

ABSTRACT
Rho GTPases are important regulators of many cellular processes. Subversion of Rho GTPases is a common infection strategy employed by many important human pathogens. Enteropathogenic Escherichia coli and enterohemorrhagic Escherichia coli (EPEC and EHEC) translocate the effector EspH, which inactivates mammalian Rho guanine exchange factors (GEFs), as well as Map, EspT, and EspM2, which, by mimicking mammalian RhoGEFs, activate Rho GTPases. In this study we found that EspH induces focal adhesion disassembly, triggers cell detachment, activates caspase-3, and induces cytotoxicity. EspH-induced cell detachment and caspase-3 activation can be offset by EspT, EspM2, and the Salmonella Cdc42/Rac1 GEF effector SopE, which remain active in the presence of EspH. EPEC and EHEC therefore use a novel strategy of controlling Rho GTPase activity by translocating one effector to inactivate mammalian RhoGEFs, replacing them with bacterial RhoGEFs. This study also expands the functional range of bacterial RhoGEFs to include cell adhesion and survival.
IMPORTANCE Many human pathogens use a type III secretion system to translocate effectors that can functionally be divided into signaling, disabling, and countervirulence effectors. Among the signaling effectors are those that activate Rho GTPases, which play a central role in coordinating actin dynamics. However, many pathogens also translocate effectors with antagonistic or counteractive functions. For example, Salmonella translocates SopE and SptP, which sequentially turn Rac1 and Cdc42 on and off. In this paper, we show that enteropathogenic E. coli translocates EspH, which inactivates mammalian RhoGEFs and triggers cytotoxicity and at the same time translocates the bacterial RhoGEFs EspM2 and EspT, which are insensitive to EspH, and so neutralizes EspH-induced focal adhesion disassembly, cell detachment, and caspase-3 activation. Our data point to an intriguing infection strategy in which EPEC and EHEC override cellular Rho GTPase signaling by disabling mammalian RhoGEFs and replacing them with with bacterial RhoGEFs that promote cell adhesion and survival.
IMPORTANCE
Many human pathogens use a type III secretion system to translocate effectors that can functionally be divided into signaling, disabling, and countervirulence effectors. Among the signaling effectors are those that activate Rho GTPases, which play a central role in coordinating actin dynamics. However, many pathogens also translocate effectors with antagonistic or counteractive functions. For example, Salmonella translocates SopE and SptP, which sequentially turn Rac1 and Cdc42 on and off. In this paper, we show that enteropathogenic E. coli translocates EspH, which inactivates mammalian RhoGEFs and triggers cytotoxicity and at the same time translocates the bacterial RhoGEFs EspM2 and EspT, which are insensitive to EspH, and so neutralizes EspH-induced focal adhesion disassembly, cell detachment, and caspase-3 activation. Our data point to an intriguing infection strategy in which EPEC and EHEC override cellular Rho GTPase signaling by disabling mammalian RhoGEFs and replacing them with with bacterial RhoGEFs that promote cell adhesion and survival.
Introduction
The Rho family of small GTPases, including RhoA, Rac1, and Cdc42, are important regulators of actin organization (1), as well as many other cellular processes, including cell adhesion and migration, vesicle trafficking, cytokinesis, and apoptosis (2). Rho GTPases switch between active GTP-bound and inactive GDP-bound forms. The cycling of these two states is regulated by guanine nucleotide exchange factors (GEFs), which promote dissociation of GDP and subsequent binding of GTP and GTPase-activating proteins (GAPs), which enhance the rate of GTP hydrolysis to GDP, while guanine nucleotide dissociation inhibitors (GDIs) maintain Rho GTPases in an inactive state in the cytosol (1, 2).
An important mediator of focal adhesion (FA) signaling is focal adhesion kinase (FAK) (3). FAK is a protein-tyrosine kinase that is implicated in formation of nascent focal complexes at lamellipodial protrusions, as well as disassembly of mature focal adhesions in a dynamic process known as “FA turnover” (3). FAK-null fibroblasts exhibit high Rho activity, reduced migration, and severe FA turnover defects (4). The major phosphorylation site of FAK is Y397, which recruits c-Src to form a FAK-Src signaling complex that then activates multiple signaling pathways (3). Recent studies have shown that FAK regulates the localized activity of Rho GTPases by recruiting RhoGEFs and RhoGAPs at focal adhesion sites to facilitate FA turnover and cell migration (5).
Several important bacterial pathogens subvert Rho GTPase signaling by utilizing a type III secretion system (T3SS) to translocate effector proteins into eukaryotic host cells (6). Enteropathogenic Escherichia coli (EPEC) and enterohemorrhagic Escherichia coli (EHEC) translocate the effector EspH, which by binding to tandem Dbl homology-pleckstrin homology (DH-PH) domains of Dbl-family RhoGEFs blocks the activation of Rho GTPases (7). EPEC and EHEC also translocate the effectors Map, EspM, and EspT, which, based on an invariant Trp-XXX-Glu motif, are grouped with IpgB1 and IpgB2 (Shigella) and SifA and SifB (Salmonella) into the WxxxE family of effectors (8–11).
Structural analysis of Map and EspM2 revealed that the WxxxE effectors share a three-dimensional fold with the Salmonella GEF SopE, which catalyzes the exchange of GDP to GTP by binding to the switch I and II regions of Rho GTPases (12–14). Map exhibits a GEF activity for Cdc42 (13) and induces transient filopodium formation at bacterial attachment sites during the early stages of EPEC infection (15, 16). EspM2 is a bacterial RhoA GEF that induces stress fiber formation (10, 14), while EspT activates both Cdc42 and Rac1, leading to formation of membrane ruffles and lamellipodia (11).
Importantly, EspH was implicated in modulating Map-induced filopodium kinetics, as deletion of espH results in filopodium persistence and overexpression of EspH inhibits filopodium formation (17). However, the overall functional relationships between these effectors during infection are poorly understood. Moreover, it is not known if EspH can inhibit the activity of the SopE-like WxxxE effectors; while structurally distinct from DH-PH eukaryotic RhoGEFs (12), they activate Rho GTPases by a similar mechanism. The aim of this study was to characterize the interplay between EspH and bacterial RhoGEFs in the context of actin remodeling, cell adhesion, and cell death.
RESULTS
EspH induces cell detachment, cytotoxicity, and focal adhesion disassembly.
EspH binds to DH-PH mammalian RhoGEFs and induces actin cytoskeletal disruption and cell rounding (7, 17). We further characterized EspH-induced cytotoxicity by assessing the levels of cell rounding, cell detachment, and lactate dehydrogenase (LDH) release. Myc-tagged EspH (Myc-EspH), or Myc-tagged green fluorescent protein (Myc-GFP) as a control, was ectopically expressed for 24 h, and the levels of cell rounding were quantified by immunofluorescence microscopy. Cells transfected with Myc-EspH showed a marked increase in cell rounding compared to those transfected with Myc-GFP (Fig. 1A).
FIG 1 .

