

Abstract
The activities of the bacterial RecA protein are involved in the de novo development and transmission of antibiotic resistance genes, thus allowing bacteria to overcome the metabolic stress induced by antibacterial agents. RecA is ubiquitous and highly conserved among bacteria, but has only distant homologs in human cells. Together, this evidence points to RecA as a novel and attractive antibacterial drug target. All known RecA functions require the formation of a complex formed by multiple adenosine 5′-O-triphosphate (ATP)-bound RecA monomers on single-stranded DNA. In this complex, RecA hydrolyzes ATP. Although several methods for assessing RecA's ATPase activity have been reported, these assay conditions included relatively high concentrations of enzyme and ATP and thereby restricted the RecA conformational state. Herein, we describe the validation of commercial reagents (Transcreener® adenosine 5′-O-diphosphate [ADP]2 fluorescence polarization assay) for the high-throughput measurement of RecA's ATPase activity with lower concentrations of ATP and RecA. Under optimized conditions, ADP detection by the Transcreener reagent provided robust and reproducible activity data (Z′=0.92). Using the Transcreener assay, we screened 113,477 small molecules against purified RecA protein. In total, 177 small molecules were identified as confirmed hits, of which 79 were characterized by IC50 values ≤10 μM and 35 were active in bioassays with live bacteria. This set of compounds comprises previously unidentified scaffolds for RecA inhibition and represents tractable hit structures for efforts aimed at tuning RecA inhibitory activity in both biochemical and bacteriological assays.
Introduction
New antibacterial strategies will be required to overcome the looming public health threat posed by the combination of an increasing prevalence of antibiotic-resistant bacterial pathogens with a dwindling pipeline of new antibiotics.1,2 Significant scientific and environmental challenges remain in the discovery and development of novel mechanism antibiotics.3 One alternative to conventional antibiotic discovery would be the development of adjuvants to enhance the outcomes of antibacterial therapy. Recent studies demonstrate that bacterial strains with inactive RecA enzyme are more susceptible than wild-type strains to killing by antibacterial agents.4–7 Moreover, loss of RecA function also attenuates the rates of induced mutagenesis and intrachromosomal recombination upon antibiotic exposure, thereby slowing the development of antibiotic resistance.6–8 RecA inactivation also diminishes the efficiency of horizontal gene transfer, hindering the acquisition and dissemination of antibiotic resistance genes.9–11 Given this evidence, we hypothesized that small-molecule RecA inhibitors could sensitize bacteria to conventional antibiotics and attenuate the frequency with which resistance genes develop and are transmitted.12 The discovery of potent and selective RecA inhibitors that modulate the target in living bacteria would be an important step in establishing RecA as a druggable target in the management of bacterial infectious diseases.
RecA's importance in the bacterial survival of and response to antibacterial exposure arise from its cardinal roles in mediating the SOS response and facilitating DNA strand exchange. All RecA activities require the formation of a helical homopolymeric filament comprising multiple adenosine 5′-O-triphosphate (ATP)-bound RecA monomers coating single-stranded DNA (ssDNA).13 Once formed, this RecA-DNA filament (RDF) hydrolyzes ATP. Thus, monitoring ATPase activity can be used as a diagnostic for small molecule inhibition of RecA by in vitro screening.14–17 These previously reported assay technologies had sensitivity limitations, requiring high concentrations of enzyme ([RecA]≥0.5 μM) and substrate ([ATP]≥0.75 mM). Under such forcing conditions, RecA exists almost exclusively in an active, DNA-bound conformational state, and inhibitors selective for this conformation (Fig. 1A) were primarily identified.
Fig. 1.

Cartoons depicting the inactive RecA monomers and active RecA-DNA filament (RDF). (A) Assay conditions, including high concentrations of RecA, DNA, and ATP, shift the filament assembly and activation equilibrium such that RecA exists almost exclusively in an active, DNA-bound conformational state. Inhibitors identified under such forcing conditions will be strongly biased toward binders of this conformational state. (B) The desired assay conditions include 10-fold lower concentrations of RecA and ATP, and RecA samples both its inactive and active conformational states during the assay. Such nonforcing conditions would facilitate the identification of additional inhibitors without bias with respect to RecA conformational preference as well as ATP-competitive inhibitors. ATP; adenosine 5′-O-triphosphate.
The development of a more sensitive screening assay with the ability to detect low ATP turnover under reduced RecA and ATP concentrations was desired. Under such nonforcing conditions, RecA could sample both its inactive and active conformational states, facilitating the identification of additional small molecule inhibitors without bias with respect to RecA conformational preference (Fig. 1B). Moreover, a reduced enzyme concentration would lower the stoichiometric limit on measured IC50 values, and reduced ATP levels would allow the identification of ATP competitive inhibitors.
Assay optimization under these desired conditions represented a challenge because the equilibrium constant for RecA self-association during filament assembly and activation is modest and its turnover number for ATP hydrolysis is low (kcat≤0.5 s−1). The robust assessment of RecA's ATPase activity under such conditions required a sensitive detection methodology.
