

Abstract
Ionizing radiation (IR) and certain chemotherapeutic drugs are designed to generate cytotoxic DNA double-strand breaks (DSBs) in cancer cells. Inhibition of the major DSB repair pathway, nonhomologous end joining (NHEJ), will enhance the cytotoxicity of these agents. Screening for inhibitors of the DNA ligase IV (Lig4), which mediates the final ligation step in NHEJ, offers a novel target-based drug discovery opportunity. For this purpose, we have developed an enzymatic assay to identify chemicals that block the transfer of [α-33P]-AMP from the complex Lig4-[α-33P]-AMP onto the 5′ end of a double-stranded DNA substrate and adapted it to a scintillation proximity assay (SPA). A screen was performed against a collection of 5,280 compounds. Assay statistics show an average Z′ value of 0.73, indicative of a robust assay in this SPA format. Using a threshold of >20% inhibition, 10 compounds were initially scored as positive hits. A follow-up screen confirmed four compounds with IC50 values ranging from 1 to 30 μM. Rabeprazole and U73122 were found to specifically block the adenylate transfer step and DNA rejoining; in whole live cell assays, these compounds were found to inhibit the repair of DSBs generated by IR. The ability to screen and identify Lig4 inhibitors suggests that they may have utility as chemo- and radio-sensitizers in combination therapy and provides a rationale for using this screening strategy to identify additional inhibitors.
Introduction
DNA damage triggers cell death when it overwhelms the DNA repair capacity. Many cancer therapeutic strategies utilize DNA damaging agents to kill cancer cells. However, mammalian cells have developed sophisticated and redundant DNA repair systems to maintain genomic integrity, including DNA damage-activated cell cycle checkpoints that increase time available for repair, and enzymatic DNA repair complexes that remove or remediate various DNA lesions.1,2 Since DNA repair-deficient cells are hypersensitive to DNA damaging agents, inhibition of DNA repair should sensitize cells, including cancer cells, to DNA damaging agents. As our understanding of the molecular mechanisms of the cellular DNA damage responses has increased, there has been growing interest in developing small molecules to inhibit these cell defense systems.
DNA-damaging anticancer treatments induce a variety of DNA lesions, such as base damage, DNA single-strand breaks, DNA double-strand breaks (DSBs), DNA crosslinks, and replication collapse.3 In general, these lesions are recognized and repaired by a lesion-specific DNA repair machinery. Two major pathways in eukaryotes, homologous recombination (HR) and nonhomologous end joining (NHEJ), counteract one of the most toxic lesions, the DSB. Thus, the toxicity of DSB-induced anticancer therapy maybe reduced by activation of HR or NHEJ, making each pathway a potential therapeutic target. While HR is restricted in the S phase of cell cycle and predominantly repairs replication-associated DSBs, NHEJ is active in all cell cycle stages and repairs the majority of DSBs generated by ionizing radiation (IR).4,5 Thus, NHEJ is an attractive target for the development of inhibitors that increase cell killing by DSB-inducing agents.
The core protein complex mediating NHEJ in mammals includes the Ku70/Ku80 heterodimer, DNA-dependent protein kinase (DNA-PKcs), Artemis, DNA ligase IV (Lig4), XRCC4, and XCRCC4-like factor.6 Once DSBs occur, Ku70/Ku80 binds DNA ends and recruits DNA-PKcs forming the DNA-PK that phosphorylates NHEJ factors including itself, and also brings and aligns two broken DNA ends together via protein–protein interactions.7,8 The aligned DNA ends are then processed by specific DNA polymerases and nucleases to generate termini that are joined by Lig4 complexed with XRCC4.9 Our strategy was to utilize the enzymatic properties of the NHEJ core complex for the discovery and development of novel NHEJ inhibitors; we focused our attention on the final ligation process of NHEJ by Lig4.
Lig4 belongs to an ATP-dependent DNA ligase family. Molecular mechanistic studies have divided ligation into three steps: (i) formation of the ligase-AMP intermediate; (ii) transfer of AMP to the 5′-phosphate group at the end of DNA; and (iii) phosphodiester bond formation with AMP release.10 ATP-dependent DNA ligases contain a conserved lysine active site residue that interacts with ATP to form the adenylate-ligase intermediate with the AMP moiety linked via a phosphoramidate bond to the ɛ amino group of a lysine.11,12 Previous studies have shown that the Lig4 purified from cell extracts of HeLa cells and baculovirus is mostly preadenylated.13 Since the AMP transfer reaction occurs in the presence of double-stranded DNA (dsDNA) in vitro,14 it appears likely that Lig4 is maintained in the activated, charged state in vivo and that the AMP transfer step specifically takes place once DNA damage has occurred.
In this report, we have designed an in vitro enzyme-based assay measuring AMP transfer by Lig4 to a 50-bp dsDNA substrate and adapted it to a scintillation proximity assay (SPA) format amenable to high-throughput screening (HTS) of large chemical libraries to identify inhibitors of Lig4. The assay was constructed in two parts: first, we utilized the adenylation reaction to generate radiolabeled Lig4 (Lig4-[α-33P]-AMP), and second, we measured the transfer of radioactive [α-33P]-AMP onto a 50-bp dsDNA substrate captured by SPA imaging beads as the assay readout in 384-well microtiter plates. Using this novel assay approach, we initially identified 10 hits that inhibited Lig4 by screening a chemical library of 5,280 compounds. We describe the initial characterization of four confirmed hits in both biochemical and live cell-based assays.
Materials and Methods
Purification of the Human DNA Ligases
Insect sf9 cells (Invitrogen) (500 mL, 2×107 cells/mL) were coinfected with baculovirus harboring two expression plasmids that express His-tagged Lig4 and XRCC4 individually for 60 h. Cells were harvested by centrifugation for 5 min at 1,000 g, resuspended in 10 mL of lysis buffer containing 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0, 0.4 μg/mL aprotinin, 1 mM benzamidine-HCl, 0.5 μg/mL chymostatin, 0.5 μg/mL leupeptin, 0.7 μg/mL pepstatin, and 1 mM PMSF, and lysed by sonication. After centrifugation for 10 min at 10,000 g at 4°C, the Lig4/XRCC4 complex was purified from the cleared lysate by Ni-NTA agarose affinity chromatography. After washing with lysis buffer containing 20 mM imidazole, His-tagged Lig4/XRCC4 was eluted from the beads with lysis buffer containing 250 mM imidazole, analyzed by sodium dodecyl sulfate (SDS)-gel electrophoresis for purity assessment, and stored at −70°C until further use. The human DNA ligase I (Lig1) and ligase III (Lig3) were purified as described previously.15
Preparation of DNA Substrates
Oligonucleotides were synthesized by Gene Link. The oligonucleotides used in the AMP transfer and the SPA-based assays for screenings were 50M with the sequence 5′- AACAAAGTTTGGATTGCTACTGACCGCTCTCGTGCTCGTCGCTGCGTT and 41M with the sequence 5′-GCGACGAGCACGAGAGCGGTCAGTAGCAATCCAAACTTTGT. The 50M and 41M DNA strands were annealed by slow temperature cooling as follows: 95°C for 5 min, 70°C for 10 min, 50°C for 10 min, 40°C for 10 min, 30°C for 10 min, and finally 4°C for 10 min to form the dsDNA substrate (50M/41M).
In the gel mobility shift assay, the 41M oligonucleotide substrate was labeled at the 5′ end with T4 polynucleotide kinase and [γ-33P]-ATP (3,000 Ci/mmol). After purification on a MERmaid spin column (MP Biomedicals) to remove unincorporated deoxynucleotide triphosphates, isotopically labeled 41M substrate was annealed with the cold 50M oligonucleotides as described previously.