EspH induces cell detachment, cytotoxicity, and focal adhesion disassembly. (A) Quantification of cell rounding of HeLa cells transfected with Myc-GFP or Myc-EspH for 24 h. One hundred transfected cells were counted in triplicate. Ectopic expression of EspH induced a significant increase in cell rounding compared to GFP expression. Results are means plus standard deviations (SD) of three independent experiments. (B) Quantification of detachment levels of HeLa cells transfected with Myc-GFP or Myc-EspH for 24 h. Ectopic expression of EspH induced a significant increase in cell detachment compared to GFP. Results are means plus SDs of three independent experiments. **, P < 0.01. (C) Quantification of detachment levels of HeLa cells infected with WT EPEC, ΔespH EPEC, or ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h and then incubated with gentamicin for 3 h. STS treatment of HeLa cells for 5 h was used as a positive control. Cell detachment was expressed as a percentage of the value for uninfected cells. Cell detachment was reduced in the ΔespH mutant compared to WT EPEC. Complementation with EspH increased cell detachment to STS levels. Results are means ± SD of three independent experiments. *, P < 0.05; **, P < 0.01. (D) Quantification of cytotoxicity to HeLa cells infected with WT EPEC, ΔespH EPEC, or ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 30 min and then incubated with gentamicin for 4 h. STS treatment of HeLa cells for 5 h was used as a positive control. Cytotoxicity was assayed by quantifying the levels of LDH release into the culture supernatant from cells 4 h postinfection. Results are means ± SD of three independent experiments. (E) Immunofluorescence microscopy of HeLa cells infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 0, 60, 120, and 180 min. Bacteria were visualized with anti-intimin antibody (magenta), vinculin was detected with anti-vinculin antibody (green), and actin was stained with TRITC-phalloidin (red). Infection with ΔespH/pEspH EPEC progressively induced focal adhesion disassembly, as cells were rounded after 180 min of infection. Bar = 10 µm.
The level of cell detachment following Myc-EspH or Myc-GFP expression was measured and quantified as the percentage of total cells. Myc-EspH was found to induce 45.8 ± 5.5% cell detachment, compared to 8.2 ± 5.0% cell detachment in the Myc-GFP control (Fig. 1B). A cell detachment assay was also performed to assess the role of EspH during EPEC infection. HeLa cells were infected with wild-type (WT) EPEC, ΔespH EPEC, or ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h and then incubated for a further 3 h with gentamicin to stop bacterial replication. Treatment of cells with the apoptotic inducer staurosporine (STS) for 5 h was used as a positive control. Quantification of cells postinfection showed that 11.0 ± 9.5% of cells infected with wild-type EPEC were detached (Fig. 1C). In contrast, only 2.9 ± 8.7% of cells were detached following infection with ΔespH EPEC. Complementation of the mutant with a plasmid-encoded EspH dramatically increased the level of cell detachment relative to that of the STS control, as 51.1 ± 1.8% of cells infected with ΔespH/pEspH EPEC were detached.
The contribution of EspH to cytotoxicity during EPEC infection was also measured by LDH release, which results from plasma membrane damage. Treatment of cells with STS for 5 h was used as a positive control. Infection with wild-type EPEC resulted in a slightly higher LDH release than did infection with the ΔespH mutant (Fig. 1D). Complementation of the ΔespH mutant significantly increased LDH release to levels comparable to those observed with the STS control. EspH is thus a potent inducer of cytotoxicity and cell detachment.
The phenotype of cell rounding and cell detachment led us to examine whether EspH had an effect on focal adhesions. To investigate this, we performed a time course study of HeLa cells infected with ΔespH/pEspH EPEC, as this strain gave the most distinct phenotype compared to wild-type EPEC or ΔespH EPEC. When coverslips were fixed hourly for 3 h and vinculin was used as a marker for focal adhesions, immunofluorescence microscopy revealed that EspH progressively induces disassembly of focal adhesions concomitantly with actin disruption; no focal adhesions were observed when cells were rounded at the end of infection (Fig. 1E).
EspH mediates focal adhesion disassembly in a caspase-independent manner.
Inhibition of Rho GTPase signaling is also a trigger for caspase activation and apoptosis (18), and the EspH-mediated focal adhesion disassembly observed could therefore be a result of caspase-mediated cleavage of focal adhesion proteins (19). The ability of EspH to induce caspase-3 activation was tested by infecting HeLa cells with WT, ΔespH, or ΔespH/pEspH EPEC for 1 h and incubating them for a further 3 h in the presence of gentamicin. Treatment of cells with STS for 5 h was used as a positive control, and cleaved caspase-3 was detected by immunofluorescence microscopy. In this assay, 61.1 ± 0.8% of STS-treated cells stained positive for active caspase-3, whereas only 2.9 ± 0.5% of untreated cells stained positive (Fig. 2A and B); 16.9 ± 3.7% of WT EPEC-infected cells stained positive for caspase-3 activation, compared to 9.7 ± 2.1% of cells infected with the ΔespH mutant (Fig. 2A and B). Complementation of the ΔespH mutant with plasmid-encoded EspH significantly increased caspase-3 activation, as 29.0 ± 1.6% of cells infected with ΔespH/pEspH EPEC stained positive for active caspase-3 (Fig. 2A and B).
FIG 2 .

EspH induces focal adhesion disassembly in a caspase-independent manner. (A) Immunofluorescence microscopy of uninfected HeLa cells or cells infected with WT EPEC, ΔespH EPEC, or ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h, then incubated with gentamicin for 3 h. Bacteria were visualized with anti-intimin antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), actin was stained with TRITC-phalloidin (red) and cell nuclei were stained with DAPI (cyan). The ΔespH mutant induced reduced levels of caspase-3 activation compared to WT EPEC. Complementation with EspH resulted in a marked increase in caspase-3 activation when compared to ΔespH alone. Bar = 20 µm. (B) Quantification of caspase-3 activation of infected cells from (A). STS treatment of HeLa cells for 5 h was used as a positive control. 100 infected cells were counted in triplicates. Results are means ± SD of three independent experiments. *, P < 0.05), ***, P < 0.001). (C) Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-EspH. Myc-tagged proteins were visualized with anti-Myc antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), actin was stained with TRITC-phalloidin (red) and nuclei were stained with DAPI (cyan). Ectopic expression of EspH, but not GFP, induced caspase-3 activation. Bar = 20 µm. (D) Quantification of caspase-3 activation of transfected cells from (C). STS treatment of HeLa cells for 5 h was used as a positive control. 100 transfected cells were counted in triplicates. Results are means ± SD of three independent experiments. **, P < 0.01). (E) Quantification of detachment levels of HeLa cells, with or without zVAD-fmk treatment, infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h, then incubated with gentamicin for 3 h. STS treatment of HeLa cells for 5 h was used as a positive control. Cell detachment was expressed as a percentage of uninfected cells. Treatment with zVAD-fmk inhibited STS-induced cell detachment, but not cell detachment following infection with ΔespH/pEspH EPEC. Results are means ± SD of three independent experiments. (F) Immunofluorescence microscopy of HeLa cells infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 180 min, with or without zVAD-fmk treatment. Bacteria were visualized with anti-intimin antibody (magenta), vinculin was detected with anti-vinculin antibody (green), and actin was stained with TRITC-phalloidin (red). Treatment with zVAD-fmk did not prevent focal adhesion disassembly. Bar = 10 µm.
To determine if EspH alone was sufficient to induce caspase-3 activation, cleaved caspase-3 was detected in cells ectopically expressing EspH (Myc-EspH) or Myc-tagged GFP (Myc-GFP) for 24 h. We observed that while 35.9 ± 8.7% of cells expressing EspH stained positive for active caspase-3, only 1.9 ± 0.8% of GFP-expressing cells exhibited the presence of cleaved caspase-3 (Fig. 2C and D).
