

Abstract
Prenyl- and pyrano-xanthones derived from 1,3,6-trihydroxy-9H-xanthen-9-one, a basic backbone of gambogic acid (GA), were synthesized and evaluated for in vitro cytotoxic effects against four human cancer cell lines (KB, KBvin, A549, and DU-145) and anti-inflammatory activity toward superoxide anion generation and elastase release by human neutrophils in response to fMLP/CB. Among them, prenylxanthones 7-13 were generally less active than pyranoxanthones 14-21 in both anticancer and anti-inflammatory assays. Furthermore, two angular 3,3-dimethypyranoxanthones (16 and 20) showed the greatest and selective activity against the KBvin multidrug resistant (MDR) cell line with IC50 values of 0.9 and 0.8 μ g/mL, respectively. An angular 3-methyl-3-prenylpyranoxanthone (17) selectively inhibited elastase release with 200 times more potency than phenylmethylsulfonyl fluoride (PMSF), the positive control.
Keywords: 1,3,6-Trihydroxy-9H-xanthen-9-one; Gambogic acid (GA); Prenylxanthones; Pyranoxanthones; Cytotoxicity; Anti-inflammatory activity
Gamboge resin, obtained from Garcinia hanburyi in Southeast Asia, has been used as a coloring agent and folk medicine in China.1 Gambogic acid (GA) is a natural product isolated from this resin. Its molecular structure features a unique 4-oxatricyclo[4.3.1.0]decan-2-one ring system built on a xanthone backbone, and this unique ring system is found only in natural products from the genus Garcinia.2 The biogenesis of GA in nature must involve two different pathways, one similar to that of caged benzophenones and the other to simple xanthones.3 Ollis and his colleagues reported that GA can be synthesized from normal xanthone.4
Aside from its striking chemical architecture, pharmacological studies have revealed that GA possesses potent antitumor activity both in vitro and in vivo,5 and GA has entered phase I clinical trials in China for tolerance testing.6 In addition, the Garcinia genus is recognized as a rich source of xanthone natural products with high pharmaceutical potential.7 GA contains many functional groups; however, this complex lead compound may have a simpler pharmacophoric moiety buried within its structure. If this pharmacophore can be clearly identified, the resulting simpler molecule may have improved synthetic tractability and be more useful. In order to elucidate the structure-activity relationship (SAR) correlations of GA’s basic xanthone skeleton, a retro-synthetic analysis (Figure 1) suggested the design and evaluation of the biological activities of 1,3,6-substituted xanthone derivatives would be reasonable.
Figure 1.

The retrosynthesis of gambogic acid.
Xanthone compounds show potent biological activities, including growth inhibition of various tumor cell lines,8 inhibition of human lymphocyte proliferation,9 and PKC modulation,10 as well as antitumor11 and anti-inflammatory activities.12 These activities have been associated with the compounds’ tricyclic scaffold depending on the nature and/or position of the different substituents.13 A previous paper also revealed that several related xanthones, including 1,3,6-trihydroxy-9H-xanthen-9-one, which is the basic skeleton of GA, showed significant activity against sarcoma 180 tumor cells.14 Furthermore, recent literature has also shown that prenylated dihydroxyxanthone derivatives exhibited tumor growth inhibitory activity.3,13
Based on the above results, we designed and synthesized prenylated derivatives structurally related to 1,3,6-trihydroxy-9H-xanthen-9-one, in an effort to find the optimal structural features required for antitumor and anti-inflammatory effects. The synthesized compounds 4–21 have never been isolated as natural products. All new xanthone compounds were assayed for in vitro cytotoxicity against four human cancer cell lines, KB (nasopharyngeal), KBvin (multidrug-resistant nasopharyngeal over-expressing P-gp), A549 (lung), and DU-145 (prostate), and for anti-inflammatory action in terms of superoxide anion generation and elastase release by human neutrophils in response to fMLP/CB.
The synthetic methodologies used to synthesize the xanthone building blocks 4 and 5, and their derivatives 6–21 are outlined in Schemes 1 and 2. 1,3,6-Trihydroxy-9H-xanthen-9-one was originally prepared by condensation and cyclization reactions between phloroglucinols and appropriately substituted salicylic acids with phosphorus oxychloride–zinc chloride as catalyst.15 Later studies provided better results by using a mixture of phosphorus pentoxide–methanesulfonic acid (Eaton’s reagent).16 Therefore, compounds 4 and 5 were synthesized in good yields (90–95%) by the intramolecular oxidative coupling reaction between phloroglucinol (3) and 2,4-dihydroxybenzoic acid (1) or 2-hydroxy-4-methoxybenzoic acid (2), respectively, in the presence of Eaton’s reagent, and were used in the next step without purification. Treatment of 4 with iodomethane in the presence of K2CO3/acetone gave 6 (84%). Prenylation of 4 with prenyl bromide in the presence of KOH furnished a mixture of compounds 7–9, which were separated by silica gel column chromatography (7: 5.8%; 8: 4.2%; 9: 16%). The O-prenylated compounds 10–13 were prepared by reaction of 4 with prenyl bromide in the presence of both KOH and KI, followed again by chromatographic separation (10: 4.2%; 11: 50%; 12: 5.8%; 13: 10%).