For the DNA joining assay by Lig4, a linear dsDNA fragment, 400-bp linear DNA with 4-nucleotide 5′ overhangs, was generated by digestion with HindIII. After separation by agarose gel electrophoresis, the DNA fragment was purified by a QIAquick gel extraction kit (QIAGEN). The desired DNA fragment was labeled at the 5′ end with T4 polynucleotide kinase and [γ-33P]-ATP (3,000 Ci/mmol). Unincorporated deoxynucleotide triphosphates were removed using a QIAquick PCR purification kit as recommended by the manufacturer.
For the DNA joining assay by Lig1 and Lig3, a linear 50-bp dsDNA with a nick was generated by annealing three oligonucleotides (50M, 20M, and 30M). The 5′ end of 20M was labeled with [γ-33P]-ATP (3,000 Ci/mmol) by T4 polynucleotide kinase. Unincorporated deoxynucleotide triphosphates were removed using a MERmaid spin kit (MP Biomedicals). The obtained and labeled 20M was annealed with 50M and 30M to generate the following radioactive DNA-based substrates: 50M with the sequence 5′-AACAAAGTTTGGATTGCTACTGACCGCTCTCGTGCTCGTCGCTGCGTT, 20M with the sequence 5′-AACGCAGCGACGAGCACGAG, and 30M with the sequence 5′-AGCGGTCAGTAGCAATCCAAACTTTGTT.
Adenylation Reaction
Lig4/XRCC4 (1 nmol) was incubated for 15 min at room temperature (RT) in a reaction mixture containing 1 μCi [α-33P]-ATP (3,000 Ci/mmol), 60 mM Tris-HCl (pH 8.0), and 10 mM MgCl2.
Reactions were stopped by the addition of SDS sample buffer (Bio-Rad) and analyzed by SDS-polyacrylamide gel electrophoresis. Gels were fixed in buffer containing 30% methanol and 10% acetic acid for 20 min at RT, followed by incubation in Amplify buffer (GE Healthcare) for 20 min at RT. Labeled Lig4-adenylate complexes were detected by autoradiography or Phosphor-Imager analysis.
AMP Transfer Assay
The adenylation reaction was performed to generate isotope-labeled Lig4/XRCC4 complexes, followed by rebinding to Ni-NTA agarose beads to remove unincorporated [α-33P]-ATP. Labeled complex-Ni-NTA agarose beads were washed and eluted with 250 μM imidazole. Labeled Lig4/XRCC4 complexes (1 nmol) were incubated for 15 min at RT in a reaction mixture (10 μL) containing 60 mM Tris-HCl (pH 8.0), 10 mM MgCl2, and 250 pmol of dsDNA substrate (50M/41M). Reactions were stopped by addition of SDS sample buffer and analyzed by SDS-PAGE. Gels were fixed and radioactive signals were amplified as described previously. Radioactive Lig4-adenylate proteins were detected on the dried gel by autoradiography or Phosphor-Imager analysis.
Design of SPA-Based High-Throughput Screen
Radioactive Lig4/XRCC4 preparation: His-tagged Lig4/XRCC4 complexes were adenylated for 15 min at RT in a reaction mixture containing [α-33P]-ATP (3,000 Ci/mmol), 60 mM Tris-HCl (pH 8.0), and 10 mM MgCl2 and then incubated with His-Bind Ni-charged beads. Proteins were eluted from the beads with a buffer containing 750 mM imidazole, 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 10% glycerol (v/v). The eluted complexes were dialyzed against 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 10% glycerol (v/v), and stored at −70°C until further use.
High-throughput assay conditions were adapted from the previous work and industrialized for use in 384-well microtiter plate format using SPA technology. One microliter of 10% dimethyl sulfoxide (DMSO) (v/v) was plated to a 384-well microtiter plate (Corning #3704; Corning) with a custom-designed 384 head on a PP-384-M Personal Pipettor (Apricot Designs). Radioactive adenylated Lig4 was diluted in the reaction buffer (60 mM Tris-HCl [pH 8.0] and 10 mM MgCl2) to a working concentration of 200 nmol/mL; 5 μL of which (equivalent to 1 nmol enzyme) was added to wells using the FlexDrop IV dispenser (Perkin Elmer) for test compound incubation step. After 15 min at RT, 6 μL of reaction buffer containing 250 pmol of the 50M/41M dsDNA substrate was added using the FlexDrop IV dispenser. Following an incubation step of 15 min at RT, 50 μL of 10 mg/mL suspension of HIS TAG PS Imaging beads (GE Healthcare) was added using the Multidrop 384 (Thermo) and sealed with a clear plastic adhesive seal (Perkin Elmer). After an overnight incubation at 4°C, the assay plates were centrifuged at 2,000 rpm for 1 min and the plates were imaged on the LEADseeker Multimodality Imaging System (GE Healthcare). The assay performance of the developed SPA was assessed in a control run involving three 384-well microtiter plates as “high control” plates containing 250 pmol 50M/41M dsDNA substrate in 1% DMSO (v/v) mimicking a “no inhibition control” condition and three 384-well microtiter plates as “low control” plates containing 1% DMSO (v/v) mimicking an “inhibition control” condition. The control run was performed as described previously.
Chemical Libraries
The library used for the screen combines 5,280 chemicals obtained from MicroSource, Prestwick, Tocris, and other commercial sources.16–18 The MicroSource Library contains 2,000 biologically active and structurally diverse compounds from known drugs, experimental bioactives, and pure natural products. The library includes a reference collection of 160 synthetic and natural toxic substances (previously characterized as being inhibitors of DNA/RNA synthesis, protein synthesis, cellular respiration, and membrane integrity); a collection of 80 compounds representing classical and experimental pesticides, herbicides, and endocrine disruptors; and a singular collection of 720 natural products and their derivatives. The latter collection includes simple and complex oxygen heterocycles, alkaloids, sequiterpenes, diterpenes, pentercyclic triterpenes, sterols, and many other diverse representatives. The Prestwick Chemical Library is a unique collection of 1,119 high-purity chemical compounds, all off patent and selected for structural diversity and broad spectrum, with established activities in therapeutic areas including neuropsychiatry, cardiology, immunology, antiinflammatory, and analgesia, with known safety, and bioavailability in humans. Approximately 90% of the library consists of marketed drugs and 10% of bioactive alkaloids or related substances.16–18 Compounds used in the follow-up tests were purchased from Santa Cruz Biotechnology (rabeprazole and U73122) or Sigma (cytochalasin A and NSC95397). About 10 mM stocks of compounds were prepared in 100% DMSO (v/v) and stored at −20°C until further use. The purity of rabeprazole, U73122, cytochalasin A, and NSC95397 was confirmed by mass spectrometry as previously described.18
Chemical Screen of Lig4 Using the Developed SPA
A library collection of 5,280 compounds containing known drugs and bioactives was screened at a final concentration of 10 μM test compound in 1% DMSO (v/v) and the screen was performed in duplicate. One microliter of 100 μM compound solution in 10% DMSO (v/v) was transferred from an intermediate 384-well polypropylene microtiter plate (Abgene) to an assay plate using the PP-384-M Personal Pipettor. Every assay plate contained control wells in columns 13 and 14. Column 13 wells mimicked “high control” conditions and column 14 mimicked “low control” conditions as described previously. The optimized assay steps are summarized in Table 1.
Table 1.
