

3-Ketosteroid Δ1-dehydrogenase from Rhodococcus erythropolis SQ1 was successfully crystallized and its initial structure was solved.
Keywords: 3-ketosteroid Δ1-dehydrogenase, Rhodococcus erythropolis SQ1
Abstract
3-Ketosteroid Δ1-dehydrogenase plays a crucial role in the early steps of steroid degradation by introducing a double bond between the C1 and C2 atoms of the A-ring of its 3-ketosteroid substrates. The 3-ketosteroid Δ1-dehydrogenase from Rhodococcus erythropolis SQ1, a 56 kDa flavoprotein, was crystallized using the sitting-drop vapour-diffusion method at room temperature. The crystals grew in various buffers over a wide pH range (from pH 5.5 to 10.5), but the best crystallization condition consisted of 2%(v/v) PEG 400, 0.1 M HEPES pH 7.5, 2.0 M ammonium sulfate. A native crystal diffracted X-rays to 2.0 Å resolution. It belonged to the primitive orthorhombic space group P212121, with unit-cell parameters a = 107.4, b = 131.6, c = 363.2 Å, and contained eight molecules in the asymmetric unit. The initial structure of the enzyme was solved using multi-wavelength anomalous dispersion (MAD) data collected from a Pt-derivatized crystal.
1. Introduction
The microbial biotransformation of steroids has attracted substantial interest in the pharmaceutical industry since the 1950s (Fernandes et al., 2003 ▶; Mahato & Garai, 1997 ▶). Through their biotransformation, a large variety of physiologically active steroid intermediates are produced (Horinouchi et al., 2003 ▶; Sedlaczek, 1988 ▶). These intermediates and their derivatives are utilized extensively as drugs and hormones because of their anti-inflammatory, diuretic, anabolic, contraceptive, anti-androgenic, progestational and anticancer properties (Donova, 2007 ▶; Mahato & Garai, 1997 ▶). The microbial steroid catabolic pathway has received even more attention since the discovery that this pathway is closely related to the pathogenicity of several pathogenic bacteria, e.g. Mycobacterium tuberculosis (van der Geize et al., 2007 ▶) and Rhodococcus equi (van der Geize et al., 2011 ▶). In particular, the degradation of cholesterol was shown to be crucial for M. tuberculosis to persist in the severe environment of the host macrophages (van der Geize et al., 2007 ▶). M. tuberculosis is able to use cholesterol as a sole carbon and energy source, converting the C atoms of the steroid nucleus to energy while the aliphatic side-chain atoms are used as a carbon source (Pandey & Sassetti, 2008 ▶). For this purpose, M. tuberculosis H37Rv contains a large gene cluster coding for enzymes catalyzing cholesterol degradation, including a 3-ketosteroid Δ1-dehydrogenase (Rv3537; van der Geize et al., 2007 ▶).
3-Ketosteroid Δ1-dehydrogenase [4-ene-3-oxosteroid:(acceptor)-1-ene-oxoreductase; EC 1.3.99.4] catalyzes the insertion of a double bond between the C1 and C2 atoms of the chemically stable 3-ketosteroid A-ring (Fig. 1 ▶). Enzymes with this activity have been discovered in several steroid-degrading bacteria, including bacteria from the genera Arthrobacter, Comamonas, Mycobacterium and Rhodococcus (formerly Nocardia; Donova, 2007 ▶; Horinouchi et al., 2003 ▶). Together with the activity of a 3-ketosteroid 9α-hydroxylase, insertion of this double bond facilitates the opening of the steroid B-ring as a first step in degradation of the steroid nucleus (Horinouchi et al., 2003 ▶). The activity of the dehydrogenase is dependent on FAD (flavin adenine dinucleotide) and requires the presence of a carbonyl group at the C3 position of the steroid substrate (Itagaki, Matushita et al., 1990 ▶; Itagaki, Wakabayashi et al., 1990 ▶). The enzyme acts on a variety of 3-ketosteroid substrates, with a preference for substrates possessing a double bond at the C4–C5 position (Itagaki, Wakabayashi et al., 1990 ▶; Knol et al., 2008 ▶) such as, for example, the main catabolic steroid intermediate 4-androstene-3,17-dione (Fig. 1 ▶).
Figure 1.

An example of the reaction catalyzed by 3-ketosteroid Δ1-dehydrogenase.
