

Abstract
Fluorescence-based endpoint detection of microarrays with 10,000 or more molecular targets is a most useful tool for high-throughput profiling of biomolecular interactions, including screening large molecular libraries for novel protein ligands. However, endpoint fluorescence data such as images of reacted microarrays contain little information on kinetic rate constants, and the reliability of endpoint data as measures of binding affinity depends on reaction conditions and postreaction processing. We here report a simultaneous measurement of binding curves of a protein probe with 10,000 molecular targets in a microarray with an ellipsometry-based (label-free) optical scanner. The reaction rate constants extracted from these curves (kon, koff, and ka=kon/koff) are used to characterize the probe-target interactions instead of the endpoints. This work advances the microarray technology to a new milestone, namely, from an endpoint assay to a kinetic constant assay platform. The throughput of this binding curve assay platform is comparable to those at the National Institutes of Health Molecular Library Screening Centers, making it a practical method in screening compound libraries for novel ligands and for system-wide affinity profiling of proteins, viruses, or whole cells against diverse molecular targets.
Introduction
Biomolecular microarrays have emerged as a leading high-throughput technology for system-wide biology such as genomics,1–3 proteomics,4–11 glycomics,12–18 and small molecule drug discovery.19–26 By immobilizing thousands or tens of thousands of molecular targets as distinct features on a solid support and simultaneously exposing all the targets to a probe solution of interest, chemical reactions of the probe with the targets are assayed at the same time. In situ synthesis is used to produce high-density peptide and oligonucleotide microarrays.27 For majority of biomolecules including cDNA, proteins, carbohydrates, lipids, and small molecule compounds, contact-printing techniques are employed to fabricate target microarrays on chemically functionalized glass slides.28,29 Printed microarrays easily have 10,000 to 35,000 features (or spots) over an area of 8–20 cm2. The significance of these large target microarrays to system-level biology is obvious considering that the human genome has 25,000 protein-encoding genes,30,31 the yeast proteome has 6,000 proteins,5,32 libraries of recombinantly expressed immunoglobulins (IgG) are typically of the order of 10,000 or smaller,33,34 and libraries of small molecule compounds typically have 104–106 molecules.
So far, most microarrays are detected with fluorescence scanners, wherein solution-phase probe molecules are modified with a fluorescent label or an affinity tag before incubation with the microarray. After the incubation under specific conditions (e.g., probe concentration, incubation time, temperature, and secondary reaction of affinity tags), the unbound probes are removed by washing before the “reacted” microarray is read with a fluorescence scanner. Such “endpoint” measurements do not provide information on reaction kinetic rate constants (the true measure of binding affinity) and the results may vary significantly when the target density on a microarray varies, a common occurrence for printed microarrays. Furthermore, depending upon the reaction rate constants, the reaction endpoints can vary as incubation conditions change. For example, relative affinity assessment based on endpoint fluorescence intensity often assumes that probe-target complexes increase linearly with time up to the end of incubation and that the complexes survive postreaction washing. Without the information on the association rate constants a priori, one needs to confirm experimentally the linear regime before the endpoint data can be so interpreted. In addition, without the information on dissociation rate constants, one cannot properly assess those probe-target complexes that do not survive post-incubation washing. The remedy to these drawbacks associated with endpoint assays is to observe binding reactions in real-time instead. In this case, one measures binding curves of the probe to immobilized targets during association and dissociation phases of reactions and extracts reaction rate constants from these curves as characterizing parameters of probe-target interactions. We will show as is expected that measurements of binding kinetic constants are independent of target density variation in a microarray. Furthermore from binding curves, one can assess other issues such as avidity, heterogeneity, mass transport, and conformational changes that are difficult to address by endpoint assays.
Fluorescence-based methods are seldom used for real-time binding curve measurements due to photobleaching suffered by many fluorescent tags and the potential effect of these tags on intrinsic association and dissociation rate constants.35 Label-free optical biosensing methods, such as surface plasmon resonance reflectometry (SPR),36–42 reflection interference spectroscopy,43–45 dielectric wave guide reflectometry,46–49 and imaging ellipsometry,50–54 complement fluorescence-based detection by doing away with labeling and providing both endpoint and kinetic measurements of binding reactions. However, these biosensors only detect a small number of reactions (no more than a few hundred reactions) at a time and often require special (and costly) sensor surfaces. As a result, they are not suitable for large microarray detection with 10,000 or more immobilized targets.
In this report we demonstrate an ellipsometry-based optical sensor platform capable of simultaneous measurement of binding curves of a probe with 10,000 targets immobilized on a functionalized glass slide. This development advances microarray technology from an endpoint assay to a true binding affinity assay platform with a potential to assay over 50,000 targets in one day.
The summary of this detection platform is illustrated in Fig. 1. Molecular targets expressing specific epitopes are immobilized in form of a microarray on an epoxy-functionalized glass slide using a contact-printing robot. The unprinted surface is blocked with macromolecules that do not express the epitopes. By incubating the microarray in a solution of protein probes that specifically recognize the epitopes, the probes become captured by the immobilized targets, causing the thickness d or coverage Θ or generally the surface mass density Γ (gm/cm2) of the immobilized target layer to change. The endpoint and/or real-time evolution of such a change lead to extra changes in magnitude and phase of an incident monochromatic light beam upon reflection from the glass surface. We directly measure the extra magnitude change Δr and extra phase change Δδ as fluorescenceless measures of the protein-target binding reactions.