The Transcreener® adenosine 5′-O-diphosphate (ADP)2 fluorescence polarization (FP) assay from BellBrook Laboratories (Madison, WI) is a screening assay kit that allows ADP detection using an ADP-antibody/tracer system with FP readout. Briefly, the Transcreener ADP2 FP assay utilizes an antibody that selectively binds ADP to quantify the ADP produced from an ATPase reaction. ADP conjugated to AlexaFluor-633 dye is used as a tracer, and the amount of tracer that is displaced from the antibody is proportional to the amount of ADP generated in the reaction. This highly sensitive, homogeneous assay technology has been used to study protein kinases,18 lipid kinases,19 and heat shock proteins.20 Moreover, the assays have proven to be suitable for high-throughput screening (HTS).21 In the present study, we report the adaptation of the Transcreener ADP2 FP assay to detect the ATP hydrolysis activity of purified Escherichia coli RecA protein. We demonstrate the ability of the assay technology to be optimized for lower enzyme and ATP concentrations and its use in HTS of a diverse collection of drug-like small molecules, leading to the identification of novel RecA inhibitor scaffolds.
Materials and Methods
Materials
The Transcreener ADP2 FP assay kit (cat. no. 3010-10K) was purchased from BellBrook Labs. Polydeoxythymidylic acid (Poly[dT]) ssDNA was purchased from The Midland Certified Reagent Company (Midland, TX). RecA was purified and stored as previously described.22 Unless otherwise stated, all other reagents used for buffers and assays were purchased from Fisher Scientific International (Ipswich, MA).
LOPAC Compound Collection
The Library of Pharmacologically Active Compounds (LOPAC) was purchased from Sigma-Aldrich as 10-mM stocks in dimethyl sulfoxide (DMSO). The library was previously prepared as 1-μL samples in 384-well V-bottom polypropylene microplates (Greiner, Monroe, NC), sealed by an ALPS 3000 microplate heat sealer (Thermo Fisher Scientific, Hudson, NH) and stored at −20°C. On the day of use, the compounds were thawed and diluted to 150 μM (10× final concentration) in reaction buffer (R buffer: 25 mM Tris-HOAc, 10 mM Mg (OAc)2, 1 mM DTT, 5% glycerol, and 0.01% TritonX-100) over two steps using a Thermo Scientific MultidropCombi Reagent Dispenser (Waltham, MA) and Multimek NSX-1536 assay workstation system fitted with a 384-well head (Nanoscreen, Charleston, SC). Finally, 1 μL of this stock was spotted into the wells of a 384-well black PerkinElmer Proxiplate (Waltham, MA) for assay use, as described below.
100k Diversity Screening Compound Collection
The 100k Diversity Collection of screening compounds was developed by structural chemists from St. Jude and the Center for Integrative Chemical Biology and Drug Discovery (CICBDD). Compounds were selected based on structural diversity at the Murcko scaffold level.23 Essentially, a compound's Murcko scaffold includes contiguous ring systems plus chains that link two or more rings. For Murcko scaffolds with more than 20 compounds, 20 compounds were randomly selected for that scaffold to maximize the diversity of scaffolds in the Diversity Collection. Compounds were also filtered to eliminate reactive function groups (REOS score >−2)24 and include compounds that obey the “rule of five”25 with slight deviations to permit slightly larger and more lipophilic compounds. Based on the above selection process, a set of 100k compounds was chosen and purchased from Enamine Ltd (Kiev, Ukraine) in powder stock. At random, around 5% of the collection was examined by LC/MS for identity and purity confirmation. The compound collection plates were prepared by resuspending the powder stock to 1 mM in DMSO in barcoded glass vials with sonication using a Covaris S2 (Covaris, Woburn, MA). Compounds were plated at 1 mM in 100% DMSO in 384-well V-bottom polypropylene microplates using a Tecan Genesis 200 (Münnedorf, Switzerland). A Multimek spotted 1 μL of the 1 mM compounds into 384-well V-bottom polypropylene microplates and plates were heat-sealed and stored at −20°C. During screening, these previously prepared plates were thawed and diluted to 150 μM by a single addition of R buffer, and 1 μL of this stock was spotted into the wells of a 384-well Proxiplate.
Kinase Focus Set
The Kinase Focus Set was designed by the CICBDD based upon a combination of kinase pharmacophore-based searching and selection from vendor kinase directed sets. The 4,727 compounds chosen for the Kinase Focus Set were distinct from any in the Diversity Collection and all were “rule of five” compliant.25 The Kinase Focus Set was acquired from several vendors in powder stock. Compound and assay plates were prepared as same as the 100k Diversity Collection.
Transcreener Assay
The assay was performed using a Multidrop dispenser to add 1 μL of 10% DMSO in the left two columns (columns 1 and 2) for positive control reactions. In the right two columns (columns 23 and 24), 1 μL of 500 mM ethylenediaminetetraacetic acid (EDTA) was spotted for negative control reactions. Assay reactions were carried out directly in the Proxiplate wells containing 1-μL compounds or controls. The reaction volume in each well was 10 μL, and the final concentration of the compound and DMSO was 15 μM and 0.1%, respectively. A 2× solution of RecA was prepared in R buffer. To all wells of the assay plates, 4.5 μL of the 2× RecA in R buffer was added using a Multidrop dispenser. RecA was allowed to preincubate with compounds and controls for 20 min in a 37°C air incubator. After preincubation, a 2× cocktail of ATP and poly(dT) ssDNA in R buffer was added in a volume of 4.5 μL to each well. Immediately upon addition of the ATP/poly(dT) mixture, assay plates were again transferred to the 37°C air incubator and the reaction was allowed to proceed for 30 min. After 30 min, the plates were removed and quenched by adding 10 μL of 2× Transcreener detection buffer (D buffer: 50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid [HEPES], pH 7.5, 400 mM NaCl, 0.02% Brij-35, and 20 mM EDTA) with 4 nM Transcreener AlexaFluor 633 ADP tracer, and 75 μg/mL ADP2 antibody. To ensure uniformity of results, no more than 20 plates were processed at a time during screening and plates were incubated at ambient temperature for 30 min before reading on an EnVision Multilabel Reader (PerkinElmer) using far-red FP filter set.