Steps of the DNA Ligase IV Assay with the Scintillation Proximity Assay Method
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Library compounds | 1 μL | 100 μM in 10% DMSO (v/v) |
| 2 | Control compounds | 1 μL | 10% DMSO (v/v) |
| 3 | Enzyme | 5 μL | 1 nmol His-Lig4-[α-33P]-AMP |
| 4 | Incubation time | 15 min | Room temperature |
| 5 | Substrate | 6 μL | 250 pmol dsDNA substrate |
| 6 | High control | 6 μL | 250 pmol dsDNA substrate |
| 7 | Low control | 6 μL | Reaction buffer (60 mM Tris-HCl [pH 8.0] and 10 mM MgCl2) |
| 8 | Incubation time | 15 min | Room temperature |
| 9 | Imaging beads | 50 μL | HIS TAG PS Imaging beads |
| 10 | Incubation time | 24 h | 4°C—cold temperature |
| 11 | Centrifugation | 1 min | 2,000 rpm |
| 12 | Assay readout | Epi mirror: empty Emission mirror: empty |
LEADseeker multimodality imaging system |
Step Notes
1–2. Dispensing on the PP-384-M Personal Pipettor using a custom 384 head
3. Dispensing with the FlexDrop IV
4. Plates incubated at room temperature for 15 min
5 to 7. Dispensing with the FlexDrop IV
8. Plates incubated at room temperature for 15 min
9. Dispensing with the Multidrop 384
10. Plates incubated at cold temperature for 24 h
11. Plates centrifuged at 2,000 rpm for 1 min
12. Radiometric signal quantified with empty epi mirror and empty emission mirror with [14C]-plate as a reference
DMSO, dimethyl sulfoxide; dsDNA, double-stranded DNA.
The screen was performed on a fully automated linear track robotic platform (CRS F3 Robot System; Thermo CRS) using several integrated peripherals for plate handling, liquid dispensing, and detection systems. Compounds were plated using a custom-designed 384 head on a PP-384-M Personal Pipettor (Apricot Designs). The addition of reagents was performed using the FlexDrop (Perkin Elmer) or the Multidrop 384 (Thermo) as described previously for an SPA-based assay.19 Screening data files were obtained from the automated LEADseeker (GE Healthcare) for the SPA. The screening data files obtained were subsequently loaded into the HTS Core Screening Data Management System, a custom-built suite of modules for compound registration, plating, and data management, powered by ChemAxon Cheminformatic tools (ChemAxon). The summary of the identified positives was exported as chemical structure data files for further analysis and reporting.
Dose–Response Study of Primary Positives with the SPA Method
Serial 12-point doubling dilutions of the compounds in 10% DMSO (v/v) were prepared in an intermediate 384-well polypropylene microtiter plate and 1 μL of each compound was transferred to the assay plate with the PP-384-M Personal Pipettor. Compounds were tested at a final concentration between 50 nM and 100 μM in 1% DMSO (v/v). For controls, 1 μL of 10% DMSO (v/v) was added to rows A and P of each assay plate. Reaction buffer containing dsDNA was added to row A mimicking “high control” conditions and only reaction buffer was added to row P mimicking “low control” conditions. Dose–response studies were performed according to the assay steps described previously and dose–response curves for each dataset were fitted using a logistic four-parameter equation from Sigmaplot (SYSTAT).
Z′ Factor
The Z′ factor was used to assess the performance of the optimized SPA-based assay as previously described.19 The Z′ factor constitutes a dimensionless parameter that ranges from 1 (infinite separation) to <0. It is defined as Z′=1 − (3σc+ + 3σc−) / |μc+ − μc−|, where σc+, σc−, μc+, and μc− are the standard deviations (σ) and averages (μ) of the high (c+) and low (c−) control. The Z′ factor between 0.5 and 1 indicates an excellent assay with good separation between controls. The Z′ factor between 0 and 0.5 indicates a marginal assay and <0 signifies a poor assay with overlap between controls.19
Electrophoretic Mobility Shift Assay
Inhibitory compounds and Lig4/XRCC4 complexes were incubated for 15 min at RT. Then the mixtures were incubated with 5′ end-radiolabeled 50M/41M dsDNA substrates for 15 min at RT in buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM KCl, 0.1% Triton X-100, 60 μg/mL bovine serum albumin (BSA), and 5% glycerol (v/v). Samples were loaded onto a 15% TBE native polyacrylamide gel (Bio-Rad) and electrophoresis was carried out at 100 V at 4°C. Labeled DNA substrates were detected at the dried gel by autoradiography or Phosphor-Imager analysis.
DNA Ligation Assay
For DNA joining by Lig4, Lig4/XRCC4 (1 nmol) was incubated with or without compounds (100 μM in 1% DMSO [v/v]) for 15 min at RT. Next, the reaction mixtures were incubated with the labeled DNA substrates (250 pmol) in ligation buffer (final volume, 10 μL) containing 60 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 5 mM dithiothreitol, 1 mM ATP, and 50 μg/mL BSA for 30 min at RT. Reactions were deproteinized by phenol/chloroform extraction and then analyzed by 1% agarose gel electrophoresis. The gel was fixed in the buffer containing methanol and acetic acid for 20 min; radioactive-labeled oligonucleotides in the dried gel were detected by autoradiography and quantitated by the Phosphor-Imager analysis.
For DNA joining by Lig1 and Lig3, Lig1 (3 nmol) and Lig3 (0.5 nmol) were incubated with or without compounds (100 μM in 1% DMSO [v/v]) for 15 min at RT. Next, the reaction mixtures were incubated with the labeled DNA substrates (250 pmol) in ligation buffer for 30 min at RT. The reaction was terminated by the addition of equal volume of 2× TBE-Urea loading buffer (Invitrogen) followed by heating at 95°C for 3 min. Reactions were analyzed by 10% TBE-Urea gel electrophoresis. Then, the gel was fixed and then incubated in the radioactive signal enhancement buffer for 20 min; radioactive-labeled oligonucleotides in the dried gel were detected by autoradiography and quantitated by the Phosphor-Imager analysis.
Cell Culture and Pulsed Field Gel Electrophoresis
HeLa cervical cancer cells were obtained from the American Type Culture Collection. HeLa cells were maintained in DMEM/HG medium (obtained from the MSKCC Media Core Facility) with 10% fetal bovine serum (Gibco). The effect of individual compounds on DNA repair was measured by pulsed field gel electrophoresis in two replicates. The HeLa cells (4×106 cells/100 ×20 mm dish) were grown overnight prior to exposure to 100 Gy of IR. Cells were harvested by trypsinization and centrifugation at different time points, washed once in serum-free medium, and then resuspended in serum-free medium (1×106 cells/60 μL). The cell suspension was mixed with an equal volume of 1% agarose incubated at 56°C, and then loaded into the plug molds. The solidified plugs were incubated with 2 mL lysis buffer (10 mM Tris-HCl [pH 8.0], 50 mM NaCl, 0.5 M EDTA, 2% N-lauryl sarcosyl, 0.1 mg/mL proteinase K) for 1 h on ice, and then for 16 h at 50°C. After washing once with washing buffer (10 mM Tris-HCl [pH 8.0] and 0.1 M EDTA), the plugs were incubated in washing buffer for 1 h at 37°C and then transferred to RNase buffer (10 mM Tris-HCl [pH 7.5], 0.1 M EDTA, and 0.1 mg/mL RNase) and incubated at 37°C for 60 min. DNA in the plugs was analyzed by 0.8% agarose (in 0.5×TBE) gel electrophoresis. This electrophoresis was performed at 200 V with 60-s pulses for the first 8 h, followed by 120-s pulses for a further 15 h. The gel was stained with SYBR Green and analyzed by FUJI Fluorescent Imager FLA5000.