The catalytic mechanism of 3-ketosteroid Δ1-dehydrogenase has been studied for a long time. Levy & Talalay (1959 ▶) suggested that the dehydrogenation proceeds directly, excluding the possibility of the formation of a hydroxylated intermediate, and proposed that the enzyme uses a flavin prosthetic group as coenzyme, which was later confirmed to be FAD (Itagaki, Wakabayashi et al., 1990 ▶). Ringold et al. (1963 ▶) showed by isotopic exchange experiments that the dehydrogenase prefers a trans-diaxial elimination of the 1α,2β H atoms from a 3-ketosteroid substrate rather than cis-elimination, and proposed a two-step mechanism starting with the enolization of the C3 carbonyl keto function in concert with a proton departing from C2 and followed by abstraction of a hydride ion from C1 by the flavin cofactor. Itagaki, Matushita et al. (1990 ▶) supported the trans-diaxial elimination idea, but put forward a slightly different catalytic mechanism by proposing the formation of a carbanion intermediate instead of the enolization of the steroid substrate. By chemical modification, mutagenesis and kinetics experiments, 3-ketosteroid Δ1-dehydrogenase from R. rhodochrous (formerly N. corallina) was shown to have one or two histidine and arginine residues that are essential for catalytic activity, Tyr121 was shown to play an important role in catalysis and both Tyr104 and Tyr116 were found to be important for binding of the steroid substrates (Matsushita & Itagaki, 1992 ▶; Fujii et al., 1999 ▶). Moreover, mutagenesis studies on 3-ketosteroid Δ1-dehydrogenase isoenzyme 2 from R. erythropolis SQ1 (Δ1-KSTD2) suggested Ser325 and Thr503 as crucial residues for catalysis (van der Geize et al., 2002 ▶).
R. erythropolis SQ1 has three 3-ketosteroid Δ1-dehydrogenase isoenzymes: Δ1-KSTD1 (van der Geize et al., 2001 ▶), Δ1-KSTD2 (van der Geize et al., 2002 ▶) and Δ1-KSTD3 (Knol et al., 2008 ▶). Δ1-KSTD1 has been expressed in Escherichia coli strain BL21 (DE3) at higher levels than was possible with the other two isoenzymes and, in contrast to the latter isoenzymes, could be purified relatively easily (Knol et al., 2008 ▶). Amino-acid sequence alignment of Δ1-KSTD1 with the sequences of structurally characterized enzymes showed that Δ1-KSTD1 has the highest homology to 3-ketosteroid Δ4-(5α)-dehydrogenase from R. jostii (van Oosterwijk et al., unpublished work) followed by flavocytochrome c fumarate reductase from Shewanella putrefaciens (PDB entry 1d4c; Leys et al., 1999 ▶), with sequence identities of 28 and 24%, respectively. In an effort to identify the nature and positions of the amino-acid residues involved in catalysis and to clarify the catalytic mechanism of 3-ketosteroid Δ1-dehydrogenase, we describe here the successful purification, crystallization and preliminary X-ray crystallographic analysis of Δ1-KSTD1. The three-dimensional structure of this enzyme will allow manipulation of its catalytic properties and will facilitate the design of inhibitors that could possibly be developed into efficacious drugs to combat pathogenic steroid-degrading bacteria.
2. Experimental
2.1. Expression and purification
Total DNA from R. erythropolis SQ1 has been isolated (van der Geize et al., 2000 ▶) and characterized to contain three genes, kstD1 (van der Geize et al., 2001 ▶), kstD2 (van der Geize et al., 2002 ▶) and kstD3 (Knol et al., 2008 ▶), that code for three different 3-ketosteroid Δ1-dehydrogenases. The kstD1 gene (1533 bp; GenBank accession No. AF096929) has been cloned into the NdeI/BamHI restriction sites of pET15b (Novagen) as pET15b-kstD1 plasmid and a protocol for the heterologous expression of the Δ1-KSTD1 protein in E. coli strain BL21 (DE3) has been established (Knol et al., 2008 ▶).