Compounds for dose–response runs were resuspended to 10 mM in DMSO in barcoded glass vials. The representative selection hits and Kinase Focus Set hits were sonicated using the Covaris S2 and plated as 3-fold dilutions over 10 points using the Tecan in 384-well V-bottom polypropylene microplates. The remaining hits were resuspended and placed in a sonicating water bath if needed for solubility. The dose–response curves of the remaining hits were prepared as 3-fold dilutions over 10 points using a multi-channel pipette. Compound titration series of all hits for IC50 evaluation were spotted at 1 μL by a Multimek instrument into 384-well V-bottom microplates and diluted 20-fold in R buffer using a Multidrop dispenser. The diluted titrations were then spotted at 1 μL into 384-well black Proxiplates in duplicate or triplicate, and reagents were added to initiate the assay as described above. The final top concentration of dose–response curves was 50 μM compound (0.5% DMSO).
Data Analysis
The FP of the ADP tracer was measured using the far-red FP filter set from PerkinElmer (optimized Cy5 FP Dual Emission label, 620/40 nm excitation filter, 688/45 nm emission filter, and D658/fp688 dual mirror) with the G factor set at 0.64. The emission values were calculated to a milli-polarization (mP) signal using the following FP fit sequence:
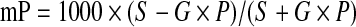 |
where G is the G-factor (0.64), S is the emission filter parallel to the excitation filter, and P is the emission filter perpendicular to the excitation filter.
Screening data were processed using ScreenAble software (Screenable Solutions, Chapel Hill, NC). The percent inhibition was determined on a plate-to-plate basis by comparing the calculated mP value per compound well with the plate-averaged control wells (n=32 for each control), using the following relationship:
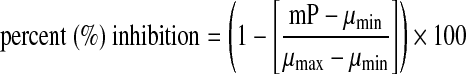 |
where mP is the compound well mP signal, and μinh and μmax are the plate-averages of the minimum signal (maximum ATPase activity, 1% DMSO added) and maximum signal (inhibited, 50 mM EDTA added) controls, respectively.
The Z′ factor was calculated for each assay plate to assess the quality and robustness of the HTS. The Z′ factor was determined using the following formula:
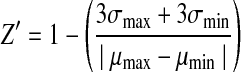 |
where σmax and σmin are standard deviations (SDs) in respective plate maximum and minimum signal controls, and μmax and μmin are the plate-averages of the respective controls, as defined above.
Compound IC50 values were calculated by first converting the mP signal to percent inhibition with respect to on-plate controls and then fitting the percent inhibition values to curve equations using ScreenAble or Prism (GraphPad Software, San Diego, CA). The IC50 values of the representative selection hits and Kinase Focus Set hits were calculated using ScreenAble with a 3- or 4-parameter curve fit. The remaining hits were analyzed in Prism using a one-site-specific binding with Hill slope and Bmin.
Selection of Representative Active Compounds
The representative compounds were selected according to the following procedure. First, all 235 actives from the 100k Diversity Collection were grouped into compact clusters of similar compounds. More specifically, the similarity between any two compounds within a cluster was at least 65% according to the Tanimoto metrics with ECFP4 fingerprints (Pipeline Pilot, ver 7.5; Accelrys Software, Inc.; 2009). The clustering procedure revealed 22 clusters (of 3–10 compounds) and 92 singletons—that is, compounds that did not have any structural analogs among the 235 hits. Despite being outliers among the hits, most of 92 singletons had numerous structural analogs that did not show any activity in the Transcreener assay. These compounds were assigned a lower priority because of a high risk of them being either false positives or intractable hits. Finally, the first priority representative subset of 78 compounds consisted of 3 compounds per cluster that showed the highest response in the primary high-throughput screening (i.e., 66 actives) and 12 true singletons (i.e., hits that did not have any structural analogs neither among other hits nor among the inactive compounds).
Results
Assay Development and Validation
Previous reaction conditions used for measuring RecA's ATPase activity required relatively high concentrations of RecA and ATP. To lower enzyme and substrate concentrations, we optimized the conditions of the Transcreener ADP2 FP assay to our desired levels of enzyme and ATP. We also assessed the robustness, reproducibility, and HTS compatibility of the Transcreener assay to ensure its success in screening collections of diverse drug-like small molecules for the inhibition of RecA.
An ADP/ATP standard curve was produced (Fig. 2A), as described in the TranscreenerADP2 FPassay technical manual.26 The experiment simulates the production of ADP during the course of a RecA-catalyzed ATP hydrolysis reaction and the expected decrease in FP signal (mP) was observed. The ATP concentration required for half-maximal steady-state velocity, or S0.5 for cooperative enzymes such as RecA,27 depends on the reaction conditions, and published values range from 2.5 to 200 μM.12,28 In the presence of poly(dT) as the activating DNA, RecA's ATPase activity was measured with initial ATP concentrations from 25 to 100 μM. It was found that 75 μM was the lowest concentration that resulted in linear (steady-state) kinetics for nearly 40 min with an acceptable signal-to-background ratio. Subsequently, under the optimized ATPase screening conditions described below, the apparent S0.5 was measured to be 88 μM (data not shown). Because a substrate concentration equal to or slightly less than the KM (S0.5) value in HTS campaigns allows the identification of all types of inhibitors, including those that are competitive with substrate,29 we selected 75 μM as the ATP concentration and optimized the other assay parameters.