Scheme 1.

The synthesis of compounds 4–13.
Reagents: (a) P2O5-CH3SO3H, 80 ° C, 1 h (4: 92%; 5: 95%). (b) MeI, K2CO3, acetone, reflux, 60 ° C, overnight (6, 85%). (c) (i) KOH, prenyl bromide, H2O, 0 ° C, 24 h (7: 5.8%; 8: 4.2%; 9: 16%;). (ii) KOH, KI, prenyl bromide, DMF, 0 ° C, 24 h (10: 4.2%; 11: 50%; 12: 5.8%; 13: 10%).
Scheme 2.
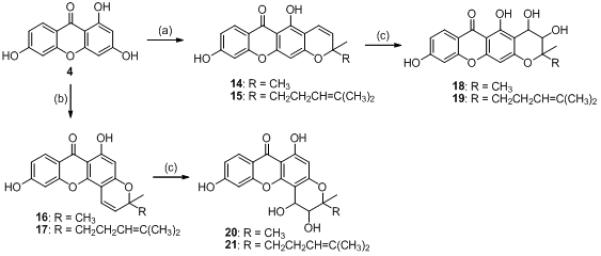
The synthesis of pyranoxanthones 14–21.
Reagents and conditions: (a) prenal/citral, Ca(OH)2, MeOH, rt, 36 h (14: 85%; 15: 70%). (b) prenal/citral, 140–150 ° C, 6 h (16: 95%; 17: 93%). (c) OsO4, NMMO, t-BuOH-THF-H2O (10:3:1), rt, 48 h (18: 85%; 19: 70%; 20: 19%; 21: 22%).
Cyclization of 4 to the desired linear pyranoxanthones 14 and 15 was accomplished by reactions with prenal (3-methyl-2-butenal) and citral (3,7-dimethyl-2,6-octadienal), respectively, in methanolic calcium hydroxide solution at rt to afford 85% and 70% yields, respectively. For the angular pyranoxanthones 16 and 17, compound 4 was reacted with prenal or citral at 140-150 ° C for 6 h resulting in yields of 95% and 93%, respectively.17 Dihydrodiolpyranoxanthones 18-21 were prepared by catalytic osmium tetroxide oxidation of 14–17, respectively, using N-methylmorpholine N-oxide to regenerate the oxidizing agent (18: 50%; 19: 45%; 20: 19%; 21: 22%).18
Cytotoxic activity
The synthesized compounds 4–21 can be divided into three classes, simple xanthones 4–6, prenylxanthones 7–13, and pyranoxanthones 14–21. Table 1 lists the IC50 values obtained with the test compounds compared to natural GA and the anticancer drug paclitaxel as a positive control. The inhibitory effect of the test drugs on cell viability was measured by the MTT colorimetric method as described previously.19
Table 1.
Cytotoxicity of compounds 4–21.
| Compound | Cancer cell line (IC50 value,a μg/mL) |
|||
|---|---|---|---|---|
| KB | KBvin | A549 | DU-145 | |
| 4 | NDb | > 10 | > 10 | > 10 |
| 5 | ND | > 10 | > 10 | > 10 |
| 6 | ND | > 10 | > 10 | > 10 |
| 7 | ND | 6.7 | 6.4 | 3.9 |
| 8 | ND | 5.4 | 6.2 | 3.8 |
| 9 | ND | 4.4 | 5.0 | 2.8 |
| 10 | ND | 5.3 | 6.9 | 3.7 |
| 11 | ND | 7.5 | 7.9 | 4.3 |
| 12 | ND | > 10 | 9.3 | > 10 |
| 13 | ND | > 10 | > 10 | 6.3 |
| 14 | 4.6 | 5.5 | 5.8 | 4.4 |
| 15 | 6.2 | 6.0 | 6.6 | 6.2 |
| 16 | 4.8 | 0.9 | 5.8 | 5.1 |
| 17 | 5.3 | 5.7 | 5.1 | 5.2 |
| 18 | 6.3 | 6.8 | 5.9 | 5.4 |
| 19 | > 10 | > 10 | > 10 | > 10 |
| 20 | > 10 | 0.8 | 9.9 | 6.7 |
| 21 | > 10 | > 10 | > 10 | > 10 |
| GA | 0.3 | 0.4 | 0.5 | 0.3 |
| Paclitaxelc | 1.6×10−3 | 1.1×10−3 | 1.9×10−3 | 2.4×10−3 |
Data are expressed as mean = 3.
ND: Not determined.
Positive control.