Fig. 1.

Summary of a scanning ellipsometry-based detection of endpoints and real-time association–dissociation curves of protein probes with surface-immobilized targets in form of a large microarray on an epoxy-functionalized glass slide. The changes in thickness d and coverage Θ or surface mass density Γ of the target layer as a result of binding reaction cause extra changes in magnitude (Δr=r−r0) and phase (Δδ=δ−δ0) of an incident light beam (E0) upon reflection from the target-covered surface. r0 and δ0 are magnitude and phase changes due to reflection from the “bare” glass surface in the absence of the target-probe layer. The present ellipsometry-based detection platform measures Δr and Δδ in real-time from all immobilized targets.
Methods and Materials
Oblique-Incidence Reflectivity Difference Scanning Microscope
Our optical sensor platform for large microarray detection is a scanning optical microscope based on polarization-modulated oblique-incidence reflectivity difference (OI-RD).26,55–57 It does not require specially structured substrates such as gold films or dielectric waveguides for detection, and has a large “field of view” (presently ∼10 cm2). It is thus fully compatible with large microarrays printed on inexpensively functionalized glass slides. Compared to imaging ellipsometers based on polarizer-compensator-sample-analyzer schemes,50–54 the OI-RD scanning microscope is inherently more sensitive to surface-bound changes (e.g., thickness and mass density) by more than one order of magnitude.58
The arrangement of our scanning OI-RD microscope used in this work is briefly described here (Fig. 2). A scan lens focuses a polarization-modulated He-Ne laser beam (λ=633 nm) to a 30-μm-diameter spot on the back surface of a glass slide printed with a target microarray. The incidence angle is θ=36.6° in the glass slide. The back surface is in contact with an aqueous solution in a fluidic chamber. Images of the microarray are obtained by raster scanning the beam across the back surface at a step size of 20 μm with a combination of a galvanometer mirror and the scan lens along the y-axis and by moving the microarray fluidic assembly relative to the beam along the x-axis with a linear stage. Image “contrast” is based upon the polarization change of the laser beam upon reflection from the back surface,50,52 described by the ratio of reflection coefficients for p- and s-polarized components of the beam, rp/rs=tanψ·exp(iδ). An OI-RD scanning microscope directly measures the polarization change. When solution-phase probes bind to immobilized targets on the glass surface, the surface mass density (mass per unit area) of the target layer Γ changes and in turn alters rp/rs. When the target, the probe, and the glass slide are transparent at the optical wavelength λ, the change in Γ primarily alters the phase δ (see Supplementary Data for detailed description of Δδ measurement; Supplementary Data are available online at www.liebertonline.com/adt) as follows,55,59,60
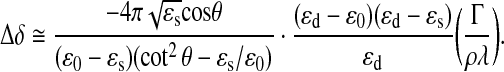 |
(1) |
Fig. 2.

(a) Optical layout of the scanning OI-RD microscope. A functionalized glass slide with a microarray printed on the bottom surface is installed in a fluidic chamber assembly. An illumination laser beam is raster swept across the microarray with a scan mirror assembly for y-scan; a linear translation stage moves the fluidic chamber assembly with respect to the illumination optics in the orthogonal direction for x-scan. (b) The fluidic chamber assembly showing the 2 cm×4 cm accessible area of the glass slide. (c) Side view of the microscope illustrating the y-scan. OI-RD, oblique-incidence reflectivity difference.
ɛs, ɛ0, and ɛd are the optical dielectric constants of the glass slide, the aqueous solution, and the probe-target layer, respectively. ρ=1.35 g/cm3 is the volume mass density of globular proteins.61 With typical values of ɛs=2.31, ɛ0=1.77, and ɛd=2.03, a probe-target layer with Γ=1 ng/mm2 yields Δδ=−2.3×10−3. The current limit of our OI-RD microscope is |Δδ|≈2×10−5, corresponding to a surface mass density of protein probes in the order of ∼10 pg/mm2. It is not yet as sensitive as surface-plasmon-resonance or waveguide-based sensors since we have not yet utilized the resonance-like enhancement factor 1/(cot2 θ−ɛs/ɛ0) in Equation (1). However, the high throughput and low operation cost of the OI-RD scanning microscope for compound screening are the major merits when compared to these other label-free detection methodologies.
For simultaneous measurement of binding curves of a probe to thousands of immobilized targets, we perform repeated scans of a subset of pixels on the microarray surface as follows. We select one pixel from each target spot as the signal channel and two pixels from the unprinted region on two sides of the target as the reference channels. Each reference channel is shared by two neighboring signal channels. The signal and reference pixels along vertical and horizontal lines form a rectangular readout grid. For a 9,216 (64×144)- spot microarray, the readout grid has 9,216 target channels and 9,360 reference channels. We scan these channels in every 20–70 s. The optical signal Δδ from a signal channel minus the averaged optical signal from the two neighboring reference channels yields the background-corrected signal for the target. This procedure compensates for instrumental drift, ambient refractive index changes, and flow-induced signal transients. The time series of the background-corrected signal from a target collected during the course of a reaction form a binding curve of the probe against the target.