An overnight preculture of the recombinant E. coli was prepared from a glycerol stock in LB (Luria–Bertani) medium supplemented with 25 µg ml−1 carbenicillin (Duchefa Biochemie) by shaking at 200 rev min−1 at 310 K. This preculture was used for a 1% inoculation of 1 l fresh LB medium containing 500 mM sorbitol, 2.5 mM betaine, 25 µg ml−1 carbenicillin and 100 µM IPTG (isopropyl β-d-1-thiogalactopyranoside; Promega). The E. coli cells were grown by shaking at 200 rev min−1 at 290 K and harvested after 48 h by centrifugation at 6000g for 15 min.
The cell pellet was resuspended in 30 ml buffer A [50 mM Tris–HCl pH 8.5, 100 mM NaCl, 10%(v/v) glycerol, 5 mM β-mercaptoethanol, 10 mM imidazole]. After supplementation with FAD (25 µmol; Sigma), complete EDTA-free protease-inhibitor cocktail (one tablet; Roche Diagnostics) and DNAse I (catalytic amount; Roche Diagnostics), the cell suspension was lysed by three passages through a French press (Fisher Scientific) at 55 MPa and centrifuged at 35 000g for 15 min to remove cell debris. For immobilized Ni2+-affinity chromatography, cleared supernatant was applied onto a 5 ml HisTrap HP (GE Healthcare) column pre-equilibrated with buffer A and washed with three column volumes of the same buffer. Elution was carried out using a linear imidazole gradient (10–300 mM) in buffer A. The yellow-coloured Δ1-KSTD1 fractions were pooled, diluted five times with buffer B {25 mM bicine [N,N-bis(2-hydroxyethyl)glycine] pH 8.5, 10%(v/v) glycerol, 5 mM β-mercaptoethanol} and loaded onto a 6 ml Resource Q (GE Healthcare) anion-exchange column pre-equilibrated with buffer B. After washing the column with 20 and 150 mM NaCl in buffer B (three column volumes each), Δ1-KSTD1 was eluted with 250 mM NaCl in the same buffer. Fractions containing Δ1-KSTD1 were combined, concentrated to about 100 mg ml−1 using an Amicon Ultra-4 30K (Millipore) filter and applied onto a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with buffer C [25 mM bicine pH 9.0, 100 mM NaCl, 10%(v/v) glycerol] for size-exclusion chromatography. Δ1-KSTD1 was eluted from the column with the same buffer at a flow rate of 0.5 ml min−1. The purified Δ1-KSTD1 was finally concentrated to 50 mg ml−1 using an Amicon Ultra-4 30K filter and stored at 253 K until use.
All chromatographic experiments were performed using an ÄKTAexplorer (GE Healthcare). Protein concentrations were determined using the Bradford protein assay kit (Bio-Rad) with BSA (bovine serum albumin) as a standard and protein purity was monitored by Coomassie Blue-stained SDS–PAGE.
2.2. ThermoFAD stability assay
To find a buffer system in which Δ1-KSTD1 is stable, a ThermoFAD (Thermofluor-adapted flavin ad hoc detection system) assay was carried out according to Forneris et al. (2009 ▶) using a buffer screen of MMT buffer {1:2:2 dl-malic acid:MES [2-(N-morpholino)ethanesulfonic acid]:Tris} ranging from pH 4.0 to 9.0. Samples of 25 µl had a typical composition (final concentration) of 100 mM MMT, 100 mM NaCl, 10%(v/v) glycerol and 2.5 mg ml−1 protein. The samples were analyzed in a 96-well thin-wall PCR plate (Bio-Rad) sealed with optical quality sealing tape (Bio-Rad). The sealed plate was inserted into a real-time PCR machine (iCycler, Bio-Rad) and was heated from 293 to 363 K with a 0.5 K increment per 20 s. The changes in the fluorescence of FAD were recorded every 0.5 K after a 10 s hold using a fluorescence detector (MyIQ single-colour RT-PCR detection system, Bio-Rad) with an excitation-wavelength range between 470 and 500 nm and a SYBR Green fluorescence emission filter (523–543 nm).
2.3. Crystallization
Prior to crystallization experiments, the protein sample was thawed on ice and its concentration was adjusted to 15 mg ml−1 with buffer C. Crystallization conditions were screened using the JCSG-plus Screen (Molecular Dimensions Ltd), Structure Screens I and II (Molecular Dimensions Ltd) and Wizard Screens I and II (Emerald BioSystems). Crystallization experiments were performed by the sitting-drop vapour-diffusion method using MRC 2-Well Crystallization Plates (Swissci). These experiments were performed with a Mosquito (TTP LabTech) crystallization robot by mixing 0.15 µl screen solution with 0.15 µl protein solution. After equilibration against 50 µl screen solution for 5–7 d, bright yellow rectangular crystals were observed in several conditions. For X-ray data collection, Δ1-KSTD1 crystals were routinely reproduced using condition No. 30 of Structure Screen I [2%(v/v) PEG (polyethylene glycol) 400, 0.1 M HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) buffer pH 7.5, 2.0 M ammonium sulfate] as the crystallization solution. All crystallization experiments were carried out at 293 K.
