

Abstract
An oxetane-substituted sulfoxide has demonstrated potential as a dimethylsulfoxide substitute for enhancing the dissolution of organic compounds with poor aqueous solubilities. This sulfoxide may find utility in applications of library storage and biological assays. For the model compounds studied, significant solubility enhancements were observed using the sulfoxide as a cosolvent in aqueous media. Brine shrimp, breast cancer (MDA-MB-231), and liver cell line (HepG2) toxicity data for the new additive are also presented, in addition to comparative IC50 values for a series of PKD1 inhibitors.
Introduction
Modern drug development relies on high-throughput screening assays. Often, these trials use compound libraries stored in solution for periods of several months to as long as 3 years.1 Dimethylsulfoxide (DMSO) 1 has been used as the storage solvent of choice, but problems, including compound degradation and precipitation, are frequently encountered. In one case study, qualitative compound precipitation was observed in 26% of test plates.2 Systematic studies of compound degradation in DMSO have indicated that ∼50% of samples degraded over 12 months when stored in anhydrous DMSO at ambient temperature.3,4 Compound storage problems are augmented by low hydrophilicity, since a large portion of screening libraries is composed of compounds designed for enhanced membrane permeability. The trend toward lipophilic, higher-molecular-weight compounds results in libraries of materials with lower intrinsic aqueous solubilities.5 Current estimates state that 30%–50% of compounds in screening libraries have aqueous solubilities of <10 μM.6 These lipophilic molecules are more likely to precipitate from DMSO stock solutions, leading to erroneously low assay concentrations when using the DMSO stock for sample preparation. Additionally, poor aqueous solubility causes precipitation from aqueous media after dilution of DMSO stock solutions.6 When compound concentrations in assay media fall below calculated concentrations, flawed conclusions regarding toxicity, efficacy, or structure–activity relationships are drawn.5,6
Aqueous dissolution of problematic compounds can be enhanced by pH adjustment,7 salt formation,8 or chemical modification of the substrate (formation of pro-drugs).8 If these methods are not applicable, complexing agents or cosolvents can be added to aid in dissolution. Some examples include cyclodextrins,9 dendrimers,10 low-molecular-weight polyethylene glycols (PEGs, e.g., PEG 400),5 and solvents such as glycerin2 and N-methyl pyrrolidone (NMP).11 Other solubilizing agents have designs based on DMSO; one example is a polymeric sulfoxide derived from poly-L-methionine 2 (Fig. 1).12
Fig. 1.

Structures of sulfoxides used for compound storage or aqueous solubility enhancement.
In this study, we examined the use of sulfoxide 3 (Fig. 1) as a solubilizer and general compound storage additive. We found that addition of sulfoxide 3 increased the aqueous solubility of several model problem compounds, including naproxen, quinine, curcumin, carbendazim, and griseofulvin. The solubility enhancement surpassed that of DMSO at mass fractions >10%. Herein, we describe our findings and hypothesis of the mode of the cosolvent effect. A comparative study of the toxicity and applicability of sulfoxide 3 to cellular and in vivo bioassays is also discussed.
Materials and Methods
General
Moisture-sensitive reactions were performed under an atmosphere of nitrogen. 3-Tosyloxymethyl-3-methyl-oxetane was prepared according to a literature protocol.13 Curcumin (Acros, 95%), naproxen (Acros, 99%), quinine (Acros, 99%), DMSO (Aldrich, 99.9+%), and high-performance liquid chromatography (HPLC)-grade water (Aldrich, Chromasolv®) were purchased from commercial suppliers and used as received. Carbendazim (Aldrich, 97%) was recrystallized from absolute ethanol (EtOH), and griseofulvin (Acros, 97%) was recrystallized from toluene. N-Methyl-2-pyrrolidone (Acros, 99%) was distilled from CaH2 under vacuum and stored over 4 Å MS. All other reagents were used as received unless otherwise stated. Analytical thin-layer chromatography was performed on precoated silica gel 60 F-254 plates (particle size 0.040–0.050 mm, 230–400 mesh) and visualized by staining with KMnO4 or p-anisaldehyde solutions. 1H nuclear magnetic resonance (NMR) spectra (CDCl3) and 13C NMR spectra (CDCl3) were referenced to residual chloroform (7.27 ppm, 1H, 77.00 ppm, 13C). Chemical shifts (δ) are reported in ppm using the following convention: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet, b=broad), coupling constants, and integration. Infrared (IR) spectra were collected as attenuated-total-reflection infrared (ATR-IR) spectra. Mass spectra were obtained on a Micromass Autospec double focusing instrument. ultraviolet/visible (UV/VIS) spectra were recorded on a Perkin Elmer Lambda EZ210 spectrophotometer. pH Determinations were made using a 3-mm Ross™ glass combination micro-pH electrode (model 8220BNWP) after calibration in standard buffer solutions (pH 4.0, 7.0, and 10.0) at room temperature (rt).