Microarray Targets and Probes
Targets
Bovine serum albumin (BSA), human IgG (HM), mouse IgG (MS), rabbit IgG (RB), and polyclonal goat IgG against human/mouse/RB (GT anti-HM, GT anti-MS, and GT anti-RB) were purchased from Jackson ImmunoResearch Laboratories. Methamphetamine-BSA, tetrahydrocannabinol-BSA, and morphine-BSA conjugates were purchased from Biodesign International. Theophylline-BSA, phenobarbital-BSA (PB-BSA), and digoxin-BSA were purchased from Fitzgerald Industries International, Inc. Metallothionein (Metal) and biotin-N-hydroxysuccinimidyl ester (NHS-biotin ester) were purchased from Sigma-Aldrich.
We prepared biotin-BSA (B-BSA) conjugates by reacting NHS-biotin ester with a BSA solution in 0.1 M NaHCO3. The loading of biotin was controlled by the molar ratio of the NHS-biotin ester to BSA that ranged from 5×, 10×, 20×, up to 40×. Excess free biotin was removed by dialysis. We prepared iminobiotin-BSA conjugates similarly. We made 2,4-dinitrophenol-BSA conjugates with a previously described method.25 We also made glucose-BSA and maltose-BSA by reductive amination at pH 8.0.62
Probes
Monoclonal mouse anti-biotin IgG was purchased from Jackson ImmunoResearch. Monoclonal mouse anti-methamphetamine, anti-tetrahydrocannabinol, anti-phenobarbital, and anti-theophylline IgG were purchased from Biodesign. Monoclonal mouse anti-morphine IgG was purchased from Fitzgerald Industries. Polyclonal goat anti-2,4-dinitrophenol was purchased from Sigma-Aldrich. Concanavalin A was purchased from Vector Laboratories.
Microarray Fabrication and Reaction
Microarrays of 9,216 (64×144) or 10,880 (68×160) targets were printed on epoxy-functionalized glass slides (ArrayIt Corporation) using an OmniGrid 100 contact-printing robot (Digilab). Each microarray covers a 2 cm×4 cm area. A printed microarray without further processing was installed in a fluidic chamber assembly (Fig. 2) and imaged with the OI-RD scanner before exposed to a buffer solution. The large optical signals from printed materials including the buffer salts were used (i) to align the microarray axes to the scan axes and (ii) to generate a rectangular readout grid for real-time binding curve measurement and image analysis (see Supplementary Data for details). It takes 18 min to acquire an OI-RD image of an 8-cm2 area with a pixel size (scan step size) of 20 μm×20 μm. The microarray was then washed in situ by passing several milliliters of 1 × phosphate buffered saline (PBS) buffer through the fluidic chamber and imaged again for a record of the target density. Next, the washed microarray was exposed to a solution of 7.6 μM BSA (0.5 mg/mL) in 1 × PBS for 30 min to quench unreacted epoxide groups to prevent nonspecific binding of subsequent probes to the unprinted surface. After BSA blocking, the microarray was kept in 1 × PBS and ready for binding reactions.
All binding reactions were performed at ambient temperature (nominally 25°C). For each reaction, we first passed 1 × PBS buffer through the fluidic chamber at 0.01 mL/min for 30 min to acquire the baseline. Next, the buffer was quickly replaced with a probe solution at 5 mL/min for 12 s. The flow rate of the solution was then reduced to 0.01 mL/min to allow the probe to react with the microarray at a constant concentration for 30–60 min (association phase of the reaction). Afterward, the probe solution was quickly replaced with 1 × PBS buffer at 5 mL/min for 12 s. The flow rate of the buffer was subsequently reduced to 0.01 mL/min to allow the captured probe to dissociate from the microarray for 60 or 90 min (dissociation phase of the reaction). We acquired OI-RD images of the microarray before and after the reaction. During the reaction, we repeatedly scanned the readout grid every 20–70 s to acquire binding curves from all targets. We note that if the association and/or dissociation for some of the reactions take minutes or less to finish, the association–dissociation curves of these reactions (much fewer than 10,000) can be revisited on a separate but same microarray in a “cherry-picking” mode with a time step as short as a few seconds, limited only by how quick the buffer is replaced by the probe solutions and vise versa.
Results and Analysis
Binding Curves of a Protein Probe to a Microarray with 9,216 Identical Targets
To demonstrate simultaneous detection of ∼10,000 binding curves with the OI-RD scanning microscope, we performed the reaction of mouse anti-biotin IgG with a microarray with 9216 identical B-BSA targets. The targets were printed from a B-BSA solution at 1.6 μM in 1 × PBS. Figure 3 shows the OI-RD image of the microarray in 1 × PBS after excess printed material was washed off. The surface mass density of the target layer averaged over the spot is 3.0±0.5 ng/mm2, corresponding to a full monolayer of B-BSA (a side-on oriented BSA monolayer has a surface mass density56 of ∼2 ng/mm2). The variation within a spot is ±1.3 ng/mm2. Figure 4a displays 288 of 9,216 simultaneously acquired binding curves at the MS concentration of C=33 nM in 1× PBS. Each curve records the change in OI-RD signal and in turn, through Equation (1), the change in surface mass density of the probe-target layer. The average surface mass density of captured IgG molecules at saturation is 5 ng/mm2, so that on average every two immobilized B-BSA molecules captured one IgG molecule. This is the first report where binding curves were recorded for ∼10,000 features in a microarray in a single measurement. By repeating the binding curve measurement at the MS concentrations of C=100 nM and 300 nM in two separate measurements, we obtained 9,216 sets of binding curves, each set corresponding to the probe reaction with a distinct target at three probe concentrations. Figure 4b displays 288 of 9,216 binding curve sets.