We then used the pan-caspase inhibitor zVAD-fmk to assess the levels of cell detachment following infection with ΔespH/pEspH EPEC (Fig. 2E). STS was again used as a control, and as expected, STS-induced cell detachment was inhibited by zVAD-fmk, as it is caspase mediated (cell detachment was reduced from 53.9 ± 3.1% to 11.6 ± 2.6% in the presence of zVAD-fmk). However, treatment with zVAD-fmk did not diminish cell detachment induced by infection with ΔespH/pEspH EPEC (62.3 ± 3.1% cell detachment without and 53.0 ± 2.2% cell detachment with zVAD-fmk). Immunofluorescence microscopy of ΔespH/pEspH EPEC-infected cells treated with zVAD-fmk further showed that cells were still rounded, and no focal adhesions (vinculin) were observed after a 3-h infection (Fig. 2F). These results suggest that EspH-induced cell detachment was a result of FA disassembly rather than caspase activation.
FAK is involved in EspH-induced cell rounding, detachment, and caspase-3 activation.
As FAK regulates localized Rho GTPase activity and focal adhesion dynamics (3), we investigated if FAK is involved in EspH-induced cell rounding and caspase-3 activation. FAK-null (FAK−/−) and FAK-reconstituted (FAK+/+) fibroblasts were transfected with Myc-tagged EspH or Myc-tagged GFP as a control and stained for F actin and cleaved caspase-3. Immunofluorescence microscopy revealed that EspH induced cell rounding and caspase-3 activation in FAK+/+ fibroblasts (Fig. 3A to C). In contrast, FAK−/− fibroblasts exhibited reduced levels of cell rounding and little caspase-3 activation (Fig. 3A to C). As FAK−/− fibroblasts were resistant to EspH-induced cell rounding, the role of FAK on cell adhesion during EPEC infection was examined. FAK−/− and FAK+/+ fibroblasts were assessed for levels of cell detachment following infection with wild-type, ΔespH, or ΔespH/pEspH EPEC. STS treatment was used as a control and showed similar levels of cell detachment in FAK−/− and FAK+/+ fibroblasts (Fig. 3D). Consistent with the results described above, FAK−/− fibroblasts did not show any significant levels of cell detachment after infection with any of the strains (Fig. 3D). In contrast, 43.2 ± 9.5% of cells were detached following infection of FAK+/+ fibroblasts with ΔespH/pEspH EPEC (Fig. 3D). Taken together, our results show that FAK is an important mediator of EspH-induced cell rounding and cell detachment and suggest that defects in focal adhesion disassembly diminish the ability of EspH to induce cell rounding and caspase-3 activation.
FIG 3 .

FAK is involved in EspH-induced cell rounding, detachment, and caspase-3 activation. (A) Immunofluorescence microscopy of FAK+/+ or FAK−/− fibroblasts transfected with Myc-GFP or Myc-EspH. Myc-tagged proteins were visualized with anti-Myc antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), actin was stained with TRITC-phalloidin (red), and cell nuclei were stained with DAPI (cyan). Ectopic expression of EspH induced cell rounding, nuclear condensation, and caspase-3 activation in FAK+/+ cells but not FAK−/− fibroblasts. Bar = 20 µm. (B) Quantification of cell rounding of FAK+/+ or FAK−/− fibroblasts transfected with Myc-GFP or Myc-EspH. 100 transfected cells were counted in triplicates. Results are means plus SD of three independent experiments. ***, P < 0.001. (C) Quantification of caspase-3 activation of FAK+/+ or FAK−/− fibroblasts transfected with Myc-GFP or Myc-EspH. One hundred transfected cells were counted in triplicate. Results are means plus SD of three independent experiments. **, P < 0.01. (D) Quantification of detachment levels of FAK+/+ or FAK−/− fibroblasts infected with WT EPEC, ΔespH EPEC, or ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h and then incubated with 200 µg ml−1 gentamicin for 3 h. Treatment of FAK+/+ or FAK−/− fibroblasts with 1 µM staurosporine (STS) for 5 h was used as a positive control. Cell detachment was expressed as a percentage of the value for uninfected cells. Cell detachment was drastically reduced in FAK−/− fibroblasts infected with WT EPEC or ΔespH/pEspH EPEC compared to that in FAK+/+ fibroblasts. Results are means plus SD of three independent experiments. **, P < 0.01.
Bacterial RhoGEFs are active in the presence of EspH.
We next investigated whether, in addition to blocking mammalian DH-PH RhoGEFs, EspH inhibits the activity of bacterial RhoGEFs. HeLa cells transfected with Myc-tagged EspH or Myc-tagged GFP were infected with Δmap/pMap EPEC for 30 min or with EPEC/pEspT or EPEC/pEspM2 for 90 min and assessed for filopodium, lamellipodium, and stress fiber formation, respectively. For consistency, we deleted endogenous map so that all three effectors were expressed exclusively from inducible genes. Staining with F actin revealed that cells infected with EPEC expressing the WxxxE effectors were able to induce formation of their respective actin structures in cells transfected with either GFP or EspH (Fig. 4A). This suggests that EspH does not inhibit the activity of the WxxxE-family RhoGEFs during EPEC infection.
FIG 4 .

Bacterial RhoGEFs are active in the presence of EspH. (A) Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-EspH and then infected with Δmap/pMap EPEC for 30 min or EPEC expressing EspT (EPEC/pEspT) or EspM2 (EPEC/pEspM2) for 90 min. Myc-tagged proteins were visualized with anti-Myc antibody (green), bacteria were visualized with anti-intimin antibody (magenta), and actin was stained with TRITC-phalloidin (red). Ectopic expression of EspH did not inhibit the formation of filopodia (Map), lamellipodia (EspT), or stress fibers (EspM2). Bar = 20 µm. (B) Immunofluorescence microscopy of HeLa cells cotransfected with Myc-p115RhoGEF or Myc-EspM2 and with RFP or RFP-EspH (red). Myc-tagged proteins were visualized with anti-Myc antibody (magenta), and actin was stained with Oregon green phalloidin (green). Cotransfection of Myc-p115RhoGEF and RFP-EspH but not Myc-EspM2 and RFP-EspH drastically reduced stress fiber formation. Bar = 20 µm. (C) Quantification of stress fiber formation in HeLa cells cotransfected with Myc-p115RhoGEF or Myc-EspM2 and with RFP or RFP-EspH. One hundred cotransfected cells were counted in triplicate. Results are means plus SD of three independent experiments. ***, P < 0.001.
To affirm this observation, cells were cotransfected with red fluorescent protein (RFP) or RFP-EspH and Myc-tagged Map, EspT, EspM2, or the catalytic region of SopE (SopE78-240); cotransfection with Myc-tagged p115-RhoGEF, a mammalian RhoA GEF which has been shown to bind EspH (7), was used as a control. After staining for F actin, we observed that EspH was able to inhibit stress fiber formation by p115-RhoGEF, and the cotransfected cells were rounded (Fig. 4B). In contrast, cotransfection of RFP-EspH with Map, EspT, EspM2, or SopE78-240 did not inhibit formation of filopodia (Map), lamellipodia (EspT/SopE), or stress fibers (EspM2) (Fig. 4B; also, see Fig. S1 in the supplemental material). When cotransfected with RFP, the RhoA GEFs p115-RhoGEF and EspM2 induced stress fiber formation in 79.7 ± 3.3% and 90.6 ± 1.1% of cells, respectively (Fig. 4C). When cotransfected with RFP-EspH, p115-RhoGEF showed markedly reduced stress fiber formation, with only 14.5 ± 2.8% of cells displaying stress fibers. In contrast, only a modest decrease in stress fiber formation was observed in cells cotransfected with EspM2 and RFP-EspH, as 76.5 ± 2.1% of cells were positive for stress fibers (Fig. 4C). Taken together, these results demonstrate that bacterial RhoGEFs are insensitive to EspH.