Fig. 2.

Assay optimization with respect to (A) ATP/ADP signal, (B) time and enzyme dependence, and (C) DMSO tolerance determined in 384-well format. (A) ATP/ADP titration, with a constant adenosine concentration of 75 μM. (B) Reaction time course in the (•) presence or (○) absence of 30 nM RecA. (C) FP (mP) signal after 30-min RecA ATPase reaction in the presence of various amounts of DMSO. The data presented are mean±standard deviation (SD) of triplicate wells (n=3). FP, Fluorescence polarization.
In the presence of 75 μM ATP and a saturating concentration of poly(dT) (5 μM-nts), RecA's ATPase activity increased monotonically with initial RecA concentration (data not shown). A RecA concentration of 30 nM was selected for further experiments because this was the lowest RecA concentration to produce approximately linear enzyme activity over a reasonable time range and sufficient conversion to achieve an adequate signal window. Optimized conditions included 30 nM RecA, 75 μM ATP, and 5 μM-nt spoly(dT). Employing these conditions, the ADP concentration produced at 37°C was measured over time by comparison with an ADP standard curve (Fig. 2B). The Transcreener assay demonstrated linear ADP production kinetics to 40 min, and we elected to use 30 min as the reaction time to remain comfortably within the assay's linear dynamic range.
It is common for small-molecule library samples to be dissolved in DMSO in preparation for screening. Therefore, the effect of DMSO on RecA activity was investigated at several different DMSO concentrations, up to 10% (vol/vol) (Fig. 2C). There was no significant reduction in RecA ATPase activity by DMSO up to the highest concentration tested and the final DMSO concentration used for screening was 0.5%–1% (vol/vol).
To assess the reproducibility of the Transcreener assay, a number of test 384-well plates were evaluated for intercolumn, interplate, and interday variability. Over a span of 3 days, eight replicate assay plates were run under the optimized conditions described above and used to compile the mean control signals (Fig. 3) for Z′ factor calculation.30 The maximum activity controls (128 per plate, even columns of a 384-well plate) consisted of wells containing RecA, DNA, ATP, and 1% DMSO in buffer. The background or inhibited controls (128 per plate, odd columns of a 384-well plate) consisted of wells containing RecA, DNA, ATP, and 50 mM EDTA in buffer. The FP signal was read at 30 min and the overall quality of the Transcreener assay was assessed using the Z′ factor. Characteristic of a high-quality assay, the average Z′ factor was 0.85 and consistent throughout the validation run.
Fig. 3.

Assay validation over multiple plates and multiple days. Each data point represents the mean FP (mP) signal of 128 wells from 3 different experiments on 3 different days (2 or 3 plates per experiment); the error bars indicate the SD of each mean value. The (○) represent the mean values for the background or inhibited controls (50 mM EDTA final) and the (•) represent the mean values for the maximum activity controls (1% DMSO final). EDTA, ethylenediaminetetraacetic acid.
Screen Design and Pilot Study
Using the protocol outlined in Table 1, the Transcreener assay was employed to screen the LOPAC as a pilot study. The LOPAC comprises 1,280 biologically active compounds and was assessed in triplicate for RecA inhibition at a final compound concentration of 15 μM. The LOPAC pilot screen was characterized by an average Z′ factor of 0.76, and 19 compounds were identified with average relative inhibition activities ≥75% inhibition as an average of three replicates (Fig. 4A). This active threshold of 75% inhibition represented 6 SD above the mean and provided a hit rate of 1.5%. As shown in Figure 4B, the pilot screen also demonstrated strong correlation between replicates. Analysis of the three replicates revealed five compounds as false positives and an estimated false positive rate of 0.4%.
Table 1.
High-Throughput Screening Assay Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Library compound | 1 μL | 10× in R buffer containing 10% (v/v) DMSO; 15 μM final |
| 2 | Controls | 1 μL | 10×; 1% DMSO and 50 mM EDTA final |
| 3 | RecA | 4.5 μL | 2× in R buffer; 30 nM final |
| 4 | Preincubation | 20 min | 37°C |
| 5 | Adenosine 5′-O-triphosphate (ATP) and polydeoxythymidylic acid (poly[dT]) | 4.5 μL | 2× in R buffer; 75 μM ATP and 5 μM poly(dT) final |
| 6 | Incubation | 30 min | 37°C |
| 7 | Antibody and tracer | 10 μL | 2× in D buffer; 37.5 μg/mL antibody & 2 nM tracer final |
| 8 | Assay readout | After 30 min | EnVision Multilabel Reader using far-red FP filter set |
1. Multimek transfer to 384-well plates (tip wash 5× with 20 μL of dH2O).
2. 10% aqueous DMSO added to columns 1 and 2; 500 mM EDTA added to columns 23 and 24.
3. 8-tip Multidrop dispense reagent to all wells.
4. Plates uncovered in nonshaking incubator.
5. 8-tip Multidrop dispense reagent to all wells.
6. Plates uncovered in nonshaking incubator.
7. 8-tip Multidrop dispense reagent to all wells.
8. Plates kept uncovered at 25°C in dark for 30 min until read on multilabel reader using the PerkinElmer Cy5 FP dual emission label (620/40 nm excitation filter, 688/45 nm emission filter, and D658/fp688 dual mirror).