Bis((3-methyloxetan-3-yl)methyl)sulfane (6)
A three-necked 3-L round-bottom flask equipped with an overhead stirrer, internal thermometer, and a third arm bearing an argon balloon was charged with 3-tosyloxymethyl-3-methyl-oxetane 4 (45.4 g, 177 mmol) and backfilled with N2 (3×). To the flask was added acetonitrile (900 mL) via cannula. The reaction apparatus was placed in a large heating mantle. The argon balloon was replaced with a 250-mL addition funnel containing a solution of Na2S·9H2O (94.5 g 386 mmol) in degassed H2O (100 mL). The solution was added drop-wise over 25 min. Once the addition was complete, the reaction mixture was heated to 70°C over 45 min and maintained at 70°C for 1 h. The mixture was cooled to 20°C (internal temp), the resulting white precipitate was filtered by gravity, and to the filtrate was added EtOAc (1 L). The resulting precipitate was removed by aspirator filtration, and the filtrate was divided into two 1-L batches. To each batch was added water (500 mL), the layers were separated, and the aqueous portion was extracted with EtOAc (2×200 mL). The combined organic layers were washed with brine (100 mL), and the EtOAc layers from the batches were combined and dried (Na2SO4) overnight, filtered, and concentrated. Kugelrohr distillation was performed on the concentrate. One fraction (T<100°C, 15 Torr) was discarded, and subsequent product collection (140°C<T<160°C) yielded a yellow distillate. The distillate was taken up in EtOAc (200 mL), washed with water (100 mL) and brine (100 mL), dried (Na2SO4), and concentrated. Kugelrohr distillation (140°C, 15 Torr) afforded 6 (14.2 g, 79%) as a yellow-green oil: IR (ATR) 2956, 2924, 2861, 1450, 1236, 973, 829 cm−1; 1H NMR (300 MHz, CDCl3) δ 4.47 (d, J=5.7 Hz, 4 H), 4.38 (d, J=6.0 Hz, 4 H), 2.93 (s, 4 H), 1.38 (s, 4 H); 13C NMR (75 MHz, CDCl3) δ 81.9, 43.7, 40.3, 23.0; HRMS electrospray ionization (ES) m/z calc for C10H18O2NaS (M+Na) 225.0925, found 225.0908.
3,3′-Sulfinylbis(methylene)bis(3-methyloxetane) (3)
A 1-L round-bottom flask was charged with a solution of 6 (14.9 g, 73.6 mmol) in methanol (MeOH; 240 mL) and cooled to 0°C. A solution of NaIO4 (16.5 g, 77.3 mmol) in water (180 mL) was added via addition funnel for ∼15 min. The ice bath was removed and the slurry was warmed to rt. MeOH (2×50 mL, added 20 min apart) was added, and the mixture was stirred for 12 h at rt. The mixture was filtered through a fritted funnel, and the white precipitate was washed with MeOH. The combined filtrate and washings were concentrated in vacuo, and the concentrate was coevaporated with toluene (200 mL). CH2Cl2 (400 mL) was added to the residue, followed by MgSO4. The mixture was filtered, and the filtrate was concentrated in vacuo to afford crude 3 (15.82 g) as a yellow solid. To the flask containing the crude solid was added toluene (200 mL), and the slurry was heated to 60°C to affect complete dissolution. Decolorizing carbon was added, and the mixture was filtered by gravity into a 1-L Erlenmeyer flask. To the colorless solution was slowly added distilled hexanes (∼100 mL total) until cloudiness/precipitation occurred. The mixture was allowed to stand at rt overnight. Upon filtration and drying under high vacuum, 3 (10.49 g) was collected as a white solid. Material recovered from the mother liquor was recrystallized to afford an additional 2.68 g of 3 as white solid for a total yield of 82%. Reported analytical data refer to that of the first crop: Mp 92.8–94.1°C; IR (ATR) 2,939, 2,863, 1,451, 1,381, 1,227, 1,026, 971 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.80 (d, J=6.0 Hz, 2 H), 4.61 (d, J=5.6 Hz, 2 H), 4.50 (d, J=5.4 Hz, 2 H), 4.45 (d, J=6.0 Hz, 2 H), 3.38 (d, J=12.9 Hz, 2 H), 2.75 (d, J=12.9 Hz, 2 H), 1.61 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 82.4, 82.0, 61.9, 38.4, 23.4; HRMS (ES) m/z calc for C10H18O3NaS (M+Na) 241.0874, found 241.0885.
To unambiguously characterize 3 as the sulfoxide, the corresponding sulfone was synthesized from sulfide 6.
3,3′-Sulfonylbis(methylene)bis(3-methyloxetane)
A suspension of oxone (650 mg, 1.06 mmol) in water (2.0 mL) was cooled to 10°C and treated (dropwise) with a solution of 6 (108 mg, 0.533 mmol) in MeOH (2.0 mL). The solution was warmed to rt and stirred for 1 h. MeOH was removed in vacuo, and the aqueous layer was diluted with water (5 mL) and extracted with CH2Cl2 (4×10 mL). The combined organic layers were washed with brine (5 mL), dried (MgSO4), and concentrated in vacuo to afford the sulfone (120 mg, 96%) as a white solid: Mp 93.4–95.1°C; IR (ATR) 2,949, 2,867, 1,456, 1,301, 1,277, 967 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.68 (d, J=6.4 Hz, 4 H), 4.46 (d, J=6.4 Hz, 4 H), 3.43 (s, 4 H), 1.69 (s, 6 H); 13C NMR (100 MHz, CDCl3) δ 82.2, 62.6, 37.9, 23.3; HRMS atmospheric pressure chemical ionization (APCI) m/z calc for C10H19O4S (M+H) 235.1004, found 235.1032.
Determination of LogP Value of 3
The logP (octanol-water partition coefficient) was determined using the shake-flask method. Three determinations were made. A representative procedure is as follows: a 250-mL separatory funnel was charged with a solution of 3 (50.0 mg) in water (50.0 mL) and n-octanol (50.0 mL). The funnel was capped and inverted 100 times. The funnel and contents were left to stand at rt (23.5°C) for 40 h. Aliquots of both phases were analyzed by UV/VIS (214 nm for the aqueous layer and 218 nm for the octanol layer), and the concentration in each layer was determined using previously generated calibration curves. In the case of the aqueous layer, a 10-fold dilution was necessary before measurement. All measurements were made in triplicate. The logP was determined as log ([3]octanol/[3]aqueous), and the average logP value from the three trials was −0.87.