EspT, EspM2, and SopE block EspH-induced cell detachment and focal adhesion disassembly.
Next, we determined if the bacterial RhoGEFs could mitigate the detrimental effects of EspH. To investigate if bacterial RhoGEFs could block EspH-induced cell detachment, we transfected HeLa cells with Myc-tagged bacterial RhoGEFs or their respective Rho GTPase dominant positives as controls and then infected them with ΔespH/pEspH EPEC for 1 h, followed by gentamicin treatment for a further 3 h. No construct induced significant levels of cell detachment by transfection alone compared to the Myc-GFP control (Fig. 5). Infection of Myc-GFP-transfected cells with ΔespH/pEspH EPEC resulted in 55.6 ± 5.1% detachment (Fig. 5). Interestingly, cells transfected with the Rac1-activating effectors EspT and SopE78-240 and dominant-positive Rac1 (Rac1L61) showed a significant reduction in cell detachment following infection with ΔespH/pEspH EPEC, as only 30.3 ± 6.7%, 30.6 ± 2.8%, and 34.4 ± 4.2% of cells were detached, respectively (Fig. 5). We also observed an intermediate reduction in cell detachment of cells transfected with dominant-positive RhoA (RhoAL63) and its corresponding GEF EspM2, as 43.1 ± 2.7% and 35.0 ± 6.4% of cells were detached, respectively (Fig. 5). Neither the Cdc42 GEF Map nor dominant positive Cdc42 (Cdc42L61) could inhibit cell detachment following infection with ΔespH/pEspH EPEC, as 53.7 ± 11.2% and 52.7% ± 4.9% of cells were detached, respectively (Fig. 5). These results imply that Cdc42 activation alone is unable to block EspH-induced cell detachment.
FIG 5 .
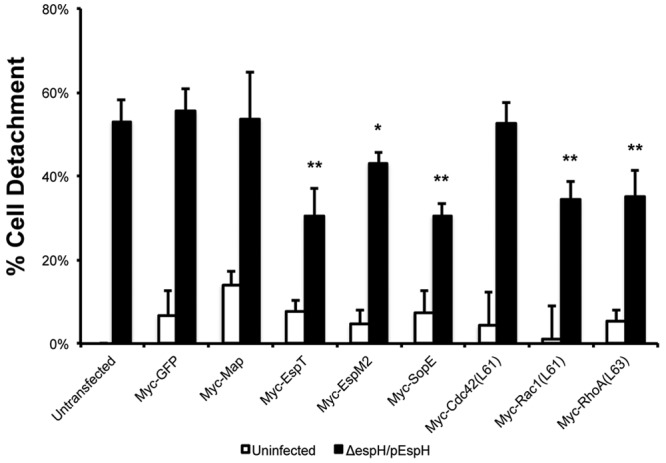
EspT, EspM2, and SopE block EspH-induced cell detachment. Detachment levels in HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs, uninfected or infected with EPEC with ΔespH complemented with EspH (ΔespH/pEspH) for 1 h, followed by incubation with 200 µg ml−1 gentamicin for 3 h, were quantified. Transfection with dominant-positive Cdc42 (Cdc42L61), Rac1 (Rac1L61), and RhoA (RhoAL63) was used as positive controls. Cell detachment was expressed as a percentage of that in nontransfected, noninfected cells. Ectopic expression of Myc-tagged EspT, EspM2, or SopE78-240 resulted in reduced cell detachment following infection with ΔespH/pEspH EPEC but not Myc-tagged GFP or Map. Results are means plus SD of three independent experiments. Statistical significance is based on a Student’s t test compared to the Myc-GFP control. *, P < 0.05; **, P < 0.01.
We next examined if EspT, EspM2, and SopE also inhibited EspH-induced focal adhesion disassembly. In contrast to GFP- and Map-transfected cells, EspT- and SopE78-240-transfected cells blocked EspH-induced cell rounding, and vinculin staining revealed that large focal adhesions were still present in a radial pattern following infection with ΔespH/pEspH EPEC (see Fig. S2 in the supplemental material). EspM2-transfected cells also retained stress fiber-associated focal adhesions following infection with ΔespH/pEspH EPEC (see Fig. S2). These results suggest that Rac1 and RhoA activation is critical for inhibition of EspH-induced cell detachment and focal adhesion disassembly.
EspT, EspM2, and SopE inhibit caspase-3 activation.
We next investigated if the bacterial RhoGEFs could also block EspH-induced caspase-3 activation. When the transfection-infection delivery strategy described above was used, ΔespH/pEspH EPEC induced caspase activation in 33.0 ± 8.5% of cells transfected with the GFP control (Fig. 6; also, see Fig. S3 in the supplemental material). Map was unable to inhibit EspH-induced caspase-3 activation, as 32.1 ± 6.7% of transfected cells were positive for caspase-3 activation. In contrast, EspT, EspM2, and SopE78-240 inhibited EspH-induced caspase-3 activation, as only 8.3 ± 2.8%, 13.9 ± 3.3%, and 9.2 ± 2.2% of transfected cells, respectively, were positive for caspase-3 activation (Fig. 6; also, see Fig. S3).
FIG 6 .
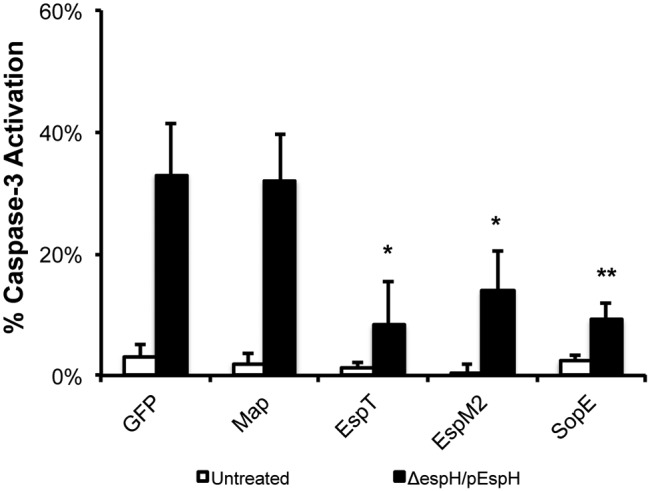
EspT, EspM2, and SopE inhibit EspH-induced caspase-3 activation. Caspase-3 activation of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs, uninfected or infected with EPEC with ΔespH complemented with EspH (ΔespH/pEspH) for 1 h, followed by incubation with 200 µg/ml gentamicin for 3 h, was quantified. One hundred transfected cells were counted in triplicate. Ectopic expression of Myc-tagged EspT, EspM2, or SopE78-240 markedly reduced caspase-3 activation in cells infected with ΔespH/pEspH EPEC but not Myc-tagged GFP or Map. Results are means plus SD of three independent experiments. Statistical significance is based on a Student’s t test compared to the Myc-GFP control. *, P < 0.05; **, P < 0.01.