Fig. 3.

OI-RD image of a biotinylated BSA microarray (64×144 spots) acquired after the microarray is washed with 1× PBS buffer. The image pixel size is 20 μm and the center-to-center spacing of the spots is 250 μm. The inset is a magnified view that displays the discrete readout points (red crosses) used during real-time experiments. PBS, phosphate buffered saline.
Fig. 4.

(a) 288 out of 9,216 simultaneously acquired OI-RD measurements of anti-biotin IgG (33 nM) binding to biotinylated-BSA microarrays. After a 30-min baseline measurement, the association reaction was monitored for 60 min (probe solution flowed starting at t=0 min) and the dissociation reaction was monitored for 90 min (buffer solution flowed starting at t=60 min). (b) Anti-biotin IgG binding curve sets (33 nM, 100 nM, and 300 nM) were acquired separately with fresh microarrays. For global curve fitting analysis of the data, binding curves from corresponding spot locations in each dataset are collected together as shown here (red curves=33 nM, green curves=100 nM, blue curves=300 nM).
The 9,216 binding curve sets were analyzed to yield the reaction kinetic rate constants using the Langmuir reaction model. In this model solution-phase probes are assumed to bind to surface-immobilized targets at a rate proportional to the probe concentration C, konC. The captured probes can dissociate from probe-target complexes at a rate koff, independent of C. When the probe solution is introduced to the microarray at t=0 and then replaced with 1 × PBS at a later time t=t0, the number of captured probes per unit target area is
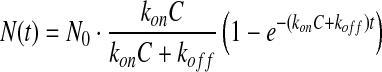 |
(2a) |
for t<t0 and
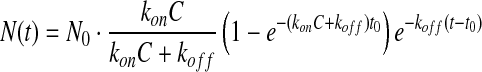 |
(2b) |
for t>t0. N0 is the maximum number of probes that can be captured per unit target area. It depends on factors such as the target density, geometric shapes and orientations of targets and probes. The surface mass density Γ in Equation (1) is proportional to N(t) and as a result Δδ(t)=γ×N(t). γ is a function of optical parameters displayed in Equation (1), and the volume mass density and molecular weight of the probe. We extract reaction rate constants kon and koff by fitting binding curve sets to Equations (2a) and (2b) simultaneously. Generally, N0 varies from spot to spot in a microarray and from microarray to microarray due to variation in wetting properties across a functionalized glass surface and in the liquid delivery of contact printing. As a result, we treat N0 as a fitting parameter that may vary from curve to curve within a set while treating kon and koff as common (global) parameters to all three curves of the set.63–65 Details are described in Supplementary Data.
We computed the equilibrium association constants Ka=kon/koff from the fitting parameters kon and koff for all 9,216 reactions. In Figure 5, we display Ka of mouse anti-biotin IgG reaction with 9216 B-BSA targets in the same layout as the target microarray (Fig. 3). This is the first equilibrium association constant map obtained from simultaneous measurement of 9216 binding curves on a microarray platform. Except for the upper-right corner of the microarray where the binding curves showed little dissociation (due to the insufficient buffer flow from left to right during the dissociation phase), the equilibrium association constants have a mean of 0.43 nM−1 and a standard error of ±0.13 nM−1, or Kd=1/Ka=2.3 nM±0.7 nM. Since the targets are of the same material, the standard error represents the uncertainty if a single B-BSA spot in a microarray is used to measure the equilibrium association constant to the probe. Given the inherent variations in contact-printed microarrays as described previously, the precision of this high-throughput kinetic constant assay is remarkably satisfactory. The slight decrease in Ka in Figure 5 from left to right is presumably the result of the depletion effect. The result can be used to quantify the depletion effect along the length of the sample chamber.
Fig. 5.

Experimental equilibrium association constants Ka of mouse IgG (MS) anti-biotin probe binding to 9216 B-BSA targets, displayed in the same layout as the target microarray (Fig. 3). The equilibrium association constant at each microarray address is determined by Ka=kon/koff, where kon and koff are the global fitting parameters determined from the corresponding binding curve set (Fig. 4). IgG, immunoglobulins.
Binding Curves of Multiple Probes to a 10,880-spot Microarray with Different Targets Printed Over a Range of Concentrations
We applied this binding kinetic constant assay to a 10,880-spot microarray with a diversity of 24 target types, each printed in concentrations ranging from 0.5 μM to 16 μM so that the target density changes intentionally by a factor of 20. Figure 6 shows the OI-RD image of such a microarray in 1 × PBS after excess printed materials were washed off. The microarray consists of 16 identical subarrays, with the 24 targets in each subarray laid out as shown in the inset of Figure 6. Each target was printed in quadruplicate and at six printing concentrations, changing two-fold from 16 μM to 0.5 μM. The targets include (i) B-BSA with different amounts of conjugated biotin, (ii) carbohydrate-BSA conjugates (glucose and maltose), (iii) drug-BSA conjugates, and (iv) whole IgG molecules. Border columns and rows were printed with BSA at 1.6 μM as controls. The real-time readout grid consisted of a total of 10,240 target channels (some BSA rows were skipped) and 10,880 reference channels, and was raster scanned every 22 s to obtain 10,240 binding curves.