None of the three simple xanthones (4–6) showed significant cytotoxic effects against the KBvin, A549, and DU-145 cancer cell lines. However, the addition of a prenyl group increased the activity. Among the prenylxanthones 7–13, the C-prenylxanthones 7–9 (IC50 2.8 ~ 6.7 μ g/mL) were generally more active than the O-prenylxanthones 10–13 (IC50 3.7 ~ >10 μ g/mL) against the three tested cancer cell lines. Compound 10 with one O-prenyl moiety was more active than compounds with two (12) or three (11, 13) groups. The DU-145 cell line was most sensitive to these compounds (7–9: IC50 2.8 ~ 3.9 μ g/mL). Among the pyranoxanthones 14–17, the angular 3-dimethylpyranoxanthone 16 showed a notable IC50 value of 0.9 μ g/mL against the KBvin cancer cell line; however, it was much less active against the KB, A549, and DU-125 cell lines (IC50 4.8 ~ 5.8 μ g/mL). The corresponding angular 3-methyl-3-prenyl compound (17) and the two linear compounds (14 and 15) exhibited the same range of activity against all four cell lines (IC 50 4.4 ~ 6.6 μ g/mL). Oxidation of the pyranoxanthones 14–17 to the dihydrodiolpyranoxanthones 18–21 generally resulted in decreased activity, with one exception. Compound 20, the dihydrodiol analog of 16, retained the high activity against the KBvin cancer cell line with an IC50 value of 0.8 μ g/mL. Overall, the 3-methyl compounds (14, 16, 18, and 20) exhibited greater activity than the corresponding 3-prenyl compounds (15, 17, 19, and 21). Although most of the synthetic compounds were inactive or much less active than the natural product GA, compounds 16 and 20 were selective for and showed comparable activity with GA against the KBvin cancer cell line, indicating that structural simplification could be a viable option in the design of new chemotherapeutic agents from this compound class. Our research also indicated that pyranoxanthones could have more potent cytotoxic effects than previously discovered, as previous literature showed that prenylated xanthones had weak cytotoxicity (IC50 values 50~ 80 μ M).20
Anti-inflammatory activity
Compounds 4–21 were also evaluated for anti-inflammatory action based on effects against superoxide anion generation and elastase release by human neutrophils in response to fMLP/CB. The assays were performed using established protocols,21 which are widely used to identify potential anti-inflammatory compounds. Table 2 lists the results for the test compounds, as well as diphenyleneiodonium (DPI) and phenylmethylsulfonyl fluoride (PMSF), included as positive controls for superoxide anion generation and elastase release, respectively.
Table 2.
Inhibitory effects of compounds on superoxide anion generation and elastase release by human neutrophils in response to fMLP/CB.
| Compound | Superoxide anion | Elastase release |
|---|---|---|
|
| ||
| IC50 (μg/mL)a or (Inh%)a | IC50 (μg/mL)a or (Inh%)a | |
| 4 | 5.84 ± 0.75 | (16.0 ± 6.57) |
| 5 | (45.2 ± 3.22) | (23.6 ± 1.24) |
| 6 | (31.3 ± 3.88)*** | (26.5 ± 5.04)*** |
| 7 | (101 ± 5.47)b | (102 ± 1.94)b |
| 8 | (45.2 ± 0.43) | (56.3 ± 3.42) |
| 9 | (31.6 ± 0.21) | (67.2 ± 5.65) |
| 10 | 20.2 ± 0.52 | (4.21 ± 5.22) |
| 11 | (10.1 ± 2.73) | (7.56±3.98) |
| 12 | (5.32 ± 0.12) | (13.3 ± 5.32) |
| 13 | (64.3 ± 0.22) | (54.9 ± 5.73) |
| 14 | 0.46 ± 0.05 | 0.64 ± 0.22 |
| 15 | 5.43 ± 0.82 | 8.75± 0.69 |
| 16 | 3.97 ± 0.45 | (18.4 ± 1.44) |
| 17 | 35.6 ± 6.83c*** | 0.49 ± 0.10 |
| 18 | 2.49 ± 0.38 | (35.3 ± 1.59)*** |
| 19 | 4.30 ± 0.33 | 5.75 ± 0.17 |
| 20 | (8.63 ± 6.95) | (6.37 ± 5.04) |
| 21 | 2.56 ± 0.28 | 8.46 ± 0.73 |
| DPId | 0.7 ± 0.4 | |
| PMSFd | 131 ± 2.91 | |
IC50 represents the 50% inhibitory concentration of the compound. If 50% inhibition was not reached at any test dose, the percentage of inhibition obtained at a test dose of 10 μg/mL is given in parentheses (Inh%). Results are presented as mean ± S.E.M. (n = 3-5).
p < 0.001 compared with the control value.
7 alone elicited superoxide anion generation and elastase release by human neutrophils in the absence of fMLP/CB.