As Rho GTPases play a role in cell survival (1), we investigated if the bacterial RhoGEFs could play an additional role in inhibiting apoptosis. To investigate this, we transfected HeLa cells with Myc-tagged Map, EspT, EspM2, and SopE78-240 and treated them with STS for 5 h. Transfection of anti-apoptotic Myc-tagged NleH1, which inhibits caspase-3 activation induced by STS (20), was used as a control. Quantification of cleaved caspase-3 by immunofluorescence revealed that cells transfected with the Myc-GFP control or Map displayed high levels of cleaved caspase-3 (Fig. 7; also, see Fig. S4 in the supplemental material). In contrast, cells transfected with Myc-tagged EspT, EspM2, or SopE78-240 all showed a marked reduction in levels of cleaved caspase-3 similar to that seen in cells transfected with anti-apoptotic effector NleH (Fig. 7; also, see Fig. S4).
FIG 7 .
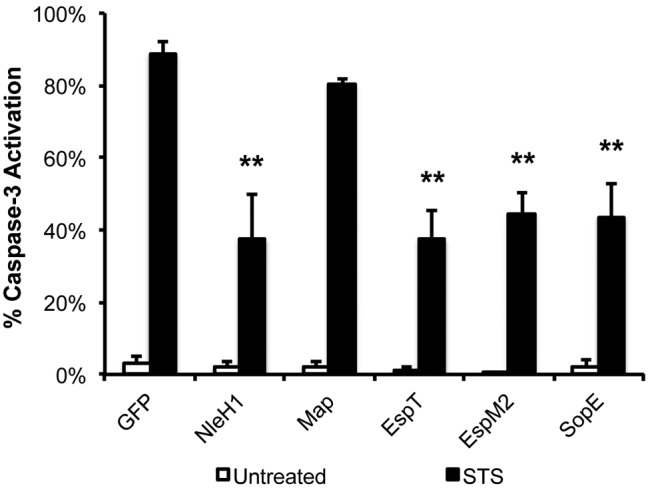
EspT, EspM2, and SopE inhibit staurosporine-induced caspase-3 activation. Caspase-3 activation of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs and then treated with 1 µM staurosporine (STS) for 5 h was quantified. Transfection of Myc-NleH1 and Myc-NleH2 was used as positive controls. One hundred transfected cells were counted in triplicate. Ectopic expression of Myc-tagged EspT, EspM2, or SopE78-240 markedly reduced caspase-3 activation in cells treated with STS compared to expression of NleH effectors but not Myc-tagged GFP or Map. Results are means plus SD of three independent experiments. Statistical significance is based on a Student’s t test compared to the Myc-GFP control. **, P < 0.01.
To demonstrate the role of EspT and EspM2 in promoting cell survival during EPEC infection, we utilized a ΔnleH1 ΔnleH2 EPEC mutant that induces high levels of apoptosis upon infection (20). We infected HeLa cells with wild-type EPEC, ΔnleH1ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing plasmid-encoded NleH1 (positive control), EspT, EspT(W63A) (defective in Rac1 activation), EspM2, or EspM2(W70A) (defective in RhoA activation) and assessed the levels of caspase-3 activation, cell detachment, and cytotoxicity (see Fig. S5 in the supplemental material). Intriguingly, EspT and EspM2, but not EspT(W63A) or EspM2(W70A), partially restored the wild-type phenotype in terms of caspase-3 activation, cell detachment, and cytotoxicity (see Fig. S5). Taken together, these results show that EspT and EspM2 are able to inhibit EspH- and STS-induced caspase-3 activation and promote cell survival dependent on Rac1 or RhoA activation during EPEC infection.
DISCUSSION
Several roles have been described for SopE and the WxxxE bacterial RhoGEFs, including actin remodeling, bacterial invasion, innate host cell response, and tight junction disruption (reviewed in reference 6). In this study, we expanded the repertoire of bacterial RhoGEF functions to include promotion of cell adhesion and survival. We also found that bacterial RhoGEFs are resistant to the DH-PH mammalian RhoGEF inhibitor EspH. EPEC and EHEC are thus able to neutralize mammalian RhoGEFs while translocating their own bacterial RhoGEFs to hijack Rho GTPase signaling for the proprietary benefit of the pathogen.
Bacterial pathogens manipulate Rho GTPase signaling in a coordinated, spatiotemporal manner. Yersinia translocates several T3SS effectors that inactivate Rho GTPases: YopE is a RhoGAP (21), YopT is a cysteine protease that cleaves Rho GTPases to prevent membrane localization (22), and YpkA/YopO has a RhoGDI activity (23). Vibrio parahaemolyticus delivers VopS, which contains a Fic domain that inactivates Rho GTPases through AMPylation (24). Salmonella enterica serovar Typhimurium temporally regulates Cdc42 and Rac1 by delivering SopE and SptP. SopE activates Cdc42/Rac1 to induce the “trigger mechanism” of bacterial invasion but is quickly degraded (25). SptP exhibits GAP activity but slower degradation kinetics, thus allowing recovery from SopE-induced membrane ruffling (25).
While EPEC and EHEC translocate several WxxxE RhoGEFs, thus far no effector with RhoGAP activity has been identified (26). Instead, EPEC uses a novel strategy by which EspH globally inhibits mammalian DH-PH RhoGEFs (7). The WxxxE effector Map triggers transient Cdc42-dependent filopodium formation at the site of bacterial attachment (15) before re-localizing to the mitochondria via its N-terminal mitochondrial targeting sequence (27). Filopodium dynamics is spatially regulated by Map interaction, through its carboxy-terminal PSD-95/Disk-large/ZO-1 (PDZ)-binding motif (DTRL), with the scaffold proteins sodium-hydrogen exchanger regulatory factors 1 and 2 (NHERF1/2) at the cell surface (8, 9, 28). In addition, the RhoA/ROCK pathway has been implicated in control of Map-induced filopodium dynamics (16). As Map was still able to induce filopodia in the presence of EspH, it is unlikely that EspH directly affects Map GEF activity for Cdc42. Rather, we speculate that EspH inhibits Map-induced filopodia by inhibiting the RhoA/ROCK pathway.
Indeed, the ability of EspH to induce cell rounding, focal adhesion disassembly, and caspase-3 activation is reminiscent of clostridial toxin B (TcdB) intoxication (18). This is not surprising given the fact that EspH, like TcdB, inactivates Rho GTPases, although TcdB directly targets Rho GTPases and inactivates them by glucosylation of Thr-37 (reviewed in reference 29). Significantly, we show that the ability of EPEC to antagonize EspH is mediated by the WxxxE effectors EspT and EspM2 and that neither Map nor Cdc42 activation was able to block EspH-induced cell detachment or caspase-3 activation. While it was previously reported that Map induces mitochondrial dysfunction (27), we did not observe any significant Map-induced cell detachment or caspase-3 activation by transfection. Consistent with our findings that Rac1 and RhoA activation are sufficient to inhibit EspH-induced cell detachment and caspase-3 activation, we also found that FAK, which coordinates the activities of Rac1 and RhoA to regulate focal adhesion dynamics, is an important mediator of EspH-induced cell rounding, cell detachment, and caspase-3 activation. As EspT and SopE are unable to trigger lamellipodium formation in FAK−/− fibroblasts (data not shown), our results support a model in which EPEC and EHEC utilize EspH and bacterial RhoGEFs to mediate focal adhesion dynamics through subversion of FAK signaling pathways.