Results
Measurement of AMP Transfer from Adenylated Lig4 onto dsDNA Substrate
We hypothesized that inhibitors of the nucleotidyl transferase activity of Lig4 will prevent the repair of DSBs by NHEJ, thereby increasing the cytotoxicity of DNA-damaging agents that induce DSBs, which are used to treat cancer (Fig. 1A). Since previous studies have shown that XRCC4 forms a complex with Lig4 and is required for Lig4 stabilization and activity,20–25 we coexpressed Lig4 and XRCC4 in insect cells and purified the resultant to near homogeneity (Fig. 1B). In accordance with what was published previously,11,12 incubation of purified Lig4/XRCC4 with [α-33P]-ATP resulted in the formation of the labeled complex Lig4-[α-33P]-AMP (Fig. 1C) that was deadenylated by incubation with linear dsDNA, but not when a single-stranded DNA (ssDNA) substrate was added (Fig. 1C); the loss of [α-33P]-AMP from Lig4 provides the basis for our proposed enzymatic assay to screen for inhibitors of Lig4 (Fig. 1D) as described in assay development.
Fig. 1.

Purification and characterization of Lig4 enzymatic function. (A) The three enzymatic reaction steps of Lig4 ligation in DNA double-strand break (DSB) repair. Step 1: Adenylation of an active site lysine residue of Lig4. The product is the Lig4-AMP intermediate. Pyrophosphate (PPi) is released. Step 2: AMP transfer to DNA ends. Once DSBs are generated, Lig4-AMP binds DNA and transfers the AMP moiety to the 5′-phosphate group at the end of DNA. Step 3: DNA joining. The AMP-activated 5′ phosphorylated DNA end attacks the 3′ hydroxyl group of an adjacent DNA strand to release AMP and rejoins the DNA strands. Our experimental design of Lig4 inhibitors targets the second step. Step 4: If Lig4 is inhibited by a compound, the breaks are no longer repaired and the consequence of which is cell death. (B) Electrophoretic analysis of purified His-tagged Lig4/XRCC4. Lig4 and XRCC4 proteins were coexpressed and copurified from insect cells as described under Materials and Methods section. The positions of molecular mass standards (Bio-Rad) are indicated on the left. (C) Lig4 transfers AMP to dsDNA and not to ssDNA. Radioactive adenylated Lig4/XRCC4 (1 nmol) was incubated without DNA (lane 1) or with ssDNA (250 pmol) (lane 2) or with dsDNA (250 pmol) (lane 3). After separation by denaturing gel electrophoresis, radioactive adenylated Lig4/XRCC4 in a dried gel was detected by autoradiography. The arrow indicates the position of radioactive adenylated Lig4/XRCC4. (D) Generation of the radioactive adenylated His-tagged Lig4/XRCC4 complex (Lig4-[α-33P]-AMP). Radioactive adenylated His-tagged Lig4/XRCC4 (1 nmol) was incubated with or without dsDNA (250 pmol). Lig4, DNA ligase IV; ssDNA, single-stranded DNA.
Assay Development and Adaptation to 384-Well Microtiter Plates
We have developed a Lig4 enzymatic assay that directly measures the deadenylation of the Lig4-[α-33P]-AMP by further incubation of the complex with a 50-bp linear dsDNA substrate and transfers the radioactive moiety to the dsDNA substrate resulting in free Lig4 devoid of radioactivity; small molecule inhibitors would, therefore, block the transfer and result in a radioactive Lig4 complex. In the absence of the dsDNA substrate, the adenylated Lig4 (Lig4-[α-33P]-AMP) can be produced in large, enough quantities as the enzymatic component to be used in the screen (Fig. 2). To measure the enzymatic activity of Lig4 to the DNA substrate in an HTS suitable assay format, we opted for the SPA technology as it is homogeneous in nature, easy to miniaturize into 384-well microtiter plates, and has been used successfully by our HTS group for a CDC7 kinase screening project.19 The assay schematic and workflow are depicted in Figure 2. After the deadenylation reaction was completed, a suspension of HIS TAG PS imaging beads will be added to the reaction mixture. The binding of His-tagged Lig4-[α-33P]-AMP to the SPA imaging beads in the absence of the dsDNA substrate results in high fluorescence emission signal in the red region (approximately at 615 nm) excited by the emitted β particles of [α-33P]-AMP (Fig. 2) and is referred to as the “low control” of the assay. However, in the presence of the 50M/41M dsDNA substrate, the [α-33P]-AMP moiety is transferred from Lig4 to the dsDNA substrate which is not captured by the SPA imaging beads, thus resulting in low fluorescence emission signal partly excited by the emitted β particles of [α-33P]-AMP through a nonproximity effect (NPE) and is referred to as the “high control” of the assay (Fig. 2).
Fig. 2.

SPA workflow. The assay workflow starts with incubating the Lig4-[α-33P]-AMP complex with compounds for 15 min followed by addition of the 50M/41M dsDNA substrate and further incubated for 15 min. After the deadenylation reaction was completed, a suspension of HIS TAG PS imaging beads is added to the reaction mixture. No inhibition of the enzyme upon binding of His-tagged Lig4-[α-33P]-AMP to the SPA imaging beads results in low fluorescence emission signal in the red region (approximately at 615 nm) excited partly by the emitted β particles of [α-33P]-AMP through a nonproximity effect and will be referred to as the “no inhibition” high control of the assay. Inhibition of the Lig4 enzyme upon binding of His-tagged Lig4-[α-33P]-AMP to the SPA imaging beads results in high fluorescence emission signal in the red region (approximately at 615 nm) excited by the emitted β particles of [α-33P]-AMP moiety in close proximity to the SPA beads and will be referred to as the “inhibition” low control of the assay. Therefore, an inhibited Lig4 enzyme would result in high fluorescence emission signal mimicking the low control values of the assay. SPA, scintillation proximity assay.
To assess the robustness of this assay for chemical screening, we performed a control run comprising of three 384-well plates that contained no dsDNA substrate and 1% DMSO (v/v) to mimic the “low control” or enzyme inhibition and three 384-well microtiter plates that contained the 50M/41M dsDNA substrate in 1% DMSO (v/v) for the “high control” or no enzymatic inhibition, and evaluated the assay performance by calculating percent coefficients of variation (% CV) and the Z′ factor. Figure 3A shows a frequency distribution plot of the fluorescence signal output from 1,152 data points of each of the low and high controls. The assay had an averaged high signal of 500 pixel densities for the low control wells and an averaged low signal of 222 pixel densities for the high control wells with an S/B ratio of 2.2. The CV of the controls was 4% and 3%, respectively, with a calculated Z′ factor of 0.74 (Fig. 3A). These statistics showed good assay robustness and the low % CV values gave us confidence to conduct a pilot screen in this assay format. In addition, a heat map analysis of the control run showed no systematic errors from dispensing, plate edge effects, and good overall consistency of data (data not shown).
Fig. 3.

Chemical screen for inhibitors of Lig4 with the SPA method. (A) Control assessment. Three 384-well microtiter plates contained 1% DMSO (v/v) mimicking the “inhibition” low control and three 384-well microtiter plates contained 250 pmol of the dsDNA substrate in 1% DMSO (v/v) mimicking the “no inhibition” high control of the assay. The resulting radiometric signals of all of the 2,304 data points are represented as a frequency distribution plot with a Z′ value of 0.74 and very tight coefficients of variations for both high and low controls. (B) Chemical screen of a collection of 5,280 compounds. Screen was conducted in duplicate, with 15 assay plates. Chart of the Z′ factor of each assay plate is presented. The Z′ factor was calculated with the average and standard deviation of the radiometric signal of 16 wells of the “no inhibition” high control and the “inhibition” low control. The results of 15 assay plates of Set 1 (□) and 15 assay plates of Set 2 (▪) are shown. Dotted line shows the average calculated Z′ factor of the 30 assay plates as 0.76. One plate (plate 1 in Set 2) was mishandled during the screen and resulted in a Z′ factor of 0.36; compound data from this plate were flagged and removed from further analysis. (C) Scatter plot analysis of the chemical screen data correlating the percent inhibition of the duplicate datasets. X-axis and Y-axis shows the percent inhibition of each compound in Set 1 and Set 2, respectively. Compounds exhibiting inhibitory activity of higher than 20% (dotted lines) in both datasets were selected as positive hits (●). Outlier compounds showing activity in either set are highlighted (▾) and not pursued in the confirmatory studies. (D) Dose–response studies of the four selected compounds rabeprazole, cytochalasin A, U73122, and NSC95397. DMSO, dimethyl sulfoxide.