2.4. Data collection and processing
For X-ray diffraction experiments, the crystals were cryoprotected by transferring them for 5 s into crystallization solution containing 40%(w/v) sucrose, followed by a 1 s transfer into a 1:1 mixture of paraffin oil and Paratone-N, before flash-cooling them in liquid nitrogen. A Pt derivative was prepared by washing a crystal with 2%(v/v) PEG 400, 0.1 M HEPES pH 7.5, 0.4 M NaH2PO4/1.6 M K2HPO4, followed by soaking the crystal overnight in the same solution but containing 10 mM Na2PtCl4. The Pt-derivatized crystal was cryoprotected in a similar way to the native crystals.
X-ray diffraction data sets were collected at 100 K on beamline ID14-1 (European Synchrotron Radiation Facility, Grenoble) using an ADSC Quantum Q210 detector (native crystal) or on beamline PXI (Swiss Light Source, Villigen) using a PILATUS detector (Pt-derivatized crystal). The native crystal data set was recorded at a wavelength of 0.93340 Å for 450 frames with an oscillation range per frame of 0.2°. Based on an XAFS (X-ray absorption fine-structure) measurement, MAD data collection was carried out from a single Pt-derivatized crystal at three wavelengths corresponding to peak (1.07240 Å), inflection point (1.07270 Å) and remote (1.06320 Å). At each wavelength, a 720-frame data set was collected to a maximum resolution of 3.1 Å with an oscillation range per frame of 0.5°.
All data sets were processed and integrated using the program XDS (Kabsch, 2010 ▶) in combination with the program SCALA (Evans, 2006 ▶) from the CCP4 package (Winn et al., 2011 ▶). Table 1 ▶ presents pertinent crystallographic details on data collection and processing. Initial phases were calculated by submitting the MAD data to autoSHARP (Vonrhein et al., 2007 ▶). Because of non-isomorphism between the native and Pt-derivatized crystals, phases for the native diffraction data were obtained with the program Phaser (McCoy et al., 2007 ▶) by placing the structure of 3-ketosteroid Δ4-(5α)-dehydrogenase from R. jostii (van Oosterwijk et al., unpublished work) in the electron-density map obtained from autoSHARP. The resulting phases were used for automatic building using the program ARP/wARP (Langer et al., 2008 ▶).
Table 1. Summary of crystallographic data collection and processing.
Values in parentheses are for the highest resolution shell.
| Pt derivative | ||||
|---|---|---|---|---|
| Data set | Native | Peak | Inflection | Remote |
| Beamline | ID14-1, ESRF | PXI, SLS | ||
| Detector | ADSC Q210 | PILATUS | ||
| Wavelength (Å) | 0.93340 | 1.07240 | 1.07270 | 1.06320 |
| Resolution (Å) | 2.00 (2.11–2.00) | 3.30 (3.48–3.30) | 3.50 (3.69–3.50) | 3.70 (3.90–3.70) |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters | ||||
| a (Å) | 107.4 | 109.5 | 110.1 | 110.4 |
| b (Å) | 131.6 | 129.8 | 130.3 | 130.6 |
| c (Å) | 363.2 | 361.5 | 362.6 | 363.3 |
| α = β = γ (°) | 90 | 90 | 90 | 90 |
| Molecules per asymmetric unit | 8 | 8 | 8 | 8 |
| Matthews coefficient (Å3 Da−1) | 2.9 | 2.9 | 2.9 | 2.9 |
| Solvent content (%) | 57 | 57 | 58 | 58 |
| Rmerge† | 0.084 (0.752) | 0.142 (0.783) | 0.156 (0.902) | 0.184 (0.957) |
| Rp.i.m.‡ | 0.050 (0.450) | 0.040 (0.216) | 0.044 (0.260) | 0.052 (0.267) |
| Total observations | 1296759 (183757) | 1057045 (156992) | 883593 (118606) | 762547 (111309) |
| Unique reflections | 345382 (49404) | 78506 (11309) | 66511 (9323) | 57113 (8220) |
| Mean I/σ(I) | 10.9 (1.8) | 15.1 (3.8) | 15.0 (3.3) | 13.1 (3.4) |
| Completeness (%) | 99.8 (98.6) | 100.0 (100.0) | 99.5 (96.9) | 100.0 (100.0) |
| Multiplicity | 3.8 (3.7) | 13.5 (13.9) | 13.3 (12.7) | 13.4 (13.5) |
R
merge = 
 .