Fig. 6.

OI-RD image of a ligand microarray (68×160 spots) acquired after washing the microarray with 1 × PBS buffer, but before further reaction of the microarray. The image pixel size is 20 μm and the center-to-center spacing of the spots is 250 μm vertically and 225 μm horizontally. The red lines in the top panel show the outlines of 16 identical subarrays and the inset shows the detailed spot layout of each. Each target type was printed in quadruplicate in six different printing concentrations ranging from 16 μM (left) to 0.5 μM (right). BSA borders and unprinted spots are also included as controls. B-BSA, biotin-BSA; Dig, digoxin; DNP, 2,4-dinitrophenol; Glc, glucose; GT a-HM, polyclonal goat IgG against human IgG; GT a-MS, polyclonal goat IgG against MS; GT a-RB, polyclonal goat IgG against rabbit IgG; HM, human IgG; IB, iminobiotin; Mal, maltose; Metal, metallothionein; Meth, Methamphetamine; Morph, morphine; PB, phenobarbital; RB, rabbit IgG; THC, tetrahydrocannabinol; THP, theophylline.
The microarray was reacted sequentially with multiple protein probes. They were, in order of reaction, anti-phenobarbital IgG, concanavalin A (lectin), anti-theophylline IgG, anti-biotin IgG, anti-tetrahydrocannabinol IgG, anti-morphine IgG, anti-dinitrophenol IgG, and anti-methamphetamine IgG. The reaction sequence was repeated on separate fresh microarrays to obtain binding curve sets for four probe concentrations of 300 nM, 100 nM, 33 nM, and 11 nM. The binding curve measurement consisted of a 30-min baseline in 1 × PBS, a 30-min association phase in a probe solution, and a 60-min dissociation phase in 1×PBS under the same flow conditions as described previously.
In Figure 7, we show the Ka map of anti-biotin IgG reactions with 10,880 targets. In Figure 8, we show the Ka map of anti-phenobarbital IgG reactions with the same 10,880 targets. Zooming in on one subarray, we note that except for targets printed at the highest concentration of 16 μM (see Sun et al.,25 who reported that at high printing concentrations and thus high immobilized target densities, the association rate of a solution-phase protein probe to the surface-bound targets deviates considerably from the rate obtained at lower printing concentrations, partly due to stereo-hindrance effect), Ka=0.53 nM−1 of anti-biotin IgG to immobilized B-BSA is essentially the same even when both the target density and the amount of captured IgG molecules vary by a factor of 10–20. The magnitude is very close to the bulk value of 0.59 nM−1 reported by Jung et al.66 and is 2.5 × the value reported by Adamczyk et al. in an SPR measurement.67 The Ka of anti-phenobarbital IgG to immobilized PB-BSA is similarly independent of the target density, and its value of 2.7 nM−1 compares well with the affinity constant of ∼20 nM−1 for monoclonal mouse anti-drug IgG molecules available from Fitzgerald Industries International, Inc. (www.fitzgerald-fii.com/Products?pId9&sId=21). This validates the notion that reaction rate constants obtained from global fitting binding curves are not and should not subject to ubiquitous target density variation in a contact-printed microarray and is independent of which pixel within a printed spot is used for binding curve measurement. It confirms that our present microarray-based kinetic constant assay is a robust platform for characterizing protein-ligand affinity. Similar findings for the other protein probes are described in Supplementary Data.
Fig. 7.

Experimental equilibrium association constants Ka of MS anti-biotin probe binding to 10,880 targets, displayed in the same layout as the target microarray (Fig. 6). The white lines show the outlines of the 16 identical subarrays and the 4×6 blocks of spots printed with different concentrations of a particular target type.
Fig. 8.

Experimental equilibrium association constants Ka of MS anti-phenobarbital probe binding to 10,880 targets, displayed in the same layout as the target microarray (Fig. 6). The white lines show the outlines of the 16 identical subarrays and the 4×6 blocks of spots printed with different concentrations of a particular target type.
Discussion and Conclusion
Microarray-based binding assays have so far been successfully applied to parallel studies of multiple biomolecular interactions,10,17,68 particularly to screening molecular libraries for protein ligands of interest.5,11,18,23,24 This platform will profit enormously from real-time binding curve detection capability that directly yields reaction rate constants (intrinsic characteristics of biomolecular interactions) with essentially the same throughput. Since extraction of kinetic rate constants from binding curve sets is insensitive to the immobilization target density, binding curve assays solve the problem of target density variation that often plagues microarray-based endpoint assays. As a bonus, OI-RD images of a target microarray acquired before and after BSA blocking in fact provide a label-free measure of the target density.56 Furthermore, binding curve assays detect those probe-target reactions with high dissociation rates that are easily missed in endpoint assays as a result of post-incubation washing. Though not elaborated here, real-time binding curves reveal other concurrent or sequential processes such as dissociation or conformational change of targets and the presence of multiple configurations of probe-target complexes (due to multiple binding pockets on a probe or multiple binding site presentation of the immobilized target) that are essentially beyond the reach of endpoint assays.
As to whether the present binding constant assay platform has the suitable throughput for large-scale molecular library screening, we observe that with one OI-RD scanning microscope, we can obtain binding curves of one protein probe to 50,000 targets (immobilized as 4 microarrays) at one concentration in one day. With three such OI-RD scanning microscopes, we can obtain 50,000 sets of binding curves at three probe concentrations to yield Ka of the probe to the 50,000 targets in a day. This means that we should be able to measure the equilibrium association constants of a single probe to 250,000 molecular targets in one week. This promises a new era for massive parallel characterization of biomolecular interactions.