Chemical Screen for Inhibitors of Lig4
To identify small molecule inhibitors of Lig4 through screening chemical libraries, the newly adapted SPA was optimized for HTS use as depicted in Table 1 and tested at a single compound concentration of 10 μM in 1% DMSO (v/v) against a library of 5,280 compounds comprised of several collections including FDA-approved drugs, known bioactives, and experimental substances.16–18 The library was plated in fifteen 384-well microtiter plates with columns 13 and 14 empty for control wells to monitor the assay's performance throughout the screen using Z′ factor. Column 13 wells contained 250 pmol of the dsDNA substrate in 1% DMSO (v/v) mimicking the “no inhibition” high control of the assay and column 14 wells contained 1% DMSO (v/v) mimicking the “inhibition” low control of the assay. The screen was performed in duplicate to assess the assay variability, reproducibility, and an initial estimation of hit rate. Plate-by-plate Z′ values for screen Set 1 and Set 2 were averaged at 0.70 and 0.68, respectively, indicating excellent performance and consistency with the previous assay development data (Fig. 3B). In particular, one plate was mishandled during the screen which resulted in a Z′ factor of 0.36; compound data from this plate were flagged and removed from further analysis.
To measure reproducibility, the datasets were plotted in a scatter graph to examine correlation for each test compound. A scatter plot of Lig4 activity as percent inhibition showed high overall correlation as depicted by the slope of the pattern (Fig. 3C) with data points centrally distributed between the X/Y axes. A large majority of compounds clustered around the X/Y intercept indicating no activity and only few outliers were present. At a threshold of 20% inhibition, 10 compounds were positive on both datasets (Table 2), with 15 and 4 compounds positive on Set 1 and Set 2, respectively. In total, we identified 10 compounds, which met our threshold and reproducibility criteria for nominating a positive (Table 2) and with an overall initial hit rate of 0.19%, as potential inhibitors of Lig4 AMP transfer activity. These compounds were structurally diverse and previously characterized activities that included signal transduction inhibitors, antifungals, chelates, cell cycle inhibitors, and ion transport inhibitors. We performed dose–response studies on four compounds repurchased from vendors and having passed quality control for chemical structural integrity and purity standards; these were rabeprazole (proton pump inhibitor [PPI]), cytochalasin A (fungal toxin), U73122 (phospholipase C inhibitor), and NSC95397 (Cdc25 phosphatase inhibitor). The compounds were tested in dose–response studies in a 12-point doubling dilution concentration series from 50 nM to 100 μM and with resulting IC50 values ranging from 1 to 30 μM as depicted in Figure 3D and Table 2. These four compounds were selected for further follow-up studies.
Table 2.
Chemical Structures and Therapeutic Usage of 10 Hits Identified Using the Scintillation Proximity Assay Method
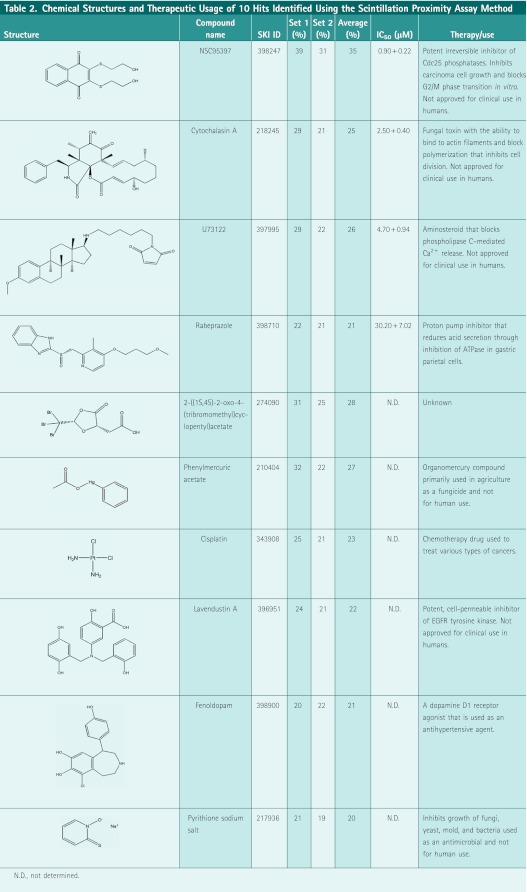 |
Effect of HTS Hits on the Enzymatic Activities of Lig4, Lig1, and Lig3
Preincubation of purified Lig4 with each of the HTS hits before the addition of [α-33P]-ATP had no significant effect on the formation of labeled Lig4-[α-33P]-AMP complex (Fig. 4A), indicating that these compounds do not inhibit the first step of the ligation reaction. Next, we examined the effect of the compounds on AMP transfer from Lig4 to dsDNA. As we have expected, all of them inhibited the transfer of [α-33P]-AMP from Lig4 to the dsDNA substrate (Fig. 4B, compare lane 2 with lanes 3–6). Rabeprazole, cytochalasin A, and U73122 were more effective than NSC95397 at inhibiting the transfer of the AMP moiety. Since the AMP moiety has to be transferred to the 5′-phosphate terminus of the DNA ends as step three of the ligation reaction, the inhibition of AMP transfer should also inhibit DNA joining. In the DNA joining assays, the amount of ligation was decreased by rabeprazole and U73122 (Fig. 4C, compare lane 2 with lanes 3 and 5), but not significantly by compounds cytochalasin A and NSC95397. The quantitative analysis further shows that compound rabeprazole and U73122 inhibited ligation by up to 35% (Fig. 4D).
Fig. 4.

Effect of selected HTS hits on the enzymatic activity of Lig4. (A) The four selected compounds (rabeprazole, cytochalasin A, U73122, and NSC95397) have no effect on Lig4 adenylation. Lig4/XRCC4 (1 nmol) was preincubated with DMSO (100%) or with indicated compound (100 μM) at RT for 15 min. Then, the reaction mixtures were incubated with α-33P-ATP at RT for 15 min. After separation by denaturing gel electrophoresis, adenylated Lig4/XRCC4 in a dried gel was detected by autoradiography. Lane 1 contains the 1% DMSO (v/v) control. Lanes 2–5 contained indicated compounds. The arrow indicates the position of adenylated Lig4/XRCC4. (B) Rabeprazole, cytochalasin A, U73122, and NSC95397 inhibit the Lig4-AMP transfer step of the reaction. A positive control, containing radioactive adenylated Lig4/XRCC4 (1 nmol) alone with no dsDNA substrate, was included in lane 1. Lanes 2–6 contain the dsDNA substrate. Lane 2 contains the 1% DMSO (v/v) control; lanes 3–6 contain indicated compounds at a final concentration of 100 μM in 1% DMSO (v/v). Reactions were incubated at RT for 15 min. After separation by denaturing gel electrophoresis, radioactive adenylated Lig4/XRCC4 in a dried gel was detected by autoradiography. The arrow indicates the position of adenylated Lig4/XRCC4. (C) The effect of rabeprazole, cytochalasin A, U73122, and NSC95397 on DNA joining by Lig4. Lane 1 contains dsDNA substrate alone. Lig4/XRCC4 (1 nmol) was preincubated with DMSO (10%) (lane 2) or with indicated compounds (100 μM) (lanes 3–6) at RT for 15 min. Then, the reaction mixtures were incubated with radioactive dsDNA at RT for 30 min. DNAs were purified and analyzed by gel electrophoresis. The arrows indicate the positions of the DNA ligation products and DNA substrate. The DNA joining results were also quantitated by phosphor imaging. (D) Obtained data from three independent experiments are quantified, represented graphically, and show no potent inhibitory effects on the ligation step of the reaction. (E) The effect of rabeprazole and U73122 on DNA binding of Lig4. A positive control containing dsDNA substrate alone was included in lane 1. Lanes 2–4 contained Lig4/XRCC4 (1 nmol) preincubated for 15 min at RT with 1% DMSO (v/v) (lane 2), 100 μM rabeprazole (lane 3), or 100 μM U73122 (lane 4), followed by a further incubation with the radioactive 50-bp dsDNA substrate (250 pmol) for 1 h and then by native TBE gel electrophoresis. The arrows indicate the positions of DNA substrate and the Lig4/XRCC4 complexed with the dsDNA substrate. RT, room temperature.
