM, Chen WJ, Sternberg PW. The L-type cyclin CYL-1 and the heat-shock-factor HSF-1 are required for heat-shock-induced protein expression in Caenorhabditis elegans. Genetics. 2004;168:1937–1949. doi: 10.1534/genetics.104.028423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V, Di Paolo G. Lipids and lipid modifications in the regulation of membrane traffic. Curr Opin Cell Biol. 2007;19:426–435. doi: 10.1016/j.ceb.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SH, Latham CF, Gee CL, James DE, Martin JL. Structure of the Munc18c/Syntaxin4 N-peptide complex defines universal features of the N-peptide binding mode of Sec1/Munc18 proteins. Proc Natl Acad Sci U S A. 2007;104:8773–8778. doi: 10.1073/pnas.0701124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LH, Collins BM, Martin JL. Possible roles for Munc18-1 domain 3a and Syntaxin1 N-peptide and C-terminal anchor in SNARE complex formation. Proc Natl Acad Sci U S A. 2011;108:1040–1045. doi: 10.1073/pnas.0914906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islas-Trejo A, Land M, Tcherepanova I, Freedman JH, Rubin CS. Structure and expression of the Caenorhabditis elegans protein kinase C2 gene: origins and regulated expression of a family of Ca2+-activated protein kinase C isoforms. J Biol Chem. 1997;272:6629–6640. doi: 10.1074/jbc.272.10.6629. [DOI] [PubMed] [Google Scholar]
- Johnson JR, Ferdek P, Lian LY, Barclay JW, Burgoyne RD, Morgan A. Binding of UNC-18 to the N-terminus of syntaxin is essential for neurotransmission in Caenorhabditis elegans. Biochem J. 2009;418:73–80. doi: 10.1042/BJ20081956. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Miyawaki A, Matsumoto K, Mori I. The C. elegans thermosensory neuron AFD responds to warming. Curr Biol. 2004;14:1291–1295. doi: 10.1016/j.cub.2004.06.060. [DOI] [PubMed] [Google Scholar]
- Kindt KS, Quast KB, Giles AC, De S, Hendrey D, Nicastro I, Rankin CH, Schafer WR. Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron. 2007;55:662–676. doi: 10.1016/j.neuron.2007.07.023. [DOI] [PubMed] [Google Scholar]
- Kuhara A, Okumura M, Kimata T, Tanizawa Y, Takano R, Kimura KD, Inada H, Matsumoto K, Mori I. Temperature sensing by an olfactory neuron in a circuit controlling behavior of C. elegans. Science. 2008;320:803–807. doi: 10.1126/science.1148922. [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gqalpha and EGL-8 PLCbeta: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. [DOI] [PubMed] [Google Scholar]
- Mahoney TR, Liu Q, Itoh T, Luo S, Hadwiger G, Vincent R, Wang ZW, Fukuda M, Nonet ML. Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol Biol Cell. 2006;17:2617–2625. doi: 10.1091/mbc.E05-12-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki M, Kunitomo H, Iino Y. Goalpha regulates olfactory adaptation by antagonizing Gqα-DAG signaling in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2006;103:1112–1117. doi: 10.1073/pnas.0506954103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- Morgan A, Burgoyne RD, Barclay JW, Craig TJ, Prescott GR, Ciufo LF, Evans GJ, Graham ME. Regulation of exocytosis by protein kinase C. Biochem Soc Trans. 2005;33:1341–1344. doi: 10.1042/BST0331341. [DOI] [PubMed] [Google Scholar]
- Mori I, Sasakura H, Kuhara A. Worm thermotaxis: a model system for analyzing thermosensation and neural plasticity. Curr Opin Neurobiol. 2007;17:712–719. doi: 10.1016/j.conb.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Narayan A, Laurent G, Sternberg PW. Transfer characteristics of a thermosensory synapse in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2011;108:9667–9672. doi: 10.1073/pnas.1106617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nili U, de Wit H, Gulyas-Kovacs A, Toonen RF, Sørensen JB, Verhage M, Ashery U. Munc18-1 phosphorylation by protein kinase C potentiates vesicle pool replenishment in bovine chromaffin cells. Neuroscience. 