General Procedure for Determination of Solubility in Solutions of 3 and HPLC-Grade Water
Preparation of sulfoxide/water solutions
Solutions of 3 and HPLC-grade water were prepared in 1-dram vials by dissolving the appropriate amount of sulfoxide 3 in HPLC-grade water (2.00 mL). In the case of the 25% solution of 3 in H2O (w/w), 3 (500 mg) was dissolved in water (1.50 mL). Each vial was placed on a platform shaker and shaken at 200 rpm for 30 min.
Solubility measurements
Eppendorf vials (1.5 mL size) were charged with model compounds in excess. Vials were charged with HPLC-grade water (0.500 mL) or the appropriate 3/water solution (0.500 mL). The vials were briefly vortexed and equilibrated in an end-over-end rotator at 30.0°C for 20 h. The vials were centrifuged (4,000 rpm, 1,300 g, 15 min, rt) directly after removal from the rotator, and aliquots of the supernatant (0.400 mL) were filtered through 0.45-μm syringe filters. The pH of each solution was measured using a ThermoScientific electrode (3 mm tip). Each solution was diluted with an appropriate volume of either absolute EtOH (quinine, naproxen, griseofulvin) or MeOH (carbendazim). Concentrations were calculated by using previously generated calibration curves. Appropriate blanks were prepared by diluting aliquots (0.400 mL) of the 3/water solutions (or HPLC-grade water in the case of the control) in an analogous fashion to the sample being measured.
General Procedure for Determination of Solubility in Solutions of DMSO and HPLC-Grade Water
Preparation of DMSO/water solutions
Solutions of DMSO and HPLC-grade water were prepared in 1-dram vials by dissolving the appropriate amount of DMSO in HPLC-grade water (2.00 mL). Each vial was placed on a platform shaker and shaken at 200 rpm for 30 min.
Solubility measurements
Eppendorf vials (1.5 mL size) were charged with model compounds in excess. Vials were charged with either HPLC-grade water (1.00 mL) or the appropriate DMSO/water solution (1.00 mL). The vials were briefly vortexed and equilibrated in an end-over-end rotator at 30.0°C for 20 h. The vials were centrifuged directly after removal from the rotator (4,000 rpm, 1,300 g, 15 min, rt), and aliquots of the supernatant (0.800 mL) were filtered through 0.45-μm syringe filters. The pH of each solution was measured using a ThermoScientific electrode (3 mm tip). Each solution was diluted with an appropriate volume of either absolute EtOH (quinine, naproxen, griseofulvin) or MeOH (carbendazim). Concentrations were calculated using previously generated calibration curves. Appropriate blanks were prepared by diluting aliquots (0.800 mL) of the DMSO/water solutions (or HPLC-grade water in the case of the control) in analogous fashion to the sample being measured.
General Procedure for Solubility Determination in Mixtures of Sulfoxide 3 in pH 7.0 Buffer
Preparation of sulfoxide/buffer solutions
In the case of a 10% w/w solution of sulfoxide 3 in 0.01 M pH 7.0 phosphate buffer, 3 (700 mg) was dissolved in 0.01 M Na2HPO4/NaH2PO4 buffer (6.30 mL) and equilibrated on a platform shaker at 200 rpm for 30 min at rt. In the case of a 25% w/w solution of sulfoxide 3 in 0.01 M pH 7.0 phosphate buffer, 3 (1.50 g) was dissolved in 0.01 M Na2HPO4/NaH2PO4 buffer (4.50 mL) and equilibrated on a platform shaker at 200 rpm for 30 min at rt.
Solubility measurements
Eppendorf vials (1.5 mL size) were charged with model compounds in excess. Vials were charged with either pH 7.0 phosphate buffer (1.00 mL) or the appropriate 3/buffer solution. The vials were briefly vortexed and equilibrated in an end-over-end rotator at 30.0°C for 20 h. The vials were centrifuged directly after removal from the rotator (4,000 rpm, 1,300 g, 15 min, rt), and aliquots of the supernatant (0.800 mL) were filtered through 0.45-μm syringe filters. The pH of each solution was measured using a ThermoScientific electrode (3 mm tip) after calibration. Each solution was diluted with an appropriate volume of either absolute EtOH (quinine, naproxen) or MeOH (carbendazim). Concentrations were determined using standard additions of a stock solution of the compound in either absolute EtOH (quinine, naproxen) or MeOH (carbendazim) to aliquots of the diluted filtrates.
General Procedure for Kinetic Solubility Measurements
A polypropylene tube was charged with phosphate-buffered saline (PBS; 490 μL). To the buffer was added a 10-mM stock solution of compound (10 μL). The tube was vortexed and equilibrated on an end-over-end rotator for 15 min at rt. Aliquots (400 μL) were filtered through 0.45-μm syringe filters and diluted to 5.0 mL with absolute MeOH. Concentrations were determined by UV/VIS analysis.
General Procedure for Brine Shrimp Toxicity Assays14
Sample preparation
Stock solutions of 3 were prepared by dissolving 3 (50.0 mg) in HPLC-grade water (5.0 mL) (solution A) and 3 (2.50 g) in HPLC-grade water (10.0 mL) (solution B). Stock solutions of DMSO were prepared by diluting DMSO (45 μL) with HPLC-grade water (5.0 mL) (solution C) and DMSO (2.27 mL) with HPLC-grade water (10.0 mL) (solution D).
In each case, five replicates were performed. Each replicate was performed in a 2-dram vial marked at the 4 and 5 mL volume points. To each vial was added artificial seawater (3 mL) followed by the appropriate volume of stock solution. For set 1 (1.0 mg/mL), solution A (0.500 mL) or solution C (0.500 mL) was added. For set 2 (5.0 mg/mL), solution B (0.100 mL) or solution D (0.100 mL) was added to each vial. For set 3 (20.0 mg/mL), solution B (0.400 mL) or solution D (0.400 mL) was added to each vial. For set 4 (50.0 mg/mL), solution B (1.00 mL) or solution D (1.00 mL) was added to each vial. Controls containing HPLC-grade water (0.100 mL, 0.400 mL, and 1.00 mL) were prepared in the same manner, and five replicates of each control were prepared.