.
R
p.i.m. = 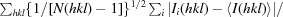 , where I
i(hkl) is the integrated intensity of a reflection, 〈I(hkl)〉 is the mean intensity of multiple corresponding symmetry-related reflections and N is the multiplicity of the given reflections.
, where I
i(hkl) is the integrated intensity of a reflection, 〈I(hkl)〉 is the mean intensity of multiple corresponding symmetry-related reflections and N is the multiplicity of the given reflections.
3. Results and discussion
Δ1-KSTD1 is a flavoprotein that contains 510 amino-acid residues. However, the recombinant protein expressed in E. coli from the pET15b-kstD1 plasmid also contains a 20-amino-acid leader sequence (MGSSHHHHHHSSGlvprgsH) which includes a 6×His tag (bold) and a thrombin cleavage site (lower case). Thus, the expressed protein contains 530 amino-acid residues with a calculated molecular mass of 55 995 Da (including one FAD molecule) and a theoretical pI (isoelectric point) of 4.73 (as calculated using http://web.expasy.org/protparam/).
Because of its relatively low pI, Δ1-KSTD1 was initially purified in a sodium phosphate buffer system at pH 7.2 and was stored prior to crystallization in 25 mM sodium phosphate buffer pH 7.2, 100 mM NaCl, 10%(v/v) glycerol. Fresh protein obtained in this way could be crystallized using Structure Screen II condition No. 29 (0.2 M potassium/sodium tartrate, 0.1 M citrate pH 5.6, 2 M ammonium sulfate). However, despite intensive efforts to optimize the crystallization conditions and procedure (e.g. by varying the concentrations of the various crystallization solution components, the pH and the crystallization method, as well as by applying various seeding techniques), the crystals grew slowly (in about three months) and the reproducibility was very low. Therefore, we considered that storage could negatively affect the quality and crystallizability of Δ1-KSTD1. Because Δ1-KSTD1 is a flavoprotein, a ThermoFAD (Forneris et al., 2009 ▶) assay was carried out to find a buffer system in which the protein was more stable. By thermally denaturing a protein and exposing (or dissociating) its buried FAD to the solvent, the assay reports on the thermal stability of a flavoprotein by way of its apparent melting temperature (T m). Although there is no quantitative correlation between protein stability and crystallizability, for a particular protein a buffer system in which the protein is more stable gives a higher probability of the protein crystallizing (Ericsson et al., 2006 ▶). Below pH 6.0 no apparent T m is observed, indicating that Δ1-KSTD1 is destabilized at low pH. In the pH 6.0–9.0 range we observed that the higher the pH value, the higher the apparent T m of Δ1-KSTD1 (Fig. 2 ▶). At pH 9.0 the apparent T m of Δ1-KSTD1 is about 7 K higher than at pH 7.0. Based on this result, Δ1-KSTD1 was purified at pH 8.5 and stored for crystallization at pH 9.0 (buffer C; see §2.1).
Figure 2.

Thermostability analysis of Δ1-KSTD1 in MMT buffer at various pH values using the ThermoFAD method (Forneris et al., 2009 ▶). The melting temperature (T m), which is defined as the midpoint temperature of the protein folding–unfolding transition (Ericsson et al., 2006 ▶), is determined as the temperature at which the first derivative d(fluorescence)/dT is maximal.