Adenylated Lig4/XRCC4 complexes bind to the DNA ends to catalyze steps 2 and 3 of the ligation reaction (Fig. 1A). We utilized an electrophoretic mobility shift assay to determine whether compounds rabeprazole and U73122 inhibit the DNA binding step to the complex Lig4/XRCC4. No significant changes in the amount or mobility of the DNA–protein complex formed by Lig4/XRCC4 were observed (Fig. 4E, compare lane 2 with lanes 3 and 4), indicating that the inhibitory effect of rabeprazole and U73122 is not due to disruption of DNA binding to the enzyme.
In addition to Lig4, there are two other human DNA ligases, Lig1 and Lig3, which function in DNA replication and repair.23 Because the human DNA ligases utilize the same enzymatic mechanism of DNA ligation, we examined the effect of the obtained Lig4 inhibitors on the ligation activities of Lig1 and Lig3. Neither rabeprazole nor U73122 inhibited ligation by Lig1 (Fig. 5A, lanes 1–8) or Lig3 (Fig. 5B, lanes 1–8). Thus, we conclude based on our mechanistic studies that rabeprazole and U73122 seem to be specific inhibitors of Lig4.
Fig. 5.

Effect of rabeprazole and U73122 on the enzymatic activities of Lig1 and Lig3. (A) The effect of rabeprazole and U73122 on DNA joining by Lig1. Recombinant Lig1 was purified as described previously.15 Lanes 1–8 contained Lig1 (3 nmol) and preincubated with indicated increasing compound concentrations in 1% DMSO (v/v) for 15 min at RT by a further incubation with the radioactive dsDNA substrates (250 pmol) for 30 min. Resulting DNA ligation products were analyzed by denaturing gel electrophoresis as described under Materials and Methods section. The arrows indicate the positions of the resulting DNA ligation products and the dsDNA substrate. (B) The effect of rabeprazole and U73122 on DNA joining by Lig3. Recombinant Lig3 was purified as described previously.15 Similar experimental design was applied to Lig3 with the exception of the Lig3 concentration in the reaction of 0.5 nmol enzyme. Lig1, DNA ligase I; Lig3, DNA ligase III.
DNA Damage Repair in Live Cells
To assess whether rabeprazole and U73122 could inhibit the repair of DSBs inside cells, we performed pulsed field gel electrophoresis studies to directly measure DNA damage and repair upon exposure of cells to IR. We found that in irradiated cells untreated with the Lig4 inhibitors, the amount of damaged chromosomal DNA decreased over time as the DSBs were being repaired. After 90 min, the amount of damaged DNA remaining was ∼40% (i.e., 40% of broken DNA was remaining in the gel as compared with time 0, or 60% of the damage was repaired). When cells were pretreated with rabeprazole or U73122 before gamma irradiation, the amount of damaged DNA remaining after 90 min was about 80% (i.e., only 20% of the damage was repaired) (Fig. 6). Based on these results, we speculate that cell exposure to either rabeprazole or U73122 inhibited Lig4 function; the consequence of which is the observed inability of the treated cells to repair efficiently the DNA damage as compared with untreated control cells.
Fig. 6.

Effect of rabeprazole and U73122 on DNA repair in live cells induced by ionizing radiation (IR). HeLa cells were pretreated with 50 μM rabeprazole or U73122 for 20 min at 37°C followed by 100 Gy IR. Cells were then harvested at indicated time points. The chromosome DNAs were analyzed by pulsed field gel electrophoresis. The DNAs were stained with SYBR Green and analyzed by FUJI Fluorescent Imager FLA5000. Damaged DNA was quantitated by the ImageJ software. The average of two independent experiments is shown.
Discussion
Major current targets for cancer therapeutic drugs include growth signaling pathways, the cellular division machinery, and cellular DNA itself. Although cytotoxic DNA-damaging agents such as IR have been used in cancer treatment for many years, there have been few attempts to target the DNA repair machinery. As the molecular mechanisms for cellular DNA damage responses have become more clearly understood, there has been increasing interest in enhancing the effects of chemotherapy by disrupting these systems.3 This approach maybe particularly important in cancers that have lost some capacity to repair DNA rendering them hypersensitive to additional loss of ordinarily redundant mechanisms. Several molecules that target the DNA damage response network have been identified, with some of them, such as PARP1 inhibitors that block the repair of DNA single-strand breaks and Chk1 and Chk2 kinase inhibitors that inhibit cell cycle progression,26,27 currently being evaluated in clinical trials.
The hypersensitivity of Lig4-defective cells to IR and DNA-damaging drugs suggests that this enzyme is a good target for the development of inhibitors that potentiate the cytotoxic effects of DNA damage. Although several small molecules inhibitors of human DNA ligases have been identified using structure-based computational design,28 none of the inhibitors were specific for Lig4. And unlike Lig1 and Lig3,23,29,30 there is no available structural information for the Lig4 catalytic domain. Chen and coworkers described a Lig4 assay with potential use in screening large chemical libraries. The assay makes use of a dual-labeled DNA strand with a donor fluorophore (D) and a quencher fluorophore (Q) within proximity to each other and separated by the ligation site. To distinguish between the ligated and unligated DNA oligonucleotides, as both exhibit 100% fluorescence intensities, a thermal denaturation step is performed using a thermocycler in which the DNA duplex is denatured to ssDNA oligonucleotides upon exposure at 95°C for 5min. Upon cooling of the reaction, the ligated ssDNA oligonucleotides adopt secondary structures bringing Q in close proximity to D resulting in a loss of fluorescence intensity.31 The described denaturation step could potentially result in many false positives as compounds would bind nonspecifically to the ssDNA oligonucleotides as the temperature is cooling down to RT at a rate of 2°C per minute for a total of 92 min. Other ligation assays using fluorescently labeled DNA substrates have also been reported for bacterial DNA ligases,32,33 and have been shown to be amenable to miniaturization and used in screening large chemical libraries; however, they do require a denaturation step using 8M urea to separate the fully ligated DNA strand harboring the fluorophore from the other strand template harboring the quencher.32,33
The SPA is a radioisotopic assay technique that has been widely used in screening chemical libraries of various therapeutic targets;34,35 it is homogeneous in nature and does not require washing steps. Like fluorescence-based assay technologies, SPA does also suffer from sensitivity to various colored compounds yielding higher hit rates due to false hits scored during screening campaigns. However, the recent introduction of the PS imaging beads results in “red-shifted” assay readouts that appear to be relatively insensitive to colored compounds, especially those absorbing in the yellow, red, and blue ranges of the light spectrum.35 Other inherent limitations of SPA are as follows: (i) the requirements for specific infrastructure to handle radioactive material usage and disposal, which is typically expensive and (ii) the NPE due to the excitation of the fluorophore in the beads by the nonspecific radiation from the tracer when using higher energy radioisotopes such as [32P], [33P], or [125I]; this NPE effect limits the specific activity of the reporter radioisotope to be used in the assay and in many instances renders the use of SPA inadequate. The ultimate advantage of SPA is the direct capture of the reaction product harboring a radioactive tracer35; this attribute was an important consideration in designing our Lig4 assay because we were monitoring the loss of an incorporated radioactive tracer in the DNA substrate upon complete ligation. Further, the preformed Lig4-[α-33P]-AMP complex offers the additional advantage for the SPA approach as it would exclude nucleotide analogs from scoring as hits in this format, but not in the fluorescence-based assay formats described previously. DNA interchelators, on the other hand, are another class of nonspecific inhibitors to which the SPA would be insensitive, but the secondary assays in place would eliminate these hits from further consideration.