We recently immobilized 8,000 drug-like small molecules (from NCI/DTP) on isocyanate-functionalized glass slide surfaces and screened these compounds for ligands of vascular endothelial growth factor (VEGF) with inhibitory effect against VEGF-KDR binding reaction using this microscope. The result demonstrated that the method described in this article would work equally well for drug-like compounds as surface immobilized targets.
Supplementary Material
Abbreviations
- B-BSA
biotin-BSA
- Dig
digoxin
- DNP
2,4-dinitrophenol
- Glc
glucose
- GT a-HM
polyclonal goat IgG against human IgG
- GT a-MS
polyclonal goat IgG against mouse IgG
- GT a-RB
polyclonal goat IgG against rabbit IgG
- HM
human IgG
- IB
iminobiotin
- IgG
immunoglobulins
- Mal
maltose
- Metal
metallothionein
- Meth
Methamphetamine
- Morph
morphine
- MS
mouse IgG
- OI-RD
oblique-incidence reflectivity difference
- PB
phenobarbital
- PB-BSA
phenobarbital-BSA
- PBS
phosphate buffered saline
- RB
rabbit IgG
- SPR
surface plasmon resonance
- THC
tetrahydrocannabinol
Acknowledgments
This work was supported by the National Institutes of Health under R01-HG003827 and by the University of California under UC Discovery Grant #bio09-156225.
Disclosure Statement
No competing financial interests exist.
References
- 1.Schena M. Shalon D. Davis RW. Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 2.“The chipping forecast.”. Nat Genet. 1999;21(1 Suppl):1–60. [Google Scholar]
- 3.“The chipping forecast II.”. Nat Genet. 2002;32(4 Suppl):461–552. [Google Scholar]
- 4.MacBeath G. Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 5.Zhu H. Bilgin M. Bangham R, et al. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 6.Haab BB. Dunham MJ. Brown PO. Protein microarrays for highly parallel detection and quantification of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:1–13. doi: 10.1186/gb-2001-2-2-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Espejo A. Cote J. Bednarek A. Richard S. Bedford MT. A protein-domain microarray identifies novel protein-protein interactions. Biochem J. 2002;367:697–702. doi: 10.1042/BJ20020860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller JC. Zhou H. Kwekel J, et al. Antibody microarray profiling of human prostate cancer sera: Antibody screening and identification of potential biomarkers. Proteomics. 2003;3:56–63. doi: 10.1002/pmic.200390009. [DOI] [PubMed] [Google Scholar]
- 9.Nielsen UB. Cardone MH. Sinskey AJ. MacBeath G. Sorger PK. Profiling receptor tyrosine kinase activation by using Ab microarrays. Proc Natl Acad Sci USA. 2003;100:9330–9335. doi: 10.1073/pnas.1633513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones RB. Gordus A. Krall JA. MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- 11.Michaud GA. Salcius M. Zhou F, et al. Analyzing antibody specificity with whole proteome microarrays. Nat Biotechnol. 2003;21:1509–1512. doi: 10.1038/nbt910. [DOI] [PubMed] [Google Scholar]
- 12.Wang D. Liu S. Trummer BJ. Deng C. Wang A. Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes, host cells. Nat Biotechnol. 2002;20:275–281. doi: 10.1038/nbt0302-275. [DOI] [PubMed] [Google Scholar]
- 13.Blixt O. Head S. Mondala T, et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc Natl Acad Sci USA. 2004;101:17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee M. Shin I. Facile preparation of carbohydrate microarrays by site-specific, covalent immobilization of unmodified carbohydrates on hydrazide-coated glass slides. Organic Lett. 2005;7:4269–4272. doi: 10.1021/ol051753z. [DOI] [PubMed] [Google Scholar]
- 15.Huang CY. Thayer DA. Chang AY, et al. Carbohydrate microarray for profiling the antibodies interacting with Globo H tumor antigen. Proc Natl Acad Sci USA. 2006;103:15–20. doi: 10.1073/pnas.0509693102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevens J. Blint O. Glaser L, et al. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol. 2006;355:1143–1155. doi: 10.1016/j.jmb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Liang PH. Wang SK. Wong CH. Quantitative analysis of carbohydrate-protein interactions using glycan microarrays: determination of surface and solution dissociation constants. J Am Chem Soc. 2007;129:11177–11184. doi: 10.1021/ja072931h. [DOI] [PubMed] [Google Scholar]
- 18.Oyelaran O. McShane LM. Dodd L. Gildersleeve JC. Profiling human serum antibodies with a carbohydrate antigen microarray. J Prot Res. 2009;8:4301–4310. doi: 10.1021/pr900515y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam KS. Lebl M. Krchnak V. The one-bead-one-compound combinatorial library method. Chem Rev. 1997;97:411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 20.MacBeath G. Koehler AN. Schreiber SL. Printing small molecules as microarrays and detecting protein-ligand interactions en masse. J Am Chem Soc. 1999;121:7967–7968. [Google Scholar]