17 induced superoxide generation in the pretreatment of cytochalasin B.
DPI and PMSF were used as positive controls.
Xanthone 4 showed a selective inhibitory effect toward superoxide anion generation with an IC50 value of 5.84 μ g/mL, while compounds 5 and 6 exhibited weak activity in both anti-inflammatory assays. Among compounds 7–21, prenylxanthones 7–13 demonstrated weaker effects than pyranoxanthones 14–21 in response to superoxide anion generation and elastase release. Linear pyranoxanthone 14 was the most active compound, with IC50 values of 0.46 and 0.64 μ g/mL against superoxide anion generation and elastase release, respectively, and angular pyranoxanthone 17 showed selective anti-inflammatory activity toward elastase release with an IC50 value of 0.49 μ g/mL. Except for 16, 18, and 20, compounds 14–21 exhibited potent activity toward elastase release and were over 15-fold more potent than the positive control PMSF.
In this investigation, we prepared a series of 1,3,6-substituted xanthones (4–6), as well as prenyl- and pyrano-xanthone analogs (7–21),22 and evaluated SAR for their cytotoxic and anti-inflammatory activities. In conclusion, among all screened compounds, prenylxanthones 7–13 were less active than pyranoxanthones 14–21 in both anticancer and anti-inflammatory assays. Two angular 3,3-dimethylpyranoxanthone analogs (16 and 20) showed notable and selective activity against a multidrug resistant (MDR) cell line (KBvin) with much lower activity against the parent cells (KB). A linear 3,3-dimethylpyranoxanthone compound (14) exhibited significant potency in both anti-inflammatory assays, and an angular 3-methyl-3-prenylpyranoxanthone compound (17) was 200-fold more potent than PMSF, the positive control, in the elastase release assay.
Acknowledgments
This investigation was supported by grant CA 17625-32 from the National Cancer Institute, NIH, USA (K. H. Lee), and by grant DOH101-TD-C-111-004 from the Department of Health, Executive Yuan, Taiwan (Y. C. Wu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1(a).Ollis WD, Redman BT, Sutherland IO, Jewers KJ. Chem. Soc. Chem. Commun. 1969;15:879. [Google Scholar]; (b) Kumar P, Baslas RK. Herba Hung. 1980;19:81. [Google Scholar]
- 2.Li NG, You QD, Huang XF, Wang JX, Guo QL, Chen XG, Li Y, Li HY. Chin. Chem. Lett. 2007;18:659. [Google Scholar]
- 3.Han Q-B, Yang N-Y, Tian H-L, Qiao C-F, Song J-Z, Chang DC, Chen S-L, Luo KQ, Xu H-X. Phytochemistry. 2008;69:2187. doi: 10.1016/j.phytochem.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 4.Ollis WD, Ramsay MVJ, Sutherland IO. Tetrahedron. 1965;21:1453. [Google Scholar]
- 5(a).Guo QL, You QD, Wu ZQ, Yuan ST, Zhao L. Acta Pharmacol. Sin. 2004;25:769. [PubMed] [Google Scholar]; (b) Liu W, Guo QL, You QD, Zhao L, Gu HY, Yuan ST. World J. Gastroenterol. 2005;11:3655. doi: 10.3748/wjg.v11.i24.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao L, Guo QL, You QD, Wu ZQ, Gu HY. Biol. Pharm. Bull. 2004;27:998. doi: 10.1248/bpb.27.998. [DOI] [PubMed] [Google Scholar]
- 6.Zhou ZT, Wang JW. Chinese J New Drugs. 2007;16:79. [Google Scholar]
- 7.Thoison O, Fahy J, Dumontet V, Chiaroni A, Riche C, Tri MV, Sevenet T. J. Nat. Prod. 2000;63:441. doi: 10.1021/np9903088. [DOI] [PubMed] [Google Scholar]