2006;143:487–500. doi: 10.1016/j.neuroscience.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Nurrish S, Ségalat L, Kaplan JM. Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron. 1999;24:231–242. doi: 10.1016/s0896-6273(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Ohnishi N, Kuhara A, Nakamura F, Okochi Y, Mori I. Bidirectional regulation of thermotaxis by glutamate transmissions in Caenorhabditis elegans. EMBO J. 2011;30:1376–1388. doi: 10.1038/emboj.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okochi Y, Kimura KD, Ohta A, Mori I. Diverse regulation of sensory signaling by C. elegans nPKC-epsilon/eta TTX-4. EMBO J. 2005;24:2127–2137. doi: 10.1038/sj.emboj.7600697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Cornelius T, Morimoto RI. Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science. 2008;320:811–814. doi: 10.1126/science.1156093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore SS, Bend EG, Yu H, Hammarlund M, Jorgensen EM, Shen J. Syntaxin N-terminal peptide motif is an initiation factor for the assembly of the SNARE-Sec1/Munc18 membrane fusion complex. Proc Natl Acad Sci U S A. 2010;107:22399–22406. doi: 10.1073/pnas.1012997108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund C, Brose N. β Phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKC. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassa T, Ogawa H, Kimoto M, Hosono R. The synaptic protein UNC-18 is phosphorylated by protein kinase C. Neurochem Int. 1996;29:543–552. doi: 10.1016/0197-0186(96)00009-5. [DOI] [PubMed] [Google Scholar]
- Sassa T, Harada S, Ogawa H, Rand JB, Maruyama IN, Hosono R. Regulation of the UNC-18-Caenorhabditis elegans syntaxin complex by UNC-13. J Neurosci. 1999;19:4772–4777. doi: 10.1523/JNEUROSCI.19-12-04772.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 (SM) proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Sieburth D, Madison JM, Kaplan JM. PKC-1 regulates secretion of neuropeptides. Nat Neurosci. 2007;10:49–57. doi: 10.1038/nn1810. [DOI] [PubMed] [Google Scholar]
- Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuse Y. Protein kinase C isotypes in C. elegans. J Biochem. 2002;132:519–522. doi: 10.1093/oxfordjournals.jbchem.a003251. [DOI] [PubMed] [Google Scholar]
- Tabuse Y, Miwa J. A gene involved in action of tumor promoters is identified and mapped in Caenorhabditis elegans. Carcinogenesis. 1983;4:783–786. doi: 10.1093/carcin/4.6.783. [DOI] [PubMed] [Google Scholar]
- Tsunozaki M, Chalasani SH, Bargmann CI. A behavioral switch: cGMP and PKC signaling in olfactory neurons reverses odor preference in C. elegans. Neuron. 2008;59:959–971. doi: 10.1016/j.neuron.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buskirk C, Sternberg PW. Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans. Nat Neurosci. 2007;10:1300–1307. doi: 10.1038/nn1981. [DOI] [PubMed] [Google Scholar]
- van der Linden AM, Moorman C, Cuppen E, Korswagen HC, Plasterk RH. Hyperactivation of the G12-mediated signaling pathway in Caenorhabditis elegans induces a developmental growth arrest via protein kinase C. Curr Biol. 2003;13:516–521. doi: 10.1016/s0960-9822(03)00164-7. [DOI] [PubMed] [Google Scholar]
- Waggoner LE, Dickinson KA, Poole DS, Tabuse Y, Miwa J, Schafer WR. Long-term nicotine adaptation in Caenorhabditis elegans involves PKC-dependent changes in nicotinic receptor abundance. J Neurosci. 2000;20:8802–8811. doi: 10.1523/JNEUROSCI.20-23-08802.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM. Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci. 2003;6:1023–1030. doi: 10.1038/nn1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Xiao C, Mileva-Seitz V, Seroude L, Robertson RM. Targeting HSP70 to motoneurons protects locomotor activity from hyperthermia in Drosophila. Dev Neurobiol. 2007;67:438–455. doi: 10.1002/dneu.20344. [DOI] [PubMed] [Google Scholar]