- 21.Xu Q. Lam KS. Protein and chemical microarrays—powerful tools for proteomics. J Biomed Biotechnol. 2003;5:257–266. doi: 10.1155/S1110724303209220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee M. Shin I. Fabrication of chemical microarrays by efficient immobilization of hydrazide-linked substances on epoxide-coated glass surfaces. Angew Chem Int Ed Engl. 2005;44:2881–2884. doi: 10.1002/anie.200462720. [DOI] [PubMed] [Google Scholar]
- 23.Bradner JE. McPherson OM. Koehler AN. A method for the covalent capture and screening of diverse small molecules in a microarray format. Nat Protoc. 2006;1:2344–2352. doi: 10.1038/nprot.2006.282. [DOI] [PubMed] [Google Scholar]
- 24.Bradner JE. McPherson OM. Mazitschek R, et al. A robust small-molecule microarray platform for screening cell lysates. Chem Biol. 2006;13:493–504. doi: 10.1016/j.chembiol.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Sun YS. Landry JP. Fei YY, et al. Macromolecular scaffolds for immobilizing small molecule microarrays in label-free detection of protein-ligand interactions on solid support. Analyt Chem. 2009;81:5373–5380. doi: 10.1021/ac900889p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fei YY. Landry JP. Sun Y, et al. Screening small-molecule compound microarrays for protein ligands without fluorescence labeling with a high-throughput scanning microscope. J Biomed Opt. 2010;15:016018. doi: 10.1117/1.3309743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipshutz RJ. Fodor SP. Gingeras TR. Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 28.Schena M. Microarray Analysis. John Wiley & Sons; Hoboken, NJ: 2003. [Google Scholar]
- 29.Schena M. Protein Microarrays. Jones and Bartlett Publishers; Sudbury, MA: 2004. [Google Scholar]
- 30.International Human Genome Sequencing Consortium: Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 31.Pennisi E. Working the (gene count) numbers: finally a firm answer. Science. 2007;316:1113–1113. doi: 10.1126/science.316.5828.1113a. [DOI] [PubMed] [Google Scholar]
- 32.Harrison PM. Kumar A. Lang N. Snyder M. Gerstein M. A question of size: the eukaryotic proteome and the problems in defining it. Nucleic Acids Res. 2002;30:1083–1090. doi: 10.1093/nar/30.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mao H. Graziano JJ. Chase TM, et al. Spatially addressed combinatorial protein libraries for recombinant antibody discovery and optimization. Nat Biotechnol. 2010;28:1195–1202. doi: 10.1038/nbt.1694. [DOI] [PubMed] [Google Scholar]
- 34.Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105–1116. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- 35.Sun YS. Landry JP. Fei YY, et al. Effect of fluorescently labeling protein probes on kinetics of protein-ligand reactions. Langmuir. 2008;24:13399–13405. doi: 10.1021/la802097z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karlsson R. Michaelsson A. Mattsson L. Kinetic analysis of monoclonal antibody-antigen interactions with a new biosensor based analytical system. J Immunol Methods. 1991;145:229–240. doi: 10.1016/0022-1759(91)90331-9. [DOI] [PubMed] [Google Scholar]
- 37.Shumaker-Parry JS. Campbell CT. Quantitative methods for spatially resolved adsorption/desorption measurements in real time by surface plasmon resonance microscopy. Analyt Chem. 2004;76:907–917. doi: 10.1021/ac034962a. [DOI] [PubMed] [Google Scholar]
- 38.Usui-Aoki K. Shimada K. Nagano M. Kawai M. Koga H. A novel approach to protein expression profiling using antibody microarrays combined with surface plasmon resonance technology. Proteomics. 2005;5:2396–2401. doi: 10.1002/pmic.200401171. [DOI] [PubMed] [Google Scholar]
- 39.Rich RL. Myszka DG. Survey of the year 2005 commercial optical biosensor literature. J Mol Recognit. 2006;19:478–534. doi: 10.1002/jmr.808. [DOI] [PubMed] [Google Scholar]
- 40.Singh BK. Hillier AC. Surface plasmon resonance imaging of biomolecular interactions on a grating-based sensor array. Anal Chem. 2006;78:2009–2018. doi: 10.1021/ac0519209. [DOI] [PubMed] [Google Scholar]
- 41.Boozer C. Kim G. Cong S. Guan H. Londergan T. Looking towards label-free biomolecular interaction analysis in a high-throughput format: a review of new surface plasmon resonance technologies. Curr Opin Biotechnol. 2006;17:400–405. doi: 10.1016/j.copbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 42.Zhu XD. Comparison of two optical techniques for label-free detection of biomolecular microarrays on solids. Opt Commun. 2006;259:751–753. [Google Scholar]
- 43.Piehler J. Brecht A. Gauglitz G. Maul C. Grabley S. Zerlin M. Specific binding of low molecular weight ligands with direct optical detection. Biosens Bioelectron. 1997;12:531–538. doi: 10.1016/s0956-5663(97)00010-9. [DOI] [PubMed] [Google Scholar]
- 44.Hanel C. Gauglitz G. Comparison of reflectometric interference spectroscopy with other instruments for label-free optical detection. Anal Bioanal Chem. 2002;372:91–100. doi: 10.1007/s00216-001-1197-3. [DOI] [PubMed] [Google Scholar]