- 8.Pedro M, Cerqueira F, Sousa ME, Nascimento MSJ, Pinto M. Bioorg. Med. Chem. 2002;10:3725. doi: 10.1016/s0968-0896(02)00379-6. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez MJ, Nascimento MSJ, Cidade HM, Pinto MMM, Kijjoa A, Anantachoke C, Silva AMS, Herz W. Planta Med. 1999;65:368. doi: 10.1055/s-2006-960790. [DOI] [PubMed] [Google Scholar]
- 10(a).Lu ZX, Hasmeda M, Mahabusarakam W, Ternai B, Ternai PC, Polya GM. Chem. Biol. Interact. 1998;114:121. doi: 10.1016/s0009-2797(98)00049-0. [DOI] [PubMed] [Google Scholar]; (b) Jinsart W, Ternai B, Buddhasukh D, Polya GM. Phytochemistry. 1992;31:3711. doi: 10.1016/s0031-9422(00)97514-9. [DOI] [PubMed] [Google Scholar]
- 11.Ho CK, Huang YL, Chen CC. Planta Med. 2002;68:975. doi: 10.1055/s-2002-35668. [DOI] [PubMed] [Google Scholar]
- 12.Nakatani K, Nakahata N, Arakawa T, Yasuda H, Ohizumi Y. Biochem. Pharmacol. 2002;63:73. doi: 10.1016/s0006-2952(01)00810-3. [DOI] [PubMed] [Google Scholar]
- 13.Castanheiro RAP, Pinto MMM, Silva AMS, Cravo SMM, Gales L, Damas AM, Nazareth N, Nascimento MSJ, Eaton G. Bioorg. Med. Chem. 2007;15:6080. doi: 10.1016/j.bmc.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 14.Finnegan RA, Merkel KE, Patel JK. J. Pharm. Sci. 1973;62:483. doi: 10.1002/jps.2600620329. [DOI] [PubMed] [Google Scholar]
- 15(a).Grover PK, Shah GD, Shah RC. J. Chem. Soc. 1955:3982. [Google Scholar]; (b) Quillinan AJ, Scheinmann F. J. Chem. Soc., Perkin Trans. 1. 1973:1329. [Google Scholar]
- 16(a).Zhou T, Shi Q, Chen C-H, Huang L, Ho P, Morris-Natschke SL, Lee K-H. Eur. J. Med. Chem. 2012;47:86. doi: 10.1016/j.ejmech.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moreau S, Varache-Lembège M, Larrouture S, Fall D, Neveu A, Deffieux G, Deffieux G, Vercauteren J, Nuhrich A. Eur. J. Med. Chem. 2002;37:237. doi: 10.1016/s0223-5234(01)01332-0. [DOI] [PubMed] [Google Scholar]
- 17.Mondal M, Puranik VG, Argade NP. J. Org. Chem. 2006;71:4992. doi: 10.1021/jo0606655. [DOI] [PubMed] [Google Scholar]
- 18.Sittisombut C, Boutefnouchet S, Van-Dufat H. Trinh, Tian W, Michel S, Koch M, Tillequin F, Pfeiffer B, Pierre A. Chem. Pharm. Bull. 2006;54:1113. doi: 10.1248/cpb.54.1113. [DOI] [PubMed] [Google Scholar]
- 19.Yen C-T, Nakagawa-Goto K, Hwang T-L, Wu PC, Morris-Natschke SL, Lai W-C, Bastow KF, Chang F-R, Wu Y-C, Lee K-H. Bioorg. Med. Chem. Lett. 2010;20:1037. doi: 10.1016/j.bmcl.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castanheiro RAP, Silva AMS, Campos NAN, Nascimento MSJ, Pinto MMM. Pharmaceuticals. 2009;2:33. doi: 10.3390/ph2020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21(a).Hwang T-L, Leu Y-L, Kao S-H, Tang M-C, Chang H-L. Free Radic. Biol. Med. 2006;41:1433. doi: 10.1016/j.freeradbiomed.2006.08.001. [DOI] [PubMed] [Google Scholar]; (b) Babior BM, Kipnes RS, Curnutte JT. J. Clin. Invest. 1973;52:741. doi: 10.1172/JCI107236. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sklar LA, McNeil VM, Jesaitis AJ, Painter RG, Cochrane CG. J. Biol. Chem. 1982;257:5471. [PubMed] [Google Scholar]