Intriguingly, EspT, EspM2, and SopE (but not Map) can also inhibit STS-induced caspase-3 activation to the same extent as the anti-apoptotic effector NleH. In addition, EspT and EspM2 promote host cell survival during EPEC infection. Indeed, several reports have demonstrated that Rac1- and RhoA-activating RhoGEFs are able to activate mitogen-activated protein kinase (MAPK) and NF-κB pathways (30–32), which have pivotal roles in the regulation of cell survival (33, 34). As FAK-mediated signaling cascades are also involved in the regulation of cell survival by bacterial RhoGEFs, more work is required to dissect the mechanisms by which WxxxE-family effectors are able to modulate the cell death response. Thus, in characterizing the interplay between EspH and bacterial RhoGEFs we found that EPEC and EHEC have evolved sophisticated mechanisms to subvert Rho GTPase signaling pathways in a functionally coordinated manner.
MATERIALS AND METHODS
Bacterial strains and plasmids.
Strains, plasmids, and primers are listed in Tables S1, S2, and S3, respectively, in the supplemental material. Bacteria were grown from single colonies in Luria-Bertani (LB) broth in a shaking incubator at 37°C. Culture medium or agar was supplemented with ampicillin (100 µg ml−1), chloramphenicol (34 µg ml−1), or kanamycin (50 µg ml−1) as appropriate. Bacterial cultures were primed for infection by dilution of 1:50 of overnight bacterial culture with prewarmed Dulbecco’s minimal Eagle medium (DMEM) with low glucose (1,000 mg liter−1) (Sigma) and incubated as static cultures at 37°C in 5% CO2 for 3 h. Static cultures were induced with 1 mM isopropyl thiogalactopyranoside (IPTG) for pSA10 effector expression 30 min prior to infection.
Molecular cloning.
The espH gene of E2348/69 was amplified by PCR using the primer pair EcoRI-EspH-E69-Fw–PstI-EspH-E69-Rv, BamHI-EspH-E69-Fw–PstI-EspH-E69-Rv, or HindIII-EspH-E69-Fw–EcoRI-EspH-E69-Rv and cloned into the pSA10, pRK5myc, or pCMV-RFP vector to generate pICC1098, pICC529, and pICC1099, respectively. All constructs were verified by DNA sequencing.
Tissue culture, transfection, and infection with EPEC E2348/69.
HeLa (ATCC), FAK−/− (ATCC), and FAK+/+ (ATCC) cells were grown at 37°C in a humidified atmosphere containing 5% CO2 in DMEM with low glucose (1,000 mg liter−1) (Sigma) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (Gibco) and 2 mM GlutaMAX (Sigma). Cells were grown in 24-well cell culture plates to 80% confluence. HeLa cells were transfected with mammalian expression vectors using Fugene 6 (Roche) according to the manufacturer’s instructions. FAK−/− or FAK+/+ cells were transfected with mammalian expression vectors using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Transfected cells were incubated at 37°C in a humidified incubator with 5% CO2 for 24 h prior to infection. For EPEC infection, HeLa cells were washed twice with phosphate-buffered saline (PBS) and then infected with primed bacterial culture (multiplicity of infection [MOI], 1:100) at 37°C in a humidified atmosphere containing 5% CO2 for the appropriate time or treatment. Coverslips were washed three times in PBS and fixed with 3% paraformaldehyde for 15 min before being processed for immunofluorescence microscopy.
Pharmacology.
Staurosporine (STS) (Calbiochem) was used at a final concentration of 1 µM for 5 h to induce apoptosis. The pan-caspase inhibitor zVAD-fmk (Promega) was used at a final concentration of 66 µM (together with bacteria or with STS).
Immunofluorescence microscopy.
Cells were quenched for 15 min with 50 mM NH4Cl and then permeabilized for 5 min in PBS–0.2% Triton X-100. Coverslips were washed three times in PBS and blocked in PBS–1% BSA for 10 min before addition of primary antibodies. Coverslips were incubated with primary antibodies diluted in PBS–1% BSA for 1 h. For caspase-3 staining, primary antibodies were incubated overnight at 4°C. After three washes with PBS, coverslips were incubated with secondary antibodies diluted in PBS–1% BSA for a further 1 h. Coverslips were washed three times with PBS and once in water before being mounted in gold Pro-Long antifade medium (Invitrogen) and examined by conventional epifluorescence microscopy using a Zeiss Axio LSM-510 microscope. Images were deconvoluted and processed using AxioVision 4.8 LE software (Zeiss) and Adobe Photoshop CS4.
Antibodies.
Actin was detected with Oregon green-conjugated phalloidin antibody (1:100 dilution) (Invitrogen) or tetramethyl rhodamine isothiocyanate (TRITC)-conjugated phalloidin (1:500 dilution) (Sigma). Intimin was labeled with polyclonal chicken anti-intimin antibody serum (1:200 dilution) (kindly provided by Roberto La Ragione). DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000 dilution). Cleaved caspase-3 (active) was detected using rabbit anti-cleaved caspase-3 antibody (1:50 dilution) (Cell Signaling). Vinculin was visualized using mouse antivinculin antibody (1:200 dilution) (Abcam). Myc-tagged proteins were labeled with mouse anti-Myc antibody (1:500 dilution) (Millipore). Cy2-, RRX-, or Cy5-conjugated donkey anti-mouse, anti-chicken, or anti-rabbit antibodies (1:200 dilution) (Jackson ImmunoResearch) were used as secondary antibodies.
Cell detachment assay.
HeLa cells grown in 24-well plates were infected for 1 h (MOI, 1:100) and then treated with 200 µg ml−1 gentamicin (Sigma) for a further 3 h. Cells were washed five times with PBS and then trypsinized; trypsin was inactivated with culture medium. Cells were counted using a Neubauer hemocytometer. Counts were expressed as a percentage of uninfected cells and plotted as a graph of percent detached cells.
Cytotoxicity assay.
HeLa cells grown in 96-well plates were infected with primed bacterial culture (MOI, 1:200) in phenol red-free DMEM and centrifuged for 4 min at 200 × g to synchronize infection. Cells were incubated for 30 min, washed once with PBS, and then incubated with 200 µg ml−1 gentamicin (Sigma) in phenol red-free DMEM for a further 4 h. LDH release during EPEC infection was assayed using a CytoTox96 cytotoxicity assay kit (Promega) according to the manufacturer’s instructions. Cytotoxicity was calculated as follows: [(experimental LDH activity − basal LDH activity)/(total LDH activity − basal LDH activity)] × 100.
Statistical analysis.
Results were expressed as means and standard deviations. Statistical significance was determined by a two-tailed Student t test. A P value of <0.05, <0.01, or <0.001 was considered significant.
SUPPLEMENTAL MATERIAL
Bacterial RhoGEFs are active in the presence of EspH. Immunofluorescence microscopy of HeLa cells cotransfected with Myc-Map, Myc-EspT or Myc-SopE, and RFP or RFP-EspH (red). Myc-tagged proteins were visualized with anti-Myc antibody (magenta), and actin was stained with Oregon green phalloidin (green). Bar = 20 µm. Download Figure S1, TIF file, 2.8 MB.