- 45.Özkumur E. Needham JW. Bergstein DA, et al. Label-free and dynamic detection of biomolecular interactions for high-throughput microarray applications. Proc Natl Acad Sci USA. 2008;105:7988–7992. doi: 10.1073/pnas.0711421105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurrat R. Textor M. Ramsden JJ. Boni P. Spencer ND. Instrumental improvements in optical waveguide light mode spectroscopy for the study of biomolecule adsorption. Rev Sci Instrum. 1997;68:2172–2176. [Google Scholar]
- 47.Höök F. Vörös J. Rodahl M, et al. A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation. Colloids Surf B. 2002;24:155–170. [Google Scholar]
- 48.Cross GH. Reeves A. Brand S, et al. The metrics of surface adsorbed small molecules on the Young's fringe dual-slab waveguide interferometer. J Phys D Appl Phys. 2004;37:74–80. [Google Scholar]
- 49.Freeman NJ. Peel LL. Swann MJ, et al. Real time, high resolution studies of protein adsorption and structure at the solid-liquid interface using dual polarization interferometry. J Phys: Condens Matter. 2004;16:S2493. [Google Scholar]
- 50.Arwin H. Ellipsometry on thin organic layers of biological interest: characterization and applications. Thin Solid Films. 2000;377:48–56. [Google Scholar]
- 51.Arwin H. Is ellipsometry suitable for sensor applications? Sensors Actuators A Phys. 2001;92:43–51. [Google Scholar]
- 52.Azzam RMA. Bashara NM. Ellipsometry, Polarized Light. North-Holland; Amsterdam, The Netherlands: 1987. [Google Scholar]
- 53.Jin G. Jansson R. Arwin H. Imaging ellipsometry revisited: developments for visualization of thin transparent layers on silicon substrates. Rev Sci Instrum. 1996;67:2930–2936. [Google Scholar]
- 54.Wang ZH. Jin G. A label-free multisensing immunosensor based on imaging ellipsometry. Analyt Chem. 2003;75:6119–6123. doi: 10.1021/ac0347258. [DOI] [PubMed] [Google Scholar]
- 55.Landry JP. Zhu XD. Gregg JP. Label-free detection of microarrays of biomolecules by oblique-incidence reflectivity difference microscopy. Opt Lett. 2004;29:581–583. doi: 10.1364/ol.29.000581. [DOI] [PubMed] [Google Scholar]
- 56.Landry JP. Sun YS. Guo XW. Zhu XD. Protein reactions with surface-bound molecular targets detected by oblique-incidence reflectivity difference microscopes. Appl Opt. 2008;47:3275–3288. doi: 10.1364/ao.47.003275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fei YY. Landry JP. Sun YS, et al. A novel high-throughput scanning microscope for label-free detection of protein and small-molecule chemical microarrays. Rev Sci Instrum. 2008;79:013708. doi: 10.1063/1.2830286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu XD. Landry JP. Sun YS. Gregg JP. Lam KS. Guo X. Oblique-incidence reflectivity difference microscope for label-free high-throughput detection of biochemical reactions in a microarray format. Appl Opt. 2007;46:1890–1895. doi: 10.1364/ao.46.001890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Landry JP. Zhu XD. Gregg JP. Guo XW. Detection of biomolecular microarrays without fluorescent-labeling agents. Proc SPIE. 2004;5328:121–128. [Google Scholar]
- 60.Thomas P. Nabighian E. Bartelt MC. Fong CY. Zhu XD. An oblique-incidence optical reflectivity difference and LEED study of rare-gas growth on a lattice-mismatched metal substrate. Appl Phys A. 2004;79:131–137. [Google Scholar]
- 61.Sober HA. Handbook of Biochemistry: Selected Data for Molecular Biology. Chemical Rubber Co.; Cleveland, OH: 1970. [Google Scholar]
- 62.Sreenath K. Prabhasankar P. Venkatesh YP. Generation of an antibody specific to erythritol, a non-immunogenic food additive. Food Addit Contam. 2006;23:861–869. doi: 10.1080/02652030600722353. [DOI] [PubMed] [Google Scholar]
- 63.Morton TA. Myszka DG. Chaiken IM. Interpreting complex binding kinetics from optical biosensors: a comparison of analysis by linearization, the integrated rate equation, and numerical integration. Analyt Biochem. 1995;227:176–185. doi: 10.1006/abio.1995.1268. [DOI] [PubMed] [Google Scholar]
- 64.Myszka DG. Arulanatham PR. Sana T. Wu Z. Morton TA. Ciardelli TL. Kinetic analysis of ligand binding to interleukin-2 receptor complexes created on an optical biosensor surface. Protein Sci. 1996;5:2468–2478. doi: 10.1002/pro.5560051209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schuck P. Reliable determination of binding affinity and kinetics using surface plasmon resonance biosensors. Curr Opin Biotechnol. 1997;8:498–502. doi: 10.1016/s0958-1669(97)80074-2. [DOI] [PubMed] [Google Scholar]
- 66.Jung H. Yang T. Lasagna MD. Shi J. Reinhart GD. Cremer PS. Impact of hapten presentation on antibody binding at lipid membrane interfaces. Biophys J. 2008;94:3094–3103. doi: 10.1529/biophysj.107.115519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adamczyk H. Mattingly PG. Shrender K. Yu Z. Surface plasmon resonance (SPR) as a tool for antibody conjugate analysis. Bioconjugate Chem. 1999;10:1032–1037. doi: 10.1021/bc990057e. [DOI] [PubMed] [Google Scholar]
- 68.MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32:526–532. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.