- 22.General: Unless stated otherwise, the chemicals were acquired from commercial sources and used without further purification. All chemicals were purchased from ACROS and Aldrich. Melting points were measured with a Fisher-John melting apparatus without correction. 1H NMR spectra were measured on 300 MHz Varian Gemini 2000 spectrometer. The solvent was CD3OD or CDCl3 or DMSO. Mass spectra were measured on PECIEX API 3000 with turbo ion spray source, Agilent-1100 LC/MSD-Trap, or Shimadzu LCMS-IT-TOF with ESI interface. Thin-layer chromatography (TLC) and preparative TLC were performed on precoated silica GF plates purchased from Merck, Inc. Biotage Flash+ or Isco Companion systems were used for flash chromatography. Silica gel (200-400 mesh) from Aldrich, Inc. was used for column chromatography.1,3,6-Trihydroxy-9H-xanthen-9-one (4). Eaton’s reagent (P2O5-CH3SO3H) (10 mL) was added slowly to a mixture of 2,4-dihydroxybenzoic acid (1; 155 mg, 1 mmol) and phloroglucinol (3; 126 mg, 1 mmol). The resulting mixture was stirred for 1 h at 80 ° C, cooled to rt, and poured onto ice. After vigorous stirring at ambient temperature for 2 h, thin slurry formed. The solid was collected by filtration, washed with water to adjust the pH to approximately 6, and dried under vacuum at 50 ° C. The residue was chromatographed on silica gel and eluted successively with hexane-EtOAc (2:3) to give the desired product (225 mg, 92%) as a yellow solid. mp: 158-160 ° C; 1H NMR (CD3OD, 300 MHz): δ 6.13 (1H, d, J = 2.1 Hz), 6.26 (1H, d, J = 2.4 Hz), 6.72 (1H, d, J = 2.4 Hz), 6.81 (1H, dd, J = 2.3, 8.9 Hz), 7.97 (1H, d, J = 9.0 Hz).1,3-Dihydroxy-6-methoxy-9H-xanthen-9-one (5). Under similar conditions to those described for 4, phloroglucinol (3; 126 mg, 1 mmol) and 2,4-dimethoxybenzoic acid (2; 182 mg, 1 mmol) afforded the desired product (245 mg, 95%) as a yellow solid, mp: 135–137 ° C; 1H NMR (CD3OD, 300 MHz): δ 3.89 (3H, s), 6.21 (1H, d, J = 2.1 Hz), 6.28 (1H, d, J = 2.4 Hz), 6.70 (1H, d, J = 2.4 Hz), 6.83 (1H, dd, J = 2.3, 8.9 Hz), 7.89 (1H, d, J = 9.0 Hz).1-Hydroxy-3,6-dimethoxy-9H-xanthen-9-one (6). Iodomethane (100 μ L, 1.59 mmol) was added to a solution of 4 (130 mg, 0.53 mmol) and K2CO3 (183 mg, 1.33 mmol) in acetone. The resulting solution was stirred at 60 ° C under reflux for 6 h. The cooled solution was filtered and concentrated in vacuo. Purification on a flash column (n-hexane/EtOAc, 85/15) yielded the desired compound (231 mg, 85%) as a yellow powder. mp: 118–120 ° C;1H NMR (CDCl3, 300 MHz): δ 3.87 and 3.92 (2 × 3H, each s), 6.31, 6.36, and 6.79 (3 × 1H, each s), 6.91 (1H, d, J = 8.7 Hz), 8.11 (1H, d, J = 8.7 Hz), 12.99 (1H, s).Prenylation of 1,3,6-trihydroxy-xanthen-9-one (4). Method 1: Prenyl bromide (243 μ L, 2 mmol) was added dropwise over 10 min to a solution of 4 (244 mg, 1 mmol) and KOH (112 mg, 2 mmol) in H2O at 0 ° C under N2 atmosphere. The resulting mixture was stirred at 0 ° C for 24 h. During this time, a yellow solid precipitated from the mixture. The reaction mixture was quenched with pH 1 (HCl) solution and extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. Purification on a flash column yielded 7, 8, and 9 (n-hexane/EtOAc, 95/5) successively.1,3,6-Trihydroxy-2,4-bis(3-methylbut-2-enyl)-9H-xanthen-9-one (7). Yield: 5.8%; yellow oil; 1H NMR (CD3OD, 300 MHz): δ 1.67 and 1.89 (2 × 6H, each s), 3.34 (2H, s), 3.43 (2H, d, J = 7.2 Hz), 5.15–5.19 (2 × 1H, m), 6.69 (1H, d, J = 2.1 Hz), 6.78 (1H, dd, J = 2.4, 8.7 Hz), 7.93 (1H, d, J = 8.7 Hz).1,3,6-Trihydroxy-2-(3-methylbut-2-enyl)-9H-xanthen-9-one (8). Yield: 4.2% yield; yellow solid; 1H NMR (CD3OD, 300 MHz): δ 1.65 and 1.84 (2 × 3H, each s), 3.35 (2H, d, J = 6.9 Hz), 