ATP, adenosine 5′-O-triphosphate; D buffer, detection buffer; DMSO, dimethyl sulfoxide; EDTA, ethylenediaminetetraacetic acid; FP, fluorescence polarization; R buffer, reaction buffer.
Fig. 4.

Results from the LOPAC pilot screen. (A) Scatter plot of the mean relative inhibition effected by 4,727 compounds from the LOPAC collection against RecA using the Transcreener assay. The data presented are mean±SD of triplicate wells (n=3). (B) Correlation plot of the mean relative inhibition from two runs of RecA LOPAC screen with solid line indicating perfect 1:1 correlation. LOPAC, library of pharmaceutically active compounds.
The LOPAC was screened previously against RecA using a different ATPase assay16 under different conditions, and 9 of the 19 active compounds were identified as RecA inhibitors by both assays. The high correspondence of the active compounds confirmed the Transcreener assay as a reliable method for identifying RecA inhibitors from large compound libraries. Importantly, however, the fact that previously unidentified RecA inhibitors were identified using the Transcreener assay substantiated our assay design and demonstrated the ability to explore new chemical space for RecA inhibitors.
Screening
Although the pilot screen provided a number of novel inhibitors of RecA, our main objective in screening the LOPAC was to validate the Transcreener assay as robust and reproducible for the evaluation of additional libraries containing compounds with greater potential for development as small-molecule therapeutics. Towards this end, we screened 113,477 compounds from collections at CICBDD at the University of North Carolina at Chapel Hill. Compounds from the 100k Diversity Collection and the Kinase Focus Set were screened against RecA in singleton at 15 μM from 1 mM DMSO stocks using the Transcreener assay. The Diversity Collection screening results demonstrated a normal distribution centered on a mean of 0% inhibition with an SD of 5% (Fig. 5A). Similarly, the Kinase Focus Set screen gave a mean of −0.9% and an SD of 7% (data not shown). Overall, the average Z′ factor was 0.92 for the 359 analyzed plates (Fig. 5B). A single plate fell below the 0.6 minimum Z′ factor accepted value and was repeated before inclusion in data analysis.
Fig. 5.

HTS results. (A) Frequency distribution of the 100K Diversity Collection relative inhibition results depicted as a histogram, where the number of compounds in each bin (±1% width) is plotted on a linear scale. Inset displays the frequency distribution of bins (±1% width) with greater than 20% inhibition. (B) Z′ factor analysis of the 359 plates assayed during 100K Diversity Collection and Kinase Focus Set HTS. 32 wells of inhibited controls (50 mM EDTA final) and 32 wells of maximum activity controls (1% DMSO final) were used to calculate the Z′ factor. Dashed line indicates Z′ factor value established as minimum threshold for accepted plates to be included in data analysis.
As summarized in Table 2, the 100k Diversity Collection primary HTS resulted in a total of 235 compounds with >30% inhibition. The activity threshold was set at 30% inhibition and defined as 6×SD above the mean. The 30% inhibition cut-off was stringent by screening standards and yielded a sufficient number of hits for follow-up (0.22% hit rate). From the 235 initial hit compounds, 78 compounds were selected based on their structural representativity23 for IC50 determination as described in Materials and Methods. The remaining 157 (of 235) hits were labeled as the “remaining hits” and given a second priority in our activity analysis.
Table 2.
Summary of Successful High-Throughput Screening for RecA Inhibitors
| Library: 100k Diversity Compound Collection | |
|---|---|
| Number of compounds tested | 108,750 |
| Overall primary screen hit rate (%) | 0.22 |
| Overall number of hit compoundsa | 235 |
| Number of hit compounds in representative selectionb | 78 |
| Validation rate from representative selection (%) | 86 |
| Number of validatedc hits from representative selection | 66 |
| Number of remaining hit compoundsd | 157 |
| Validation rate of remaining hits (%) | 64 |
| Number of validated hits from remaining hits | 99 |
| Overall validation rate (%) | 71 |
| Overall number of validated compounds | 165 |
| Overall number of compounds with IC50≤10 μM | 74 |
| Library: Kinase Focus Set | |
| Number of compounds tested | 4,727 |
| Hit rate (%) | 0.30 |
| Number of initial hitse | 14 |
| Validation rate (%) | 86 |
| Number of validatedf hits | 12 |
| Number of compounds with IC50≤10 μM | 5 |
All compounds with ≥30% inhibition (6×SD above the mean).
Compounds with ≥30% inhibition and selected based on their scaffold group. See text for selection details.
Compounds with IC50≤35 μM with visual inspection of curve shape and max % inhibition ≥60%.
Remaining compounds with ≥30% inhibition.
Compounds with ≥43% inhibition (6×SD above the mean).
Compounds with IC50≤35 μM.
We performed a concentration–response study of all the available HTS initial hits using the Transcreener assay. From the representative selection, 77 compounds were available in powder stock for evaluation. The concentration–response measurements were performed in triplicate and 66 compounds were characterized by IC50≤35 μM (Table 2). The validation rate of the representative selection set was 86%, and nearly half of the inhibitors were characterized by IC50≤10 μM.
Of the remaining hits, 154 compounds were obtained in powder form and were similarly evaluated by IC50 measurement in triplicate. The concentration–response curves allowed 84 compounds to be characterized by IC50≤35 μM (Table 2). The validation rate of the remaining hits (55%) was lower than that of the representative selection set, but still confirmed over half as validated biochemical RecA inhibitors. The high validation rate of the representative selection set was a result of our structural grouping analysis of the HTS results to prioritize the more potent inhibitors.