Brine shrimp hatching
Brine shrimp eggs (San Francisco Bay Brand) were hatched in a commercial salt mixture (Instant Ocean). Constant aeration was provided using a pump and airstone, and illumination was maintained using a desk lamp. The shrimp were collected in a separate tank after 48 h and used within 3–4 h of collection.
Assay
Brine shrimp (10) were added to each vial using a plastic transfer pipet. After the shrimp were transferred, artificial seawater was added until the volume reached the 5-mL mark. One drop of a yeast suspension prepared by suspending 11 mg yeast in seawater (20 mL) was added to each vial. The shrimp were counted at t=24 h. Another drop of freshly prepared yeast solution (6 mg in 10 mL seawater) was added, the vials were maintained under illumination, and shrimp were counted at t=48 h.
General Procedure for Calculation of Water Absorption
Oven-dried flasks capped with septa were cooled under an N2 atmosphere and charged with a volume of the appropriate solutions (DMSO, 3.0 mL; NMP, 600 μL; 25% 3/NMP, 600 μL). The water content was determined by Karl Fischer titration using ∼100-μL aliquots (t=0 measurement). Each septum was pierced with a 1.5-inch 18-gauge needle and left to stand at rt for 7 days. The water content was measured at the end of this period (t=7 days measurement) by Karl Fischer titration. All measurements were made in duplicate.
Results and Discussion
Water-soluble sulfoxide 3 was prepared in three steps from alcohol 4, which is commercially available or readily prepared from inexpensive 2-(hydroxymethyl)-2-methylpropane-1,3-diol.15 Briefly, treatment of 4 with p-toluenesulfonyl chloride in pyridine afforded tosylate 5.13 Dimeric sulfide formation using Na2S followed by oxidation with NaIO4 provided sulfoxide 3 (Fig. 2). The synthesis is amenable to large-scale preparation and requires no chromatography.
Fig 2.

Synthesis of water-soluble sulfoxide 3.
Initially, we aimed to use sulfoxide 3 as a substitute for DMSO in chemical transformations such as modified Swern and Kornblum oxidations. We reasoned the resulting sulfide 6, being less volatile and odorous than dimethyl sulfide, would be a more tractable byproduct on industrial scale. Additionally, bisoxetanyl sulfoxide 3 is water soluble, and we found that the sulfide byproduct could be removed from reaction mixtures by an oxidative work-up and aqueous washing procedure. Surprisingly, however, sulfoxide 3 was not a viable oxidizing agent in Kornblum oxidations. For example, α-bromoacetophenone was stable to an excess of 3 when monitored in CD3CN over the course of several weeks; under the same conditions, α-bromoacetophenone was reactive with DMSO within 2 days of treatment.
We took advantage of the stability and hydrophilicity of 3 by exploring its use for the solubility enhancement of poorly aqueous-soluble compounds. Oxetanes are attractive functional groups for increasing aqueous solubility due to their rigid geometries and exposed polar surface area at the ring ether. Among unsubstituted cyclic ethers, oxetane has the greatest basicity, which may be attributed to a smaller carbon-to-oxygen ratio and a larger dipole moment than its larger counterparts (e.g., tetrahydrofuran and tetrahydropyran).16 Recently, Müller, Carreira, and coworkers demonstrated that replacing a gem-dimethyl group with an oxetane moiety can increase a scaffold's aqueous solubility by up to three orders of magnitude while also enhancing metabolic stability.17 We reasoned that sulfoxide 3 may have sufficient aqueous solubility to be completely miscible with water at useful cosolvent concentrations while also disrupting the hydrogen-bonding network of water, thus aiding in solubilization of lipophilic drug candidate compounds.
To evaluate the utility of sulfoxide 3 as a solubilizing agent, we chose a test set of poorly aqueous-soluble compounds, beginning with curcumin 7 (Fig. 3). Curcumin has shown promise in treating colon cancer and various other disorders, but its use is limited in part by its low aqueous solubility (0.6 μg/mL at ambient temperature, as high as 7.4 μg/mL upon heating) and consequently limited bioavailability.18 We measured the solubility of curcumin by UV/VIS spectroscopy after equilibration for 20 h at ambient temperature in an end-over-end rotator. We were pleased to see an increase in aqueous solubility at ambient temperature to 60±20 μg/mL using a 25 wt% solution of 3 in water. Although the aqueous solubility remained low, the ∼100-fold enhancement factor was encouraging.
Fig. 3.

Compounds screened in the aqueous solubility study.
We further examined the solubility enhancement of 8–12 by equilibrating the compounds in solutions of increasing amounts of sulfoxide 3 in water. Gratifyingly, up to 10-fold increases in aqueous solubility were observed for 8–11, and almost twofold improvements in aqueous solubility were observed in comparison to equivalent DMSO/water solutions (Figs. 4–7).
Fig. 4.

Solubility of quinine 8 in aqueous solutions with sulfoxide 3 (△) and DMSO (+) as cosolvents. Each trial was run in duplicate and each point represents the average of the duplicate trials. In the case of sulfoxide 3, the pH ranged from 8.7 (with no additive) to 9.4 (with a 0.25 weight fraction additive). In the case of DMSO, the pH ranged from 8.9 (with no additive) to 9.5 (with a 0.25 weight fraction additive). DMSO, dimethylsulfoxide.
Fig. 7.

Solubility of griseofulvin 11 in aqueous solutions with sulfoxide 3 (△) and DMSO (+) as cosolvents. Each trial was run in duplicate and each point represents the average of the duplicate trials. In the case sulfoxide 3, the pH ranged from 6.3 (with no additive) to 6.8 (with a 0.20 weight fraction additive). In the case of DMSO, the pH ranged from 6.3 (with no additive) to 6.8 (with a 0.20 weight fraction additive).