For this purpose, we have successfully developed a scintillation proximity-based assay to screen for inhibitors of the enzymatic activity of Lig4 where our strategy was to block the deadenylation step of the Lig4-AMP complex and potentially resulting in an extra selectivity for actives in chemical libraries. A chemical screen was completed against a library of 5,280 compounds, containing known bioactives and FDA-approved drugs, resulting in four confirmed hits (rabeprazole, cytochalasin A, U73122, and NSC95397) with IC50 values ranging from 1 to 30 μM (Table 2); though not particularly potent, these compounds have proven useful nonetheless in follow-up mechanistic studies and in their potential use as tool compounds.
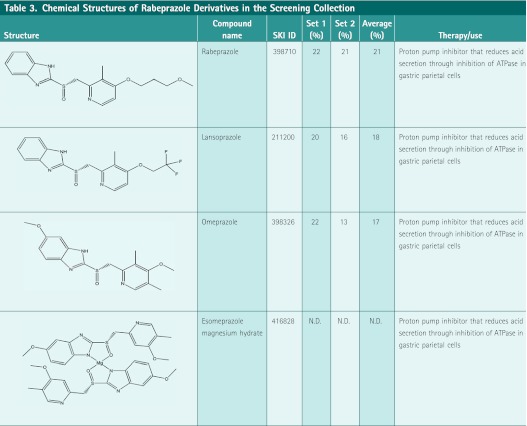
The Lig4 inhibitors found in this study are known bioactives. Rabeprazole is a PPI. The PPIs suppress gastric acid secretion by inhibiting the final transport of hydrogen ions into the gastric lumen in the gastric H+, K+-ATPase system.36,37 The PPIs become active forms upon entering the lower pH environment (e.g., inside parietal cells), and then they can bind to H+, K+-ATPase and inhibit ATPase function. Because the pH of our designed assay is 8.0, rabeprazole remains in its inactive form. Two other PPIs were included in the screening library and were initially selected as outliers in the screen and not pursued (Table 3); this suggests that this chemical scaffold maybe important for binding to the Lig4 complex. It is unlikely that the inhibitory mechanism of Lig4 by rabeprazole is the same as the inhibition of the H+, K+-ATPases. The U73122 is a phospholipase C inhibitor.38,39 Phospholipase C cleaves phosphatidylinositol 4,5-bisphosphate into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). The IP3 works to increase concentration of cytosolic calcium, and DAG and calcium work together to activate protein kinase C that triggers the signal transduction pathway. The U73122 also shows inhibitory effects on a variety of receptor-mediated signal transductions.40,41 The mechanism of inhibition of Lig4 by U73122 remains unclear.
Table 3.
Chemical Structures of Rabeprazole Derivatives in the Screening Collection
 |
Rabeprazole, cytochalasin A, U73122, and NSC95397 were found to inhibit the AMP transfer step of the ligation process by Lig4, though at a high compound concentration of 100 μM, supporting the argument that such Lig4 inhibitors could be found through screening against larger chemical libraries. Both rabeprazole and U73122, when tested in an in vitro DNA joining assay, were found to inhibit the formation of ligation products by up to 35% (Fig. 4), whereas cytochalasin A and NSC95397 had little or no effect on the DNA joining reaction. Close examination of the inhibitory effects revealed that rabeprazole and U73122 observed effects were not mediated by blocking dsDNA substrate to the enzyme thus ruling out their effects as nonspecific DNA intercalators (Fig. 4E). Although the three human DNA ligases share a related catalytic domain and a common catalytic mechanism, the two most active Lig4 inhibitors, rabeprazole and U73122, identified in the screen had no detectable inhibitory activity against both Lig1 and Lig3. It is conceivable that these inhibitors interact with the enzyme–substrate complex and that there are significant conformational constraints between the enzyme–substrate complexes formed by the three human DNA ligases. Thus, it may also be possible to identify specific inhibitors of Lig1 and Lig3 by adapting our scintillation proximity-based assay platform to screen for inhibitors of Lig1 and Lig3.
Although rabeprazole and U73122 only partially inhibited specifically the Lig4 function in vitro, they exhibited intended cellular activity by slowing down the repair of gamma irradiation–induced DNA breaks in HeLa cells. This is a good indication for their potential use in combination therapy with DNA-damaging agents and IR to treat cancer. While rabeprazole and U73122 are not potent enough for use effectively in cancer xenograft models, as a starting point followed conceptually by human clinical trials, our experiments can provide the basis for a proof-of-concept and rationale to target Lig4 as a druggable entity, identify inhibitors and develop them as drugs to treat cancer in combination therapies, or for additional screens against larger chemical libraries to identify more potent and novel Lig4 inhibitors.
Conclusion
We report on the development of a high-throughput scintillation proximity-based assay to screen for inhibitors of Lig4. Using this assay, we have successfully screened a library of 5,280 compounds and identified four specific Lig4 inhibitors that appear to work by disrupting the second step of the Lig4 function, namely, the transfer of AMP from the Lig4-AMP complex to dsDNA substrate. Additional screening and use of the assay with other substrates may lead to a better understanding of the mechanisms of this enzyme's action, as well as other human ligases, and also to potential new therapeutic agents.
Abbreviations
- BSA
bovine serum albumin
- CV
coefficients of variation
- DAG
diacyl glycerol
- DMSO
dimethyl sulfoxide
- DNA-PK
DNA-dependent protein kinase
- DSBs
double-strand breaks
- dsDNA
double-stranded DNA
- HR
homologous recombination
- HTS
high-throughput screening
- IP3
inositol 1,4,5-trisphosphate
- IR
ionizing radiation
- Lig4
DNA ligase IV
- Lig1
DNA ligase I
- Lig3
DNA ligase III
- N.D.
not determined
- NHEJ
nonhomologous end joining
- NPE
nonproximity effect
- PPI
proton pump inhibitor
- RT
room temperature
- SDS
sodium dodecyl sulfate
- SPA
scintillation proximity assay
- ssDNA
single-stranded DNA
Acknowledgments
The authors thank Dr. Dale Ramsden for providing baculoviruses containing the expression constructs of Lig4 and XRCC4, Jonathan Seideman for technical support of pulsed field gel electrophoresis, and members of the HTS Core Facility for their help during the course of this study, especially Dr. Christophe Antczak for critically reading the manuscript. The work was supported by NIH P01 CA33049, R01 CA55349, and T32 CA062948. The HTS Core Facility is partially supported by Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of the Memorial Sloan-Kettering Cancer Center, the William Randolph Hearst Fund in Experimental Therapeutics, the Lillian S. Wells Foundation, and by an NIH/NCI Cancer Center Support Grant 5 P30 CA008748-44.
Disclosure Statement
No competing financial interests exist.