The Kinase Focus Set was collected by the CICBDD and comprised 4,727 kinase-directed drug-like small molecules not found in the 100k Diversity Collection. Given that both kinases and RecA are ATP-dependent enzymes, a compound library with small molecules directed at kinases was screened with hopes that it might present compounds active against RecA ATPase activity. The screen yielded 14 compounds with ≥43% inhibition (6×SD above the mean). The 0.3% hit rate was similar to the Diversity Collection screen. The IC50 determination of the 14 initial hits was performed in duplicate from powder stocks and validated 12 compounds as having IC50≤35 μM (Table 2).
Raw data for three replicates of a validated hit compound are depicted in Fig. 6. There is a clear dose-dependent inhibition of enzyme activity over 6 concentrations tested. A reproducible IC50 of 5±1 μM confirms the robustness and reproducibility of the assay.
Fig. 6.

Reproducible concentration–response measurements for validated inhibitor UNC10036220 from the 100k Diversity Collection screen. The curve representing the mean IC50 value of 5±1 μM was consistent with data from three independent runs (circle, square and triangle symbols, respectively).
Follow-up Evaluation of Select Inhibitors
Further evaluation revealed that many of the inhibitors were active in biological assays with live E. coli and were characterized by biochemical mechanisms of inhibition that differed from those of known RecA inhibitors. Of the 165 validated hits from the 100k Diversity Collection, 35 were active in one or more bacteriological assays for SOS activation or fluoroquinolone antibacterial potentiation. We then selected 15 inhibitors that were active in bioassays with live bacteria, were characterized by IC50 values <10 μM in the Transcreener assay, or both. For the 15 selected inhibitors, ATPase rates were measured as a function of substrate ATP concentration at different inhibitor concentrations. Analysis of resulting velocity–ATP concentration curves unexpectedly revealed that none of the compounds exhibited competition with ATP; however, 13 compounds were noncompetitive inhibitors and 2 were mixed-type inhibitors with α>1.31 Similar characterization of RecA inhibitors identified by previous assays14–17 revealed only ATP-uncompetitive and DNA-competitive inhibitors, results that were consistent with restricted specificity for the active conformational state of the RecA filament. Although we expected some of the tested compounds from the Transcreener assay to competitively bind the ATP site, the discovery of inhibitors that target both free RecA enzyme and the substrate-bound conformation of RecA confirmed the successful identification of RecA inhibitors with varied specificity for RecA conformation. A full accounting of these results will be reported elsewhere.
Discussion
RecA facilitates important biological processes that allow bacteria to survive and respond to antibacterial exposure. Because RecA is ubiquitous32 and highly conserved33 among bacteria, but has only distant human homologs,13,34–38 potent and selective RecA inhibitors may serve as novel chemotherapeutic adjuvants to enhance conventional antibiotics. Although we have previously developed biochemical assay technologies that allowed target-based screening to identify RecA inhibitors, the prior assay conditions restricted both the available conformational state of RecA and the inhibitor mechanistic types that could be explored. A major conclusion of this work is that the Transcreener ADP2 FP assay method could be adapted for a robust and reproducible HTS (Z′=0.92) of 113,477 small molecules for inhibition of RecA ATPase activity. The sensitivity of the TranscreenerADP2 FP technology allowed the enzyme and substrate concentrations to be reduced by more than an order of magnitude. From the compounds identified as active in the primary HTS assay, 246 compounds were evaluated in concentration–response format and 79 inhibitors characterized by IC50≤10 μM were identified. Importantly, the evaluation of a subset of the inhibitors revealed 35 that were active in one or more assays with live E. coli and that a number of new mechanistic types and selectivities for RecA conformational states were identified. These novel RecA inhibitors represent a variety of chemotypes and 33 unique scaffold groups (group size>1) that will serve as synthetically tractable hit series for medicinal chemistry efforts aimed at optimizing biochemical and bacteriological inhibition of RecA activities.
In part, the continued search for new RecA inhibitors is motivated by the increasingly urgent public health threat posed by antibiotic-resistant pathogens. It is now clear that the use and misuse of antibiotics has played major roles in the selection and spread of resistant pathogens. The exposure of bacteria to antibacterial compounds results in the selection of resistant variants that ultimately dominate the population. Importantly, however, recent studies suggest that bacteria are not merely passively subjected to natural selection but can actively promote genetic diversification. The origins of genetic variation include local changes in the DNA sequence (mutation), intrachromosomal shuffling of DNA sequences (recombination), and the acquisition of DNA sequences from other organisms (horizontal gene transfer). Induced mutagenesis in response to antibiotic exposure dramatically accelerates bacterial mutation.7,39–41 Likewise, recombination plays a major role in bacterial evolution,7,42–44 and may be a more frequent source of nucleotide changes in E. coli than de novo mutation.45 Finally, horizontal gene transfer is an important source of genetic diversity in bacteria.10,46–48
The genetic diversification processes described above are regulated by the bacterium and depend on the function of select proteins and enzymes, including RecA. Indeed, RecA has multiple functions that contribute to induced mutation, intragenomic recombination, and horizontal gene transfer, including induction of the SOS response by stimulating autocleavage of the LexA repressor, activation of error-prone DNA polymerase V by stimulating autocleavage of UmuD, and facilitation of homologous genetic recombination. The biochemical dependency of these phenomena on RecA activities suggests that potent, selective RecA inhibitors could be developed to attenuate the development, acquisition, and dissemination of resistance, affording an opportunity to inhibit evolutionary processes and enhance antibacterial chemotherapy. Current experiments are underway to evaluate the inhibitors of RecA's DNA-dependent ATPase activity in bacteriological assays.