5.18–5.19 (1H, m), 6.14 (1H, s), 6.68 (1H, d, J = 2.1 Hz), 6.76 (1H, dd, J = 2.1, 8.9 Hz), 7.89 (1H, d, J = 9.0 Hz).1,3,6-Trihydroxy-2,5-bis(3-methylbut-2-enyl)-9H-xanthen-9-one (9). Yield: 16%; yellow solid; 1H NMR (CD3OD, 300 MHz): δ 1.67 (6H, s), 1.78 (2 × 3H, d, J = 4.2 Hz), 3.45 (2H, d, J = 6.3 Hz), 3.55 (2H, d, J = 6.3 Hz), 5.21–5.24 (2H, m), 6.19 (1H, s), 6.83 (1H, d, J = 8.7 Hz), 7.84 (1H, d, J = 9.0 Hz).Method 2: To a solution of 4 (244 mg, 1 mmol), KI (332 mg, 2 mmol), and KOH (112 mg, 2 mmol) in DMF at 0 ° C under N2 atmosphere, prenyl bromide (243 μ L, 2 mmol) was added dropwise over 10 min. The resulting mixture was then treated under conditions similar to Method 1. Purification on a flash column yielded a mixture of 10 + 11 + 12 (n-hexane/EtOAc, 95/5) and pure 13 (n-hexane/EtOAc, 90/10). The components of the mixture were then separated by preparative TLC (n-hexane/EtOAc, 4/1).1,6-Dihydroxy-3-(3-methylbut-2-enyloxy)-9H-xanthen-9-one (10). Yield: 4.2%; yellow oil; 1H NMR (CDCl3, 300 MHz): δ 1.67 (2 × 3H, each s), 3.30 (2H, s), 3.43 (2H, d, J = 7.2 Hz), 5.15-5.19 (1H, m), 6.13 (1H, d, J = 2.1 Hz), 6.26 (1H, d, J = 2.4 Hz), 6.69 (1H, d, J = 2.1 Hz), 6.78 (1H, dd, J = 2.4, 8.7 Hz), 7.93 (1H, d, J = 8.7 Hz).1-Hydroxy-2-(3-methylbut-2-enyl)-3,6-bis(3-methylbut-2-enyloxy)-9H-xanthen-9-on e (11). Yield: 50%; yellow solid;1H NMR (CDCl3, 300 MHz): δ 1.77–1.82 (12H, m), 1.85–1.90 (6H, m), 4.55–4.61 (6H, m), 5.49–5.51 (4H, m), 6.35 (1H, d, J = 1.8 Hz), 6.40 (1H, d, J = 1.8 Hz), 6.78 (1H, d, J = 1.8 Hz), 6.89 (1H, dd, J = 2.1, 9.0 Hz), 8.08 (1H, d, J = 8.7 Hz), 13.19 (1H, s).1-Hydroxy-3,6-bis(3-methylbut-2-enyloxy)-9H-xanthen-9-one (12). Yield: 5.8%; yellow solid; 1H NMR (CDCl3, 300 MHz): δ 1.77–1.82 (12H, m), 4.55–4.61 (4H, m), 5.49–5.51 (2H, m), 6.30 (1H, d, J = 1.8 Hz), 6.35 (1H, d, J = 1.8 Hz), 6.78 (1H, d, J = 1.8 Hz), 6.89 (1H, dd, J = 2.1, 9.0 Hz), 8.08 (1H, d, J = 8.7 Hz), 13.27 (1H, s).1,3,6-Tris(3-methylbut-2-enyloxy)-9H-xanthen-9-one (13). Yield: 10%; yellow solid; 1H NMR (CD3OD, 300 MHz): δ 1.78 (18H, s), 4.57 (4H, t, J = 6.5 Hz), 4.64 (2H, d, J = 6.3 Hz), 5.44–5.52 (3H, m), 6.28 (1H, d, J = 1.8 Hz), 6.38 (1H, d, J = 2.1 Hz), 6.73 (1H, d, J = 2.1 Hz), 6.82 (1H, dd, J = 2.1, 8.7 Hz), 7.97 (1H, d, J = 8.7 Hz).5,9-Dihydroxy-2,2-dimethylpyrano[3,2-b]xanthen-6(2H)-one (14). Prenal (480 μ L, 5 mmol) was added to a stirring mixture of 4 (244 mg, 1 mmol) and Ca(OH)2 (150 mg, 2 mmol) in MeOH at rt. After continued stirring for 36 h at rt, MeOH was removed under vacuum, and the reaction mixture was diluted with EtOAc. The organic layer was washed with 2 N HCl, water, and brine, and dried over MgSO4. Purification on a flash column (n-hexane/EtOAc, 90/10) afforded the desired compound (14, 85%) as a yellow solid. mp: 228–230 ° C;1H NMR (CDCl3 + CD3OD, 300 MHz): δ 1.43 (6H, s), 5.63 and 6.27 (2 × 1H, each s), 6.65 (1H, d, J = 10.2 Hz), 6.74 (1H, d, J = 1.8 Hz), 6.81 (1H, dd, J = 2.3, 8.9 Hz), 7.98 (1H, d, J = 8.7 Hz).5,9-Dihydroxy-2-methyl-2-(4-methylpent-3-enyl)pyrano[3,2-b]xanthen-6(2H)-one (15). Under conditions similar to those described for the preparation of 14, compound 4 (244 mg, 1 mmol) and citral (860 μ L, 5 mmol) afforded 15 (70%) as a yellow solid. 