Fig. 5.

Solubility of naproxen 9 in aqueous solutions with sulfoxide 3 (△) and DMSO (+) as cosolvents. Each trial was run in duplicate and each point represents the average of the duplicate trials. In the case of sulfoxide 3, the pH ranged from 4.6 (with no additive) to 4.2 (with a 0.25 weight fraction additive). In the case of DMSO, the pH ranged from 4.8 to 4.4.
Fig. 6.

Solubility of carbendazim 10 in aqueous solutions with sulfoxide 3 (△) and DMSO (+) as cosolvents. Each trial was run in duplicate and each point represents the average of the duplicate trials. In the case of sulfoxide 3, the pH ranged from 6.7 (with no additive) to 7.0 (with a 0.15 weight fraction additive). In the case of DMSO, the pH ranged from 6.8 (with no additive) to 7.4 (with a 0.25 weight fraction additive).
Based on the solubility curves (Figs. 4–7), the sulfoxide 3 and DMSO function as cosolvents rather than complexing agents. According to the model derived by Yalkowsky,7 an exponential increase in observed solubilities occurs with increasing the volume fraction solvent according to equation 1, where Smix and Sw are the solubility of the solute in the cosolvent mixture and water, respectively, σ is the solubilizing power of the cosolvent, and fc is the volume fraction of the cosolvent. The slope of a semi-log plot (σ) is related to the cosolvent's ability to disrupt the intermolecular hydrogen bond network of water and form a less polar solvent mixture. Thus, the solubilizing power of sulfoxide 3 and DMSO can be accounted for when comparing the difference in the experimental log P of sulfoxide 3 (−0.87) and the reported log P of DMSO (−1.3).19
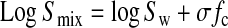 |
(1) |
We can tentatively rule out complexation as a solubilization mechanism assuming that additive–solute complexes would form in a 1:1 ratio. If that were the case, a linear correlation between additive fraction and solubility would be observed.
The aqueous solubility of estrone 12 is low (0.8 μg/mL),20,21 and we were unable to quantify the aqueous solubility or observe an increase in solubility in the case of a 25% (w/w) mixture of 3 and water; however, increasing the pH of the media using a pH 9.0 buffer (Borax) as well as addition of NMP to generate a ternary mixture was useful. In this case, the ternary mixture of 3:1:1 pH 9.0 buffer:NMP:DMSO was more effective at solubilization than the 3:1:1 pH 9.0 buffer:NMP:3 mixture (Table 1, entries 3 and 4).
Table 1.
Solubility of Estrone 12 in Media Buffered at pH 9.0
| Entry | Medium | Solubility (μg/mL)a |
|---|---|---|
| 1 | 3:1 pH 9.0 buffer/3 | 30±10 |
| 2 | 3:1 pH 9.0 buffer/NMP | 63±5 |
| 3 | 3:1:1 pH 9.0 buffer/NMP/3 | 160±20 |
| 4 | 3:1:1 pH 9.0 buffer/NMP/DMSO | 230±30 |
Solubility was determined after equilibration for 20 h at 30°C. Data were obtained by UV/VIS analysis of the saturated solutions and were confirmed by analysis of independently prepared estrone standards. Each entry represents a mean solubility±standard deviation (n=3).
DMSO, dimethylsulfoxide; NMP, N-methylpyrrolidinone.
We also explored the solubility of test compounds with ionizable functionalities in 0.01 M pH 7.0 phosphate buffer (Table 2). In the case of naproxen, the sulfoxide had little effect on the ionized substrate even at 25% w/w concentrations. The effect on the solubility of quinine was diminished at 10% w/w, but an advantage in using 3 was observed in 25% w/w solutions. The solubility of carbendazim in the buffered medium was the same as in solutions made from unbuffered HPLC-grade water.
Table 2.
Solubility of Selected Model Substrates in Solutions of Sulfoxide 3 in 0.01 M pH 7.0 Phosphate Buffer
| Entry | Compound | Weight percent 3 in pH 7.0 buffer | Solution pHa | Measured solubility (μg/mL)b |
|---|---|---|---|---|
| 1 | 8 | 0 | 6.0 | 1,100 (780)c |
| 2 | 8 | 10 | 5.8 | 1,900 (1,300)c |
| 3 | 8 | 25 | 5.2 | 6,200 (3,900)c |
| 4 | 9 | 0 | 7.6 | 950d |
| 5 | 9 | 10 | 8.0 | 1,100 |
| 6 | 9 | 25 | 8.2 | 1,400 |
| 7 | 10 | 0 | 6.8 | 10 |
| 8 | 10 | 10 | 6.9 | 34 |
| 9 | 10 | 25 | 7.1 | 77 |
The pH was measured electrochemically after excess compound had been filtered from the solution.
Measurements were performed in duplicate unless otherwise noted.
The two numbers represent data from separate trials where entries 1–3 were run in parallel. There was some variability in the results from the two trials.
Average of five trials.
We also determined the kinetic solubility of two test compounds, carbendazim 10 and griseofulvin 11, in PBS solution after adding stock solutions prepared in three media: DMSO, NMP, and 25% 3/NMP (Table 3). The test compounds were prepared at concentrations of 10 mM and added to the buffer at room temperature such that cosolvent concentration was fixed at 2%. The measured solubility was consistent for both test compounds across the three stock solutions. While the kinetic solubility of griseofulvin was found to be higher than reported22 (and nearing the threshold solubility of 200 μM), there was no statistical difference in the kinetic solubilities of the test compounds among the three different media.
Table 3.