Abbreviations
- ADP
adenosine 5′-O-diphosphate
- aq
aqueous
- ATP
adenosine 5′-O-triphosphate
- CICBDD
Center for Integrative Chemical Biology and Drug Discovery
- D buffer
detection buffer
- FP
fluorescence polarization
- HEPES
4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
- HTS
high-throughput screening
- LOPAC
library of pharmaceutically active compounds
- poly(dT)
polydeoxythymidylic acid
- R buffer
reaction buffer
- RDF
RecA-DNA filament
- SD
standard deviation
- ssDNA
single-stranded DNA.
Acknowledgments
The authors thank Dr. Anthony Klink for valuable advice and Dr. Tim Wigle, Emily Hull-Ryde, Amy VanDeusen, and Adam Cheely for their technical assistance. BellBrook Labs graciously provided two Transcreener ADP2 FP Assay Kits for evaluation. This work was supported by a grant to S.F.S. from the National Institutes of Health (GM058114).
Disclosure Statement
S.F.S. is the president and chief scientific officer of Synereca Pharmaceuticals, Inc., which has licensed RecA inhibitor technology from The University of North Carolina at Chapel Hill.
References
- 1.Boucher HW. Talbot GH. Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! An update from the infectious diseases society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Devasahayam G. Scheld WM. Hoffman PS. Newer antibacterial drugs for a new century. Expert Opin Investig Drugs. 2010;19:215–234. doi: 10.1517/13543780903505092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Payne DJ. Gwynn MN. Holmes DJ. Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 4.Miller C. Thomsen LE. Gaggero C. Mosseri R. Ingmer H. Cohen SN. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science. 2004;305:1629–1631. doi: 10.1126/science.1101630. [DOI] [PubMed] [Google Scholar]
- 5.Kohanski MA. Dwyer DJ. Hayete B. Lawrence CA. Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 6.Singh R. Ledesma KR. Chang KT. Tam VH. Impact of recA on levofloxacin exposure-related resistance development. Antimicrob Agents Chemother. 2010;54:4262–4268. doi: 10.1128/AAC.00168-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thi TD. Lopez E. Rodriguez-Rojas A, et al. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J Antimicrob Chemother. 2011;66:531–538. doi: 10.1093/jac/dkq496. [DOI] [PubMed] [Google Scholar]
- 8.Lu TK. Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc Natl Acad Sci USA. 2009;106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloyd RG. Buckman C. Conjugational recombination in Escherichia coli: genetic analysis of recombinant formation in Hfr x F-crosses. Genetics. 1995;139:1123–1148. doi: 10.1093/genetics/139.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaber JW. Hochhut B. Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2004;427:72–74. doi: 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 11.Babic A. Lindner AB. Vulic M. Stewart EJ. Radman M. Direct visualization of horizontal gene transfer. Science. 2008;319:1533–1536. doi: 10.1126/science.1153498. [DOI] [PubMed] [Google Scholar]
- 12.Lee AM. Ross CT. Zeng BB. Singleton SF. A molecular target for suppression of the evolution of antibiotic resistance: inhibition of the Escherichia coli RecA protein by N(6)-(1-naphthyl)-ADP. J Med Chem. 2005;48:5408–5411. doi: 10.1021/jm050113z. [DOI] [PubMed] [Google Scholar]
- 13.Roca AI. Cox MM. RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- 14.Lee AM. Wigle TJ. Singleton SF. A complementary pair of rapid molecular screening assays for RecA activities. Anal Biochem. 2007;367:247–258. doi: 10.1016/j.ab.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wigle TJ. Singleton SF. Directed molecular screening for RecA ATPase inhibitors. Bioorg Med Chem Lett. 2007;17:3249–3253. doi: 10.1016/j.bmcl.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wigle TJ. Sexton JZ. Gromova AV, et al. Inhibitors of RecA activity discovered by high-throughput screening: cell-permeable small molecules attenuate the sos response in Escherichia coli. J Biomol Screen. 2009;14:1092–1101. doi: 10.1177/1087057109342126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sexton JZ. Wigle TJ. He Q, et al. Novel Inhibitors of E. coli RecA ATPase Activity. Curr Chem Genomics. 2010;4:34–42. doi: 10.2174/1875397301004010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huss KL. Blonigen PE. Campbell RM. Development of a Transcreener kinase assay for protein kinase A and demonstration of concordance of data with a filter-binding assay format. J Biomol Screen. 2007;12:578–584. doi: 10.1177/1087057107300221. [DOI] [PubMed] [Google Scholar]
- 19.Klink TA. Kleman-Leyer KM. Kopp A. Westermeyer TA. Lowery RG. Evaluating PI3 kinase isoforms using Transcreener ADP assays. J Biomol Screen. 2008;13:476–485. doi: 10.1177/1087057108319864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowlands M. McAndrew C. Prodromou C, et al. Detection of the ATPase activity of the molecular chaperones Hsp90 and Hsp72 using the TranscreenerTM ADP assay kit. J Biomol Screen. 2010;15:279–286. doi: 10.1177/1087057109360253. [DOI] [PubMed] [Google Scholar]