EspT, EspM2, and SopE block EspH-induced focal adhesion disassembly. Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs, uninfected or infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h, followed by incubation with 200 µg ml−1 gentamicin for 3 h. Myc-tagged proteins were visualized with anti-Myc antibody (green), vinculin was detected with anti-vinculin antibody (cyan), and actin was stained with TRITC-phalloidin (red). Ectopic expression of Myc-tagged EspT, EspM2, or SopE retained focal adhesions following infection with ΔespH/pEspH EPEC but not Myc-tagged GFP or Map. Bar = 20 µm. Download Figure S2, TIF file, 1.4 MB.
EspT, EspM2 and SopE inhibit EspH-induced caspase-3 activation. Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs, uninfected or infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h, followed by incubation with 200 µg/ml gentamicin for 3 h. Myc-tagged proteins were visualized with anti-Myc antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), and cell nuclei were stained with DAPI (cyan). Bar = 20 µm. Download Figure S3, TIF file, 0.7 MB.
EspT, EspM2, and SopE inhibit staurosporine-induced caspase-3 activation. Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs and then treated with 1 µM staurosporine (STS) for 5 hours. Myc-tagged proteins were visualized with anti-Myc antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), and nuclei were stained with DAPI (cyan). Bar = 50 µm. Download Figure S4, TIF file, 1 MB.
EspT and EspM2 promote host cell survival during EPEC infection. (A) Immunofluorescence microscopy of HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, EspT, EspT(W63A), EspM2, or EspM2(W70A) for 1 h and then incubated with 200 µg/ml gentamicin for 3 h. Bacteria were visualized with anti-intimin antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), actin was stained with TRITC-phalloidin (red), and cell nuclei were stained with DAPI (cyan). Complementation with NleH1 or expression of EspT or EspM2 significantly reduced caspase-3 activation induced by the ΔnleH1 ΔnleH2 mutant. This is dependent on an intact WxxxE motif, as expression of EspT(W63A) and EspM2(W70A) point mutants did not reduce caspase-3 activation. Bar = 20 µm. (B) Quantification of caspase-3 activation of HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, EspT, EspT(W63A), EspM2 or EspM2(W70A) for 1 h and then incubated with 200 µg/ml gentamicin for 3 h. One hundred infected cells were counted in triplicate. Results are means ± SD of three independent experiments. **, P < 0.01. (C) Quantification of detachment levels of HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, EspT, EspT(W63A), EspM2, or EspM2(W70A) for 1 h and then incubated with 200 µg/ml gentamicin for 3 h. Cell detachment was expressed as a percentage of the value for uninfected cells. Expression of EspT and EspM2 significantly reduced cell detachment induced by the ΔnleH1 ΔnleH2 mutant to the same extent as NleH1, dependent on an intact WxxxE motif. Results are means ± SD of three independent experiments. **, P < 0.01. (D) Quantification of cytotoxicity for HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, NleH2 EspT, EspT(W63A), EspM2, or EspM2(W70A) for 30 min, then incubated with 200 µg ml−1 gentamicin for 4 h. Cytotoxicity was assayed by quantifying the levels of LDH release into the culture supernatant from cells 4 h postinfection. Expression of EspT and EspM2 significantly reduced cytotoxicity of the ΔnleH1 ΔnleH2 mutant to the same extent as NleH complementation, dependent on an intact WxxxE motif. Results are means ± SD of three independent experiments. **, P < 0.01. Download Figure S5, TIF file, 0.9 MB.
List of strains.
List of plasmids.
List of primers.
ACKNOWLEDGMENTS
We thank Rey Carabeo (Imperial College London, London, United Kingdom) for the FAK-null and FAK-reconstituted cell lines, Alan Hall (Sloan-Kettering Institute, New York, New York) for the Myc-p115RhoGEF and Myc-Rac(L61) expression vectors, Miguel Seabra (Imperial College London, London, United Kingdom) for the pCMV-RFP expressions vector, and the late Emmanuelle Caron for the Myc-Cdc42(L61) and RhoA(L63) expression vectors. We also thank Abderrahman Hachani, Ana Arbeloa, Leah Ensell, and Tristan Thwaites for technical assistance. This work was supported by grants from the Wellcome Trust.
Footnotes
Citation Wong ARC, Clements A, Raymond B, Crepin VF, Frankel G. 2012. The interplay between the Escherichia coli Rho guanine nucleotide exchange factor effectors and the mammalian RhoGEF inhibitor EspH. mBio 3(1):e00250-11. doi:10.1128/mBio.00250-11.
REFERENCES
- 1. Hall A. 1998. Rho GTPases and the actin cytoskeleton. Science 279:509–514 [DOI] [PubMed] [Google Scholar]
- 2. Etienne-Manneville S, Hall A. 2002. Rho GTPases in cell biology. Nature 420:629–635 [DOI] [PubMed] [Google Scholar]
- 3. Mitra SK, Hanson DA, Schlaepfer DD. 2005. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6:56–68 [DOI] [PubMed] [Google Scholar]
- 4. Ilić D, et al. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377:539–544 [DOI] [PubMed] [Google Scholar]
- 5. Tomar A, Schlaepfer DD. 2009. Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr. Opin. Cell Biol. 21:676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bulgin RR, et al. 2010. Bacterial guanine nucleotide exchange factors SopE-like and WxxxE effectors. Infect. Immun. 78:1417–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dong N, Liu L, Shao F. 2010. A bacterial effector targets host DH-PH domain RhoGEFs and antagonizes macrophage phagocytosis. EMBO J. 29:1363–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alto NM, et al. 2006. Identification of a bacterial type III effector family with G protein mimicry functions. Cell 124:133–145 [DOI] [PubMed] [Google Scholar]
- 9. Simpson N, et al. 2006. The enteropathogenic Escherichia coli type III secretion system effector Map binds EBP50/NHERF1: implication for cell signalling and diarrhoea. Mol. Microbiol. 60:349–363 [DOI] [PubMed] [Google Scholar]
- 10. Arbeloa A, et al. 2008. Subversion of actin dynamics by EspM effectors of attaching and effacing bacterial pathogens. Cell. Microbiol. 10:1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bulgin RR, Arbeloa A, Chung JC, Frankel G. 2009. EspT triggers formation of lamellipodia and membrane ruffles through activation of Rac-1 and Cdc42. Cell. Microbiol. 11:217–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buchwald G, et al. 2002. Structural basis for the reversible activation of a Rho protein by the bacterial toxin SopE. EMBO J. 21:3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang Z, et al. 2009. Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat. Struct. Mol. Biol. 16:853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Arbeloa A, et al. 2010. EspM2 is a RhoA guanine nucleotide exchange factor. Cell. Microbiol. 12:654–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kenny B, et al. 2002. Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol. Microbiol. 44:1095–1107 [DOI] [PubMed] [Google Scholar]
- 16. Berger CN, Crepin VF, Jepson MA, Arbeloa A, Frankel G. 2009. The mechanisms used by enteropathogenic Escherichia coli to control filopodia dynamics. Cell. Microbiol. 11:309–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tu X, Nisan I, Yona C, Hanski E, Rosenshine I. 2003. EspH, a new cytoskeleton-modulating effector of enterohaemorrhagic and enteropathogenic Escherichia coli. Mol. Microbiol. 47:595–606 [DOI] [PubMed] [Google Scholar]