Kinetic Solubility Measurements in Phosphate-Buffered Saline
| Entry | Compound | Medium of stock solution | Kinetic solubility (μM)a |
|---|---|---|---|
| 1 | 10 | DMSO | 140±13b |
| 2 | 10 | NMP | 160±5 |
| 3 | 10 | 25% 3/NMP | 150±9 |
| 4 | 11 | DMSO | 170±16 |
| 5 | 11 | NMP | 180±13 |
| 6 | 11 | 25% 3/NMP | 190±7 |
Data are reported as mean±standard deviation (n=3).
Data are reported as mean±standard deviation (n=5).
Recently, we disclosed a series of compounds with nanomolar to micromolar inhibitory activity against the serine/threonine protein kinase D isoform 1 (PKD1) (Fig. 8).23–25 Several lead structures, especially those containing a pyrimidine moiety, suffered from poor solubility not only in aqueous media but also in DMSO. To test the feasibility of using sulfoxide 3 as a cosolvent for in vitro assays, we examined its use in the radiometric PKD1 inhibition23–25 assay. Inhibitory activity at two concentrations (1 μM and 10 μM) was measured using compound stock solutions in three formulations: DMSO, NMP, and 25% 3/NMP (Figs. 9 and 10). Comparable biological efficacy was observed for stock solutions in 3/NMP versus DMSO. Most significantly, sulfoxide 3 did not interfere with the standard PKD1 inhibition assay.
Fig. 8.

Compounds assayed for PKD1 inhibitory activity. The structures have been reported previously.23–25 PKD1, protein kinase D isoform 1.
Fig. 9.

Plot of PKD1 activity with compound concentrations of 1 μM. Stock solutions were prepared in three different media [DMSO ( ), NMP (□), and 25% 3/NMP (▪)] at concentrations of 10 mM, and dilutions were performed using the same media. The % PKD1 activity is reported as the mean, and error bars represent SEM (n=3). The % PKD1 activity was determined as previously described.24 NMP, N-methylpyrrolidinone; SEM, standard error of the mean.
), NMP (□), and 25% 3/NMP (▪)] at concentrations of 10 mM, and dilutions were performed using the same media. The % PKD1 activity is reported as the mean, and error bars represent SEM (n=3). The % PKD1 activity was determined as previously described.24 NMP, N-methylpyrrolidinone; SEM, standard error of the mean.
Fig. 10.

Plot of PKD1 activity with compound concentrations of 10 μM. Stock solutions were prepared in three different media [DMSO (), NMP (□), and 25% 3/NMP (▪)] at concentrations of 10 mM, and dilutions were performed using the same media. The % PKD1 activity is reported as the mean, and error bars represent SEM (n=3). The % PKD1 activity was determined as previously described.24
Toxicity
The strain energy of the oxetane ring (25.2 kcal/mol)26 raises concerns about the electrophilicity and related toxicity and mutagenicity of molecules containing an oxetane moiety. Computational models have indicated that despite having comparable strain energy to oxirane (26.8 kcal/mol),26 oxetane is 106 times less susceptible to nucleophilic addition than oxetane.27 Animal studies have implicated the potential carcinogenicity of oxetane and 3,3-dimethyloxetane; it was shown that both compounds induce tumor formation at the site of injection in rats.28,29 However, a recent report studying the alkylating ability of oxetane, 3,3-dimethyloxetane, and 3-methyl-3-oxetanemethanol (1) demonstrated that these oxetanes are neither mutagenic nor genotoxic.30 Furthermore, alkylation of 4-(p-nitrobenzyl)pyridine was only observed at acidic pH, implicating that oxetanes do not act as alkylating agents at physiological pH.30
To assess the systemic toxicity of oxetane-substituted sulfoxide 3, we performed a brine shrimp assay (Table 4).14 Brine shrimp floating in water containing concentrations of 3 up to 20 mg/mL showed <10% mortality after 48 h. Shrimp incubated in water containing 50 mg/mL of 3 had 85% mortality after 24 h and 100% mortality within 48 h. These data indicate an LC50 of ∼32 mg/mL (i.e., at 147 mM). In comparison, brine shrimp treated with DMSO at the same concentrations showed no mortality after 24 h and only 15% mortality after 48 h at 50 mg/mL of DMSO. These results indicate that sulfoxide 3 may be tolerated in biological assays in concentrations by mass of up to 2%.
Table 4.
Results from a Brine Shrimp Assay to Assess the Toxicity of Sulfoxide 3
| Entry | Compound | Concentration (mg/mL) | % Mortalitya after 24 h | % Mortalitya after 48 h |
|---|---|---|---|---|
| 1 | 3 | 0 | <10 | 10 |
| 2 | 3 | 1 | 0 | 0 |
| 3 | 3 | 5 | 0 | 0 |
| 4 | 3 | 20 | 0–10 | 0–10 |
| 5 | 3 | 50 | 85 | 100 |
| 6 | DMSO | 0 | <10 | 10 |
| 7 | DMSO | 1 | 0 | 0–10 |
| 8 | DMSO | 5 | 0 | 0–6 |
| 9 | DMSO | 20 | 0 | 0 |
| 10 | DMSO | 50 | 0 | 15 |
Percent mortality was determined for an average of five trials. Percent mortality was determined by estimating the number of shrimp showing no motility after several minutes of observation.
The cellular toxicity of sulfoxide 3 was further determined for a breast cancer cell line (MDA-MB-231) and a liver cell line (HepG2). In both cases, the 50% growth inhibition (GI50) for 3 was ∼200 mM, whereas the GI50 for DMSO was determined as ∼800 mM. Interestingly, both cell lines exhibited the same threshold effect as observed in the case of the brine shrimp, showing only very limited toxicity at concentrations up to ∼100 mM.