- 21.Kleman-Leyer KM. Klink TA. Kopp AL, et al. Characterization and optimization of a red-shifted fluorescence polarization ADP detection assay. Assay Drug Dev Technol. 2009;7:56–67. doi: 10.1089/adt.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singleton SF. Simonette RA. Sharma NC. Roca AI. Intein-mediated affinity-fusion purification of the Escherichia coli RecA protein. Protein Expr Purif. 2002;26:476–488. doi: 10.1016/s1046-5928(02)00571-5. [DOI] [PubMed] [Google Scholar]
- 23.Bemis GW. Murcko MA. The properties of known drugs. 1. Molecular frameworks. J Med Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 24.Walters WP. Murcko MA. Prediction of ‘drug-likeness'. Adv Drug Deliv Rev. 2002;54:255–271. doi: 10.1016/s0169-409x(02)00003-0. [DOI] [PubMed] [Google Scholar]
- 25.Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discovery Today. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 26.Transcreener ADP2 FP Technical Manual. www.bellbrooklabs.com/PDFs/Tech%20Man_ADP2%20FP%20EZ_v073009.pdf. [Jun 6;2011 ]. www.bellbrooklabs.com/PDFs/Tech%20Man_ADP2%20FP%20EZ_v073009.pdf
- 27.Neet KE. Cooperativity in enzyme function: equilibrium and kinetic aspects. Methods Enzymol. 1980;64:139–192. doi: 10.1016/s0076-6879(80)64009-9. [DOI] [PubMed] [Google Scholar]
- 28.Kowalczykowski SC. Interaction of recA protein with a photoaffinity analogue of ATP, 8-azido-ATP: determination of nucleotide cofactor binding parameters and of the relationship between ATP binding and ATP hydrolysis. Biochemistry. 1986;25:5872–5881. doi: 10.1021/bi00368a006. [DOI] [PubMed] [Google Scholar]
- 29.Macarron R. Hertzberg RP. Design and implementation of high throughput screening assays. Mol Biotechnol. 2011;47:270–285. doi: 10.1007/s12033-010-9335-9. [DOI] [PubMed] [Google Scholar]
- 30.Zhang JH. Chung TD. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 31.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: a Guide for MedicinalChemists and Pharmacologists. Wiley; Hoboken, NJ: 2005. [PubMed] [Google Scholar]
- 32.Roca AI. Cox MM. The RecA protein: structure and function. Crit Rev Biochem Mol Biol. 1990;25:415–456. doi: 10.3109/10409239009090617. [DOI] [PubMed] [Google Scholar]
- 33.Rocha EP. Cornet E. Michel B. Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 2005;1:e15. doi: 10.1371/journal.pgen.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shinohara A. Ogawa H. Matsuda Y. Ushio N. Ikeo K. Ogawa T. Cloning of human, mouse and fission yeast recombination genes homologous to RAD51 and recA. Nat Genet. 1993;4:239–243. doi: 10.1038/ng0793-239. [DOI] [PubMed] [Google Scholar]
- 35.Yoshimura Y. Morita T. Yamamoto A. Matsushiro A. Cloning and sequence of the human RecA-like gene cDNA. Nucleic Acids Res. 1993;21:1665. doi: 10.1093/nar/21.7.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brendel V. Brocchieri L. Sandler SJ. Clark AJ. Karlin S. Evolutionary comparisons of RecA-like proteins across all major kingdoms of living organisms. J Mol Evol. 1997;44:528–541. doi: 10.1007/pl00006177. [DOI] [PubMed] [Google Scholar]
- 37.Leipe DD. Aravind L. Grishin NV. Koonin EV. The bacterial replicative helicase DnaB evolved from a RecA duplication. Genome Res. 2000;10:5–16. [PubMed] [Google Scholar]
- 38.Yu X. Jacobs SA. West SC. Ogawa T. Egelman EH. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc Natl Acad Sci USA. 2001;98:8419–8424. doi: 10.1073/pnas.111005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hersh MN. Ponder RG. Hastings PJ. Rosenberg SM. Adaptive mutation and amplification in Escherichia coli: two pathways of genome adaptation under stress. Res Microbiol. 2004;155:352–359. doi: 10.1016/j.resmic.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 40.Foster PL. Stress responses and genetic variation in bacteria. Mutat Res. 2005;569:3–11. doi: 10.1016/j.mrfmmm.2004.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cirz RT. Chin JK. Andes DR. de Crecy-Lagard V. Craig WA. Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spratt BG. Hanage WP. Feil EJ. The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr Opin Microbiol. 2001;4:602–606. doi: 10.1016/s1369-5274(00)00257-5. [DOI] [PubMed] [Google Scholar]
- 43.Lopez E. Elez M. Matic I. Blazquez J. Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol Microbiol. 2007;64:83–93. doi: 10.1111/j.1365-2958.2007.05642.x. [DOI] [PubMed] [Google Scholar]
- 44.Lopez E. Blazquez J. Effect of subinhibitory concentrations of antibiotics on intrachromosomal homologous recombination in Escherichia coli. Antimicrob Agents Chemother. 2009;53:3411–3415. doi: 10.1128/AAC.00358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guttman DS. Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 46.Lawrence JG. Roth JR. Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics. 1996;143:1843–1860. doi: 10.1093/genetics/143.4.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawrence JG. Ochman H. Molecular archaeology of the Escherichia coli genome. Proc Natl Acad Sci USA. 1998;95:9413–9417. doi: 10.1073/pnas.95.16.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hastings PJ. Rosenberg SM. Slack A. Antibiotic-induced lateral transfer of antibiotic resistance. Trends Microbiol. 2004;12:401–404. doi: 10.1016/j.tim.2004.07.003. [DOI] [PubMed] [Google Scholar]