1H NMR (CDCl3, 300 MHz): δ 1.35, 1.56, and 1.64 (3 × 3H, each s), 1.76–1.83 (2H, m), 2.06–2.10 (2H, m), 5.07–5.10 (1H, m), 5.52 (1H, d, J = 10.2 Hz), 6.27 (1H, s), 6.72–6.85 (3H, m), 8.05 (1H, d, J = 9.0 Hz), 13.21 (1H, s).6,10-Dihydroxy-3,3-dimethylpyrano[2,3-c]xanthen-7(3H)-one (16). A stirring mixture of 4 (244 mg, 1 mmol) and prenal (960 μ L, 10 mmol) was heated at 140–150 ° C for 6 h. After cooling, the residue was purified on a flash column (n-hexane/EtOAc, 90/10) to afford 16 (295 mg, 95%) as a yellow powder. 1H NMR (CDCl3 + CD3OD, 300 MHz): δ 1.48 (6H, s), 5.66 (1H, d, J = 9.6 Hz), 6.16 (1H, s), 6.79–6.87 (3H, m), 8.02 (1H, d, J = 8.7 Hz).6,10-Dihydroxy-3-methyl-3-(4-methylpent-3-enyl)pyrano[2,3-c]xanthen-7(3H)-one (17). Under conditions similar to those described for the preparation of 16, compound 4 (244 mg, 1 mmol) and citral (1.7 mL, 10 mmol) afforded 17 (351.9 mg, 93%) as an amber-brown oil. 1H NMR (CDCl3, 300 MHz): δ 1.43, 1.56 and 1.64 (3 × 3H, each s), 1.78–1.83 (2H, m), 2.05–2.13 (2H, m), 5.05–5.10 (1H, m), 5.53 (1H, d, J = 10.2 Hz), 6.23 (1H, s), 6.80–6.86 (3H, m), 8.09 (1H, d, J = 9.0 Hz), 13.07 (1H, s).3,4,5,9-Tetrahydroxy-2,2-dimethyl-3,4-dihydropyrano[3,2-b]xanthen-6(2H)-one (18). Compound 14 (84 mg, 0.22 mmol) was added to a solution of osmium tetroxide (2.5% in 2-methyl-2-propanol, 210 μ L) and 4-methylmorpholine N-oxide monohydrate (30 mg, 0.22 mmol) in t-BuOH-THF-H2O (10:3:1, 14 mL). The reaction mixture was stirred at rt for 48 h. After addition of saturated aqueous sodium bisulfate solution (30 mL), the mixture was stirred for 1 h and then extracted with CH2Cl2. The combined organic layers were dried with MgSO4, filtered, and evaporated in vacuo. Purification on a flash column (CH2Cl2/MeOH, 98/2) gave the desired product (172.2 mg, 50%) as a yellow oil. 1H NMR (DMSO, 300 MHz): δ 1.41 (6H, s), 3.67 (1H, d, J = 8.5 Hz), 5.02 (1H, d, J = 8.3 Hz), 6.68 (1H, d, J = 10.2 Hz), 6.77 (1H, d, J = 1.8 Hz), 6.80 (1H, dd, J = 2.3, 8.9 Hz), 7.94 (1H, d, J = 8.7 Hz).3,4,5,9-Tetrahydroxy-2-methyl-2-(4-methylpent-3-enyl)-3,4-dihydropyrano[3,2-b]xa nthen-6(2H)-one (19). Using similar conditions to those described for the preparation of 18, compound 15 (150 mg, 0.4 mmol) afforded 19 (186 mg, 45%) as a yellow solid. 1H NMR (DMSO, 300 MHz): δ 1.35, 1.56 and 1.64 (3 × 3H, each s), 1.76-1.83 (2H, m), 2.06–2.10 (2H, m), 5.07–5.10 (1H, m), 3.37 (1H, d, J = 8.5 Hz), 5.05 (1H, d, J = 8.7 Hz), 5.52 (1H, d, J = 10.2 Hz), 6.27 (1H, s), 6.72-6.85 (3H, m), 8.05 (1H, d, J = 9.0 Hz), 13.17 (1H, s).1,2,6,10-Tetrahydroxy-3,3-dimethyl-2,3-dihydropyrano[2,3-c]xanthen-7(1H)-one (20). Under similar conditions to those described for the preparation of 18, compound 16 (58.6 mg, 0.19 mmol) afforded 20 (172 mg, 50%) as an off-white solid. 1H NMR (DMSO, 300 MHz): δ 1.44 (6H, s), 3.26 (1H, d, J = 8.7 Hz), 5.03 (1H, d, J = 8.4 Hz), 6.14 (1H, s), 6.79–6.87 (3H, m), 8.03 (1H, d, J = 8.7 Hz).1,2,6,10-Tetrahydroxy-3-methyl-3-(4-methylpent-3-enyl)-2,3-dihydropyrano[2,3-c]xa nthen-7(1H)-one (21). Under similar conditions to those described for the preparation of 18, compound 17 (84 mg, 0.22 mmol) afforded 21 (194 mg, 47%) as yellow oil.1H NMR (DMSO, 300 MHz): δ 1.45, 1.60 and 1.64 (3 × 3H, each s), 1.78-1.83 (2H, m), 2.05–2.13 (2H, m), 3.78 (1H, d, J = 8.7 Hz), 5.04 (1H, d, J = 8.7 Hz), 5.53 (1H, d, J = 10.2 Hz), 6.23 (1H, s), 6.80–6.86 (3H, m), 8.09 (1H, d, J = 9.0 Hz), 13.10 (1H, s).