Properties of 3/NMP Solutions
Because sulfoxide 3 is a solid, it would have to be mixed with an appropriate water-soluble cosolvent to act as a compound storage additive. We chose NMP for exploring this potential application due to its thermal stability and low toxicity. Furthermore, it had been demonstrated that NMP had a greater solubilizing power than ethanol and propylene glycol,11 and NMP was previously used for solubility enhancements both in bioassays31 and commercial pharmaceutical applications such as the Eligard® formulation for delivery of leuprolide to prostate cancer patients. We stored the compound test set (Fig. 2) in 25% w/w solutions of 3 and NMP for 6 weeks at −20°C. During this time, we noted that 3 partly precipitated from the solution at this temperature, but no change in model compound concentration was observed after thawing of the storage vessels.
A study performed at Abbott indicated that water absorption might induce more significant compound degradation than oxygen exposure.32 To ascertain the degree of water absorption, we monitored a 25% w/w 3/NMP solution for 1 week at ambient temperature (Table 5). Although significant water absorption was observed (∼7,000 ppm over the course of 7 days), the 3/NMP solution absorbed less water than NMP alone. This result indicates that the hygroscopicity of sulfoxide 3 is low relative to NMP.
Table 5.
Comparative Water Absorption Measurements of Possible Compound Storage Media
| Entry | Medium | Water content at t=0 (ppm)a | Water content at t=7 d (ppm)a |
|---|---|---|---|
| 1 | DMSO | 160, 170b | 1,080, 1,130b |
| 2 | NMP | 150, 130c | 12,090, 17,000c |
| 3 | 25% 3/NMP (w/w) | 160, 200c | 6,500, 8,600c |
Water content was analyzed by Karl Fischer titration. The two values listed represent individual vessels, the first value in each column corresponding to the same vessel at each time-point.
Average of three measurements.
Average of two measurements.
Conclusion
We have demonstrated the utility of oxetane-substituted sulfoxide 3 as a cosolvent for enhancing the aqueous solubility of model drug compounds. Although the relative acute toxicity of 3 was higher than that of DMSO in brine shrimp and cell-based assays, it was sufficiently low to permit its use in cellular and in vivo assay development in up to 2% final concentrations. Furthermore, sulfoxide 3 proved experimentally to be far less oxidizing than DMSO, and this property could provide greater stability to long-term compound storage solutions. The amount of water absorption will likely depend on the choice of cosolvent, but the nature of 3 (being a solid) may allow the assay developer to choose cosolvents that either do not absorb as much water as DMSO (or NMP) or do not undergo the dramatic changes in physical properties observed in the case of wet DMSO solutions. As shown in our PKD1 assays, 3 does not alter the biochemical readout in standard in vitro assays.
To the best of our knowledge, this is the first report of the incorporation of an oxetane moiety into a cosolvent structure for solubility enhancement. The oxetane motif allows for the design of more lipophilic cosolvents that still maintain good aqueous miscibility due to the dipole moment at the oxetane oxygen. This study on the utility of sulfoxide 3 as an aqueous-soluble cosolvent prompts further development of additives bearing oxetanes or related heterocycles.
Abbreviations
- ATR
attenuated total reflection
- DMSO
dimethylsulfoxide
- ES
electrospray
- EtOH
ethanol
- GI50
50% growth inhibition
- HPLC
high-performance liquid chromatography
- HRMS
high resolution mass spectrometry
- IR
infrared
- MeOH
methanol
- NBP
4-(p-nitrobenzyl)pyridine
- NMP
N-methylpyrrolidinone
- NMR
nuclear magnetic resonance
- PBS
phosphate-buffered saline
- PEG
polyethylene glycol
- PKD1
protein kinase D isoform 1
- rt
room temperature
- TsCl
p-toluenesulfonyl chloride
- UV/VIS
ultra-violet/visible spectra.
Acknowledgments
This work was supported by a grant from the National Institutes of Health: NIGMS Centers for Chemical Methodologies and Library Development (P50GM067082). The authors thank Profs. Andreas Vogt and Q. Jane Wang and Dr. Manuj Tandon (Department of Pharmacology and Chemical Biology, University of Pittsburgh) for PKD1, MDA-MB-231, and HepG2 assays.
Disclosure Statement
No competing financial interests exist.