References
- 1.Harrison JC. Haber JE. Surviving the breakup: the DNA damage checkpoint. Ann Rev Genet. 2006;40:209–235. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- 2.Harper JW. Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 3.Helleday T. Petermann E. Lundin C. Hodgson B. Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 4.Aylon Y. Liefshitz B. Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ira G. Pellicioli A. Balijja A, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahaney BL. Meek K. Lees-Miller SP. Repair of ionizing radiation-induced DNA double- strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lieber MR. Grawunder U. Wu X. Yaneva M. Tying loose ends: roles of Ku and DNA-dependent protein kinase in the repair of double-strand breaks. Curr Opin Genet Dev. 1997;7:99–104. doi: 10.1016/s0959-437x(97)80116-5. [DOI] [PubMed] [Google Scholar]
- 8.Weterings E. Chen DJ. DNA-dependent protein kinase in nonhomologous end joining: a lock with multiple keys? J Cell Biol. 2007;179:183–186. doi: 10.1083/jcb.200705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lieber MR. Lu H. Gu J. Schwarz K. Flexibility in the order of action and in the enzymology of the nuclease, polymerases, and ligase of vertebrate non-homologous DNA end joining: relevance to cancer, aging, and the immune system. Cell Res. 2008;18:125–133. doi: 10.1038/cr.2007.108. [DOI] [PubMed] [Google Scholar]
- 10.Tomkinson AE. Vijayakumar S. Pascal JM. Ellenberger T. DNA ligases: structure, reaction mechanism, and function. Chem Rev. 2006;106:687–699. doi: 10.1021/cr040498d. [DOI] [PubMed] [Google Scholar]
- 11.Tomkinson AE. Totty NF. Ginsburg M. Lindahl T. Location of the active site for enzyme-adenylate formation in DNA ligases. Proc Natl Acad Sci USA. 1991;88:400–404. doi: 10.1073/pnas.88.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doherty AJ. Suh SW. Structural and mechanistic conservation in DNA ligases. Nucleic Acids Res. 2000;28:4051–4058. doi: 10.1093/nar/28.21.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robins P. Lindahl T. DNA ligase IV from HeLa cell nuclei. J Biol Chem. 1996;271:24257–24261. doi: 10.1074/jbc.271.39.24257. [DOI] [PubMed] [Google Scholar]
- 14.Riballo E. Doherty AJ. Dai Y, et al. Cellular and biochemical impact of a mutation in DNA ligase IV conferring clinical radiosensitivity. J Biol Chem. 2001;276:31124–31132. doi: 10.1074/jbc.M103866200. [DOI] [PubMed] [Google Scholar]
- 15.Chen X. Pascal J. Vijayakumar S. Wilson GM, et al. Human DNA ligases I, III, and IV-purification and new specific assays for these enzymes. Methods Enzymol. 2006;409:39–52. doi: 10.1016/S0076-6879(05)09003-8. [DOI] [PubMed] [Google Scholar]
- 16.Shelton CC. Tian Y. Shum D, et al. A miniaturized 1536-well format gamma-secretase assay. Assay Drug Dev Technol. 2009;7:461–470. doi: 10.1089/adt.2009.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shum D. Smith JL. Hirsch AJ, et al. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev Technol. 2010;8:553–570. doi: 10.1089/adt.2010.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seideman JH. Shum D. Djaballah H. Scheinberg DA. A high-throughput screen for alpha particle radiation protectants. Assay Drug Dev Technol. 2010;8:602–614. doi: 10.1089/adt.2010.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takagi T. Shum D. Parisi M, et al. Comparison of Luminescence ADP production assay and radiometric scintillation proximity assay for Cdc7 kinase. Comb Chem High Throughput Screen. 2011;14:669–687. doi: 10.2174/138620711796504442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grawunder U. Wilm M. Wu X, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 21.Critchlow SE. Bowater RP. Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol. 1997;7:588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- 22.Bryans M. Valenzano MC. Stamato TD. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat Res. 1999;433:53–58. doi: 10.1016/s0921-8777(98)00063-9. [DOI] [PubMed] [Google Scholar]
- 23.Ellenberger T. Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grawunder U. Zimmer D. Fugmann S, et al. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 25.Frank KM. Sekiguchi JM. Seidl KJ, et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 26.Durkacz BW. Omidiji O. Gray DA. Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283:593–596. doi: 10.1038/283593a0. [DOI] [PubMed] [Google Scholar]
- 27.Ashwell S. Zabludoff S. DNA damage detection and repair pathways–recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res. 2008;14:4032–4037. doi: 10.1158/1078-0432.CCR-07-5138. [DOI] [PubMed] [Google Scholar]
- 28.Chen X. Zhong S. Zhu X, et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–3177. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pascal JM. O'Brien PJ. Tomkinson AE. Ellenberger T. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004;432:473–478. doi: 10.1038/nature03082. [DOI] [PubMed] [Google Scholar]
- 30.Cotner-Gohara E. Kim I. Tomkinson AE. Ellenberger T. Two DNA-binding and nick recognition modules in human DNA ligase III. J Biol Chem. 2008;283:10764–10772. doi: 10.1074/jbc.M708175200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X. Pascal J. Vijayakumar S. Wilson GM. Ellenberger T. Tomkinson AE. Human DNA ligases I, II, and IV-purification and new specific assays for these enzymes. Methods Enzymol. 2006;409:39–52. doi: 10.1016/S0076-6879(05)09003-8. [DOI] [PubMed] [Google Scholar]
- 32.Shapiro AB. Eakin AE. Walkup GK. Rivin O. A high-throughput fluorescence resonance energy transfer-based assay for DNA ligase. J Biomol Screen. 2011;16:486–493. doi: 10.1177/1087057111398295. [DOI] [PubMed] [Google Scholar]
- 33.Mills SD. Eakin AE. Buurman ET. Newman JV. Gao N. Huynh H. Johnson KD. Lahiri S. Shapiro AB. Walkup GK. Yang W. Stokes SS. Novel bacterial NAD+-dependent DNA ligase inhibitors with broad-spectrum activity and antibacterial efficacy in vivo. Antimicrob Agents Chemother. 2011;55:1088–10896. doi: 10.1128/AAC.01181-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cook ND. Scintillation proximity assay: a versatile high-throughput screening technology. DDT. 1996;1:287–294. [Google Scholar]
- 35.Glickman JF. Schmid A. Ferrand S. Scintillation proximity assays in high-throughput screening. Assay Drug Dev Technol. 2008;6:433–455. doi: 10.1089/adt.2008.135. [DOI] [PubMed] [Google Scholar]
- 36.Shin JM. Sachs G. Gastric H,K-ATPase as a drug target. Dig Dis Sci. 2006;51:823–833. doi: 10.1007/s10620-005-9042-8. [DOI] [PubMed] [Google Scholar]
- 37.Morii M. Takata H. Fujisaki H. Takeguchi N. The potency of substituted benzimidazoles such as E3810, omeprazole, Ro 18-5364 to inhibit gastric H+, K(+)-ATPase is correlated with the rate of acid-activation of the inhibitor. Biochem Pharmacol. 1990;39:661–667. doi: 10.1016/0006-2952(90)90143-9. [DOI] [PubMed] [Google Scholar]
- 38.Thompson AK. Fisher SK. Preferential coupling of cell surface muscarinic receptors to phosphoinositide hydrolysis in human neuroblastoma cells. J Biol Chem. 1991;266:5004–5010. [PubMed] [Google Scholar]
- 39.Bleasdale JE. Thakur NR. Gremban RS, et al. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther. 1990;255:756–768. [PubMed] [Google Scholar]
- 40.Walker EM. Bispham JR. Hill SJ. Nonselective effects of the putative phospholipase C inhibitor, U73122, on adenosine A1 receptor-mediated signal transduction events in Chinese hamster ovary cells. Biochem Pharmacol. 1998;56:1455–1462. doi: 10.1016/s0006-2952(98)00256-1. [DOI] [PubMed] [Google Scholar]
- 41.Hughes SA. Gibson WJ. Young JM. The interaction of U-73122 with the histamine H1 receptor: implications for the use of U-73122 in defining H1 receptor-coupled signaling pathways. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:555–558. doi: 10.1007/s002100000326. [DOI] [PubMed] [Google Scholar]