- 18. Fiorentini C, et al. 1998. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells. Infect. Immun. 66:2660–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gervais FG, Thornberry NA, Ruffolo SC, Nicholson DW, Roy S. 1998. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J. Biol. Chem. 273:17102–17108 [DOI] [PubMed] [Google Scholar]
- 20. Hemrajani C, et al. 2010. NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc. Natl. Acad. Sci. U. S. A. 107:3129–3134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andor A, et al. 2001. YopE of Yersinia, a GAP for Rho GTPases, selectively modulates Rac-dependent actin structures in endothelial cells. Cell. Microbiol. 3:301–310 [DOI] [PubMed] [Google Scholar]
- 22. Shao F, et al. 2003. Biochemical characterization of the Yersinia YopT protease: cleavage site and recognition elements in Rho GTPases. Proc. Natl. Acad. Sci. U. S. A. 100:904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prehna G, Ivanov MI, Bliska JB, Stebbins CE. 2006. Yersinia virulence depends on mimicry of host Rho-family nucleotide dissociation inhibitors. Cell 126:869–880 [DOI] [PubMed] [Google Scholar]
- 24. Yarbrough ML, et al. 2009. AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323:269–272 [DOI] [PubMed] [Google Scholar]
- 25. Kubori T, Galán JE. 2003. Temporal regulation of Salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115:333–342 [DOI] [PubMed] [Google Scholar]
- 26. Wong AR, et al. 2011. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol. Microbiol. 80:1420–1438 [DOI] [PubMed] [Google Scholar]
- 27. Ma C, et al. 2006. Citrobacter rodentium infection causes both mitochondrial dysfunction and intestinal epithelial barrier disruption in vivo: role of mitochondrial associated protein (Map). Cell. Microbiol. 8:1669–1686 [DOI] [PubMed] [Google Scholar]
- 28. Martinez E, et al. 2010. Binding to Na+/H+ exchanger regulatory factor 2 (NHERF2) affects trafficking and function of the enteropathogenic Escherichia coli type III secretion system effectors Map, EspI and NleH. Cell. Microbiol. 12:1718–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18:247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fukazawa A, et al. 2008. GEF-H1 mediated control of NOD1 dependent NF-κB activation by Shigella effectors. PLoS Pathog. 4:e1000228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruno V, et al. 2009. Salmonella typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog. 5:e1000538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raymond B, Crepin VF, Collins JW, Frankel G. 2011. The WxxxE effector EspT triggers expression of immune mediators in an Erk/JNK and NF-κB-dependent manner. Cell. Microbiol. 13:1881–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karin M, Lin A. 2002. NF-κB at the crossroads of life and death. Nat. Immunol. 3:221–227 [DOI] [PubMed] [Google Scholar]
- 34. Wada T, Penninger JM. 2004. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23:2838–2849 [DOI] [PubMed] [Google Scholar]
- 35. Levine MM, et al. 1978. Escherichia coli that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet 1:119–122 [DOI] [PubMed] [Google Scholar]
- 36. Marchès O, et al. 2008. EspJ of enteropathogenic and enterohaemorrhagic Escherichia coli inhibits opsono-phagocytosis. Cell. Microbiol. 10:1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial RhoGEFs are active in the presence of EspH. Immunofluorescence microscopy of HeLa cells cotransfected with Myc-Map, Myc-EspT or Myc-SopE, and RFP or RFP-EspH (red). Myc-tagged proteins were visualized with anti-Myc antibody (magenta), and actin was stained with Oregon green phalloidin (green). Bar = 20 µm. Download Figure S1, TIF file, 2.8 MB.
EspT, EspM2, and SopE block EspH-induced focal adhesion disassembly. Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs, uninfected or infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h, followed by incubation with 200 µg ml−1 gentamicin for 3 h. Myc-tagged proteins were visualized with anti-Myc antibody (green), vinculin was detected with anti-vinculin antibody (cyan), and actin was stained with TRITC-phalloidin (red). Ectopic expression of Myc-tagged EspT, EspM2, or SopE retained focal adhesions following infection with ΔespH/pEspH EPEC but not Myc-tagged GFP or Map. Bar = 20 µm. Download Figure S2, TIF file, 1.4 MB.
EspT, EspM2 and SopE inhibit EspH-induced caspase-3 activation. Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs, uninfected or infected with ΔespH EPEC complemented with EspH (ΔespH/pEspH) for 1 h, followed by incubation with 200 µg/ml gentamicin for 3 h. Myc-tagged proteins were visualized with anti-Myc antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), and cell nuclei were stained with DAPI (cyan). Bar = 20 µm. Download Figure S3, TIF file, 0.7 MB.
EspT, EspM2, and SopE inhibit staurosporine-induced caspase-3 activation. Immunofluorescence microscopy of HeLa cells transfected with Myc-GFP or Myc-tagged bacterial RhoGEFs and then treated with 1 µM staurosporine (STS) for 5 hours. Myc-tagged proteins were visualized with anti-Myc antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), and nuclei were stained with DAPI (cyan). Bar = 50 µm. Download Figure S4, TIF file, 1 MB.
EspT and EspM2 promote host cell survival during EPEC infection. (A) Immunofluorescence microscopy of HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, EspT, EspT(W63A), EspM2, or EspM2(W70A) for 1 h and then incubated with 200 µg/ml gentamicin for 3 h. Bacteria were visualized with anti-intimin antibody (green), active caspase-3 was detected with anti-caspase-3 (cleaved) antibody (magenta), actin was stained with TRITC-phalloidin (red), and cell nuclei were stained with DAPI (cyan). Complementation with NleH1 or expression of EspT or EspM2 significantly reduced caspase-3 activation induced by the ΔnleH1 ΔnleH2 mutant. This is dependent on an intact WxxxE motif, as expression of EspT(W63A) and EspM2(W70A) point mutants did not reduce caspase-3 activation. Bar = 20 µm. (B) Quantification of caspase-3 activation of HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, EspT, EspT(W63A), EspM2 or EspM2(W70A) for 1 h and then incubated with 200 µg/ml gentamicin for 3 h. One hundred infected cells were counted in triplicate. Results are means ± SD of three independent experiments. **, P < 0.01. (C) Quantification of detachment levels of HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, EspT, EspT(W63A), EspM2, or EspM2(W70A) for 1 h and then incubated with 200 µg/ml gentamicin for 3 h. Cell detachment was expressed as a percentage of the value for uninfected cells. Expression of EspT and EspM2 significantly reduced cell detachment induced by the ΔnleH1 ΔnleH2 mutant to the same extent as NleH1, dependent on an intact WxxxE motif. Results are means ± SD of three independent experiments. **, P < 0.01. (D) Quantification of cytotoxicity for HeLa cells infected with WT EPEC, ΔnleH1 ΔnleH2 EPEC, or ΔnleH1 ΔnleH2 EPEC expressing NleH1, NleH2 EspT, EspT(W63A), EspM2, or EspM2(W70A) for 30 min, then incubated with 200 µg ml−1 gentamicin for 4 h. Cytotoxicity was assayed by quantifying the levels of LDH release into the culture supernatant from cells 4 h postinfection. Expression of EspT and EspM2 significantly reduced cytotoxicity of the ΔnleH1 ΔnleH2 mutant to the same extent as NleH complementation, dependent on an intact WxxxE motif. Results are means ± SD of three independent experiments. **, P < 0.01. Download Figure S5, TIF file, 0.9 MB.
List of strains.
List of plasmids.
List of primers.