References
- 1.Janzen WP. Popa-Burke IG. Advances in improving the quality and flexibility of compound management. J Biomol Screen. 2009;14:444–451. doi: 10.1177/1087057109335262. [DOI] [PubMed] [Google Scholar]
- 2.Waybright TJ. Britt JR. McCloud TG. Overcoming problems of compound storage in DMSO: solvent and process alternatives. J Biomol Screen. 2009;14:708–715. doi: 10.1177/1087057109335670. [DOI] [PubMed] [Google Scholar]
- 3.Talaga P. Compound decomposition: a new drug discovery tool? Drug Discov Today. 2004;9:51–53. doi: 10.1016/S1359-6446(03)02910-6. [DOI] [PubMed] [Google Scholar]
- 4.Kozikowski BA. Burt TM. Tirey DA, et al. The effect of room-temperature storage on the stability of compounds in DMSO. J Biomol Screen. 2003;8:205–209. doi: 10.1177/1087057103252617. [DOI] [PubMed] [Google Scholar]
- 5.Stegemann S. Leveiller F. Franchi D. de Jong H. Lindén H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31:249–261. doi: 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- 6.Di L. Kerns EH. Application of pharmaceutical profiling assays for optimization of drug-like properties. Curr Opin Drug Discov Devel. 2005;8:495–504. [PubMed] [Google Scholar]
- 7.Yalkowsky SH. Solubility, Solubilization in Aqueous Media. Oxford University Press; New York: 1999. [Google Scholar]
- 8.Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004;21:201–230. doi: 10.1023/b:pham.0000016235.32639.23. [DOI] [PubMed] [Google Scholar]
- 9.Oda M. Saitoh H. Kobayashi M. Aungst BJ. β-Cyclodextrin as a suitable solubilizing agent for in situ absorption study of poorly water-soluble drugs. Int J Pharm. 2004;280:95–102. doi: 10.1016/j.ijpharm.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Ouyang L. Ma L. Jiang B. Li Y. He D. Guo L. Synthesis of novel dendrimers having aspartate grafts and their ability to enhance the aqueous solubility of model drugs. Eur J Med Chem. 2010;45:2705–2711. doi: 10.1016/j.ejmech.2010.01.069. [DOI] [PubMed] [Google Scholar]
- 11.Sanghvi R. Narazaki R. Machatha SG. Yalkowsky SH. Solubility improvement of drugs using N-methyl pyrrolidone. AAPS PharmSciTech. 2008;9:366–376. doi: 10.1208/s12249-008-9050-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitha J. Szente A. Greenberg J. Poly-L-methionine sulfoxide: a biologically inert analogue of dimethyl sulfoxide with solubilizing potency. J Pharm Sci. 1983;72:665–668. doi: 10.1002/jps.2600720618. [DOI] [PubMed] [Google Scholar]
- 13.Rose NG. Blaskovich MA. Evindar G, et al. Preparation of 1-[N-benzyloxycarbonyl-(1S)-1-amino-2-oxoethyl]-4-methyl-2,6,7-trioxabicyclo[2.2.2]octane. Org Synth. 2002;79:216–221. [Google Scholar]
- 14.Meyer BN. Ferrigni NR. Putnam JE. Jacobsen LB. Nichols DE. McLaughlin JL. Brine shrimp: a convenient general bioassay for active plant constituents. Planta Med. 1982;45:31–34. [PubMed] [Google Scholar]
- 15.Zhdanko AG. Nenajdenko VG. Nonracemizable isocyanoacetates for multicomponent reactions. J Org Chem. 2009;74:884–887. doi: 10.1021/jo802420c. [DOI] [PubMed] [Google Scholar]
- 16.Bellon L. Taft RW. Abboud JM. Structural effects on the reactivity of ethers in donor-acceptor reactions. J Org Chem. 1980;45:1166–1168. [Google Scholar]
- 17.Wuitschik G. Carreira EM. Wagner B, et al. Oxetanes in drug discovery: structural and synthetic insights. J Med Chem. 2010;53:3227–3246. doi: 10.1021/jm9018788. [DOI] [PubMed] [Google Scholar]
- 18.Kurien BT. Singh A. Matsumoto H. Scofield RH. Improving the solubility and pharmacological efficacy of curcumin by heat treatment. Assay Drug Dev Technol. 2007;5:567–576. doi: 10.1089/adt.2007.064. [DOI] [PubMed] [Google Scholar]
- 19.Yu X. Li Y. Wu D. Enzymatic synthesis of gallic acid esters using microencapsulated tannase: effect of organic solvents and enzyme specificity. J Mol Catal-B Enzym. 2004;30:69–73. [Google Scholar]
- 20.Yalkowsky SH. He Y. Handbook of Aqueous Solubility Data. CRC Press; New York: 2003. [Google Scholar]
- 21.Shareef A. Angove MJ. Wells JD. Johnson BB. Aqueous solubilities of estrone, 17β-estradiol, 17α-ethynylestradiol, and bisphenol A. J Chem Eng Data. 2006;51:879–881. [Google Scholar]
- 22.BioFocus. http://biofocus.com/offerings/adme-pk-laboratory/solubility. [Sep 21;2011 ]. http://biofocus.com/offerings/adme-pk-laboratory/solubility
- 23.Bravo-Altamirano K. George KM. Frantz MC, et al. Synthesis and structure-activity relationships of benzothienothiazepinone inhibitors of protein kinase D. ACS Med Chem Lett. 2011;2:154–159. doi: 10.1021/ml100230n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.George KM. Frantz MC. Bravo-Altamirano K, et al. Design, synthesis, and biological evaluation of PKD inhibitors. Pharmaceutics. 2011;3:186–228. doi: 10.3390/pharmaceutics3020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharlow ER. Giridhar KV. LaValle CR, et al. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–33526. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eigenmann HK. Golden DM. Benson SW. Revised group additivity parameters for the enthalpies of formation of oxygen-containing organic compounds. J Phys Chem. 1973;77:1687–1691. [Google Scholar]
- 27.Banks HD. A computational study to elucidate the extraordinary reactivity of three-membered heterocycles in nucleophilic substitution reactions. J Org Chem. 2003;68:2639–2644. doi: 10.1021/jo0268411. [DOI] [PubMed] [Google Scholar]
- 28.Druckrey H. Kruse H. Preussmann R. Ivankovic S. Cancerogene Alkylierende Substanzen. Z Krebsforsch. 1970;74:241–273. [PubMed] [Google Scholar]
- 29.Dickens F. Jones HE. Further studies on the carcinogenic and growth-inhibitory activity of lactones and related substances. Br J Cancer. 1963;17:100–108. doi: 10.1038/bjc.1963.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gómez-Bombarelli R. Palma BB. Martins C, et al. Alkylating potential of oxetanes. Chem Res Toxicol. 2010;23:1275–1281. doi: 10.1021/tx100153w. [DOI] [PubMed] [Google Scholar]
- 31.Uch AS. Hesse U. Dressman JB. Use of 1-methyl-pyrrolidone as a solubilizing agent for determining the uptake of poorly soluble drugs. Pharm Res. 1999;16:968–971. doi: 10.1023/a:1012120210530. [DOI] [PubMed] [Google Scholar]
- 32.Cheng X. Hochlowski J. Tang H, et al. Studies on repository compound stability in DMSO under various conditions. J Biomol Screen. 2003;8:292–304. doi: 10.1177/1087057103008003007. [DOI] [PubMed] [Google Scholar]