Δ1-KSTD1 in buffer C could indeed be crystallized more quickly and the crystallization could be reproduced more easily than with protein stored in the initial storage buffer. Crystallization trials using protein purified and stored in this new buffer produced Δ1-KSTD1 crystals in 5–7 d in several crystallization conditions (Fig. 3 ▶). Except for one condition that contained 0.4 M NaH2PO4/1.6 M K2HPO4, all crystallization conditions contained 1.6 or 2.0 M ammonium sulfate as precipitant. The crystals grew in various buffers in a broad pH range (from pH 5.5 to 10.5) either with or without salts/additives (e.g. NaCl, potassium/sodium tartrate, Li2SO4, PEG 400 or dioxane). However, of all of the crystallization conditions, the most reproducible for crystallizing Δ1-KSTD1 appeared to be Structure Screen I solution No. 30, which consists of 2%(v/v) PEG 400, 0.1 M HEPES pH 7.5, 2 M ammonium sulfate.
Figure 3.

Δ1-KSTD1 crystals obtained from various crystallization screens. (a) JCSG-plus condition No. 50 (0.2 M NaCl, 0.1 M sodium cacodylate pH 6.5, 2 M ammonium sulfate). (b) JCSG-plus condition No. 83 (0.1 M bis-Tris pH 5.5, 2 M ammonium sulfate). (c) Structure Screen I condition No. 30 [2%(v/v) PEG 400, 0.1 M HEPES pH 7.5, 2 M ammonium sulfate]. (d) Structure Screen I condition No. 32 (0.1 M Tris–HCl pH 8.5, 2 M ammonium sulfate). (e) Structure Screen I condition No. 44 (2 M ammonium sulfate). (f) Structure Screen II condition No. 23 [10%(v/v) dioxane, 0.1 M MES pH 6.5, 1.6 M ammonium sulfate]. (g) Structure Screen II condition No. 29 (0.2 M potassium/sodium tartrate, 0.1 M citrate pH 5.6, 2 M ammonium sulfate). (h) Wizard Screen I condition No. 20 (0.2 M NaCl, 0.1 M imidazole pH 8.0, 0.4 M NaH2PO4/1.6 M K2HPO4). (i) Wizard Screen I condition No. 33 [0.2 M Li2SO4, 0.1 M CAPS (N-cyclohexyl-3-aminopropanesulfonic acid) pH 10.5, 2 M ammonium sulfate].
Typically, Δ1-KSTD1 crystals grew in rectangular shapes with maximum dimensions of approximately 100 × 100 × 300 µm and, as they contain FAD, were coloured bright yellow. A complete data set was collected from a native crystal (Fig. 4 ▶) and processed to 2.0 Å resolution (Table 1 ▶). The data could be indexed in the primitive orthorhombic space group P212121, with unit-cell parameters a = 107.4, b = 131.6, c = 363.2 Å. With this large unit cell, the crystal contained eight copies of the 56 kDa Δ1-KSTD1 molecule (including its 20-amino-acid leader sequence and one FAD molecule) per asymmetric unit, corresponding to a Matthews coefficient (V M) of 2.9 Å3 Da−1 and a crystal solvent content of 57%.
Figure 4.

X-ray diffraction image obtained from a Δ1-KSTD1 crystal. (a) Diffraction pattern from a native crystal obtained on beamline ID14-1 at the ESRF. The resolution at the edge is 1.8 Å. (b) A close-up view of an area in the frame where the spots are close together, showing the long cell axis.
A structural homology search using the Fold & Function Assignment (FFAS) server (Jaroszewski et al., 2005 ▶) with the Δ1-KSTD1 sequence as a query resulted in the flavocytochrome c fumarate reductase from S. putrefaciens (PDB entry 1d4c; Leys et al., 1999 ▶) as the top hit, with a primary-structure identity of 24% to Δ1-KSTD1. However, several attempts to solve the Δ1-KSTD1 structure by molecular replacement using the crystal structure of this protein as a starting model were not successful, most likely because its structural similarity to Δ1-KSTD1 is too low and/or because there are too many molecules in the asymmetric unit of the Δ1-KSTD1 crystal. Moreover, molecular replacement also failed when the structure of the 3-ketosteroid Δ4-(5α)-dehydrogenase from R. jostii (van Oosterwijk et al., unpublished work), which shares 28% sequence identity with Δ1-KSTD1, was used as input. Therefore, to solve the phase problem, a MAD experiment was conducted using a Δ1-KSTD1 crystal soaked in a solution containing Na2PtCl4. Since the crystal initially grew from a solution containing ammonium sulfate, which may compete with the protein to bind the platinum ions (Drenth, 2007 ▶), the ammonium sulfate was removed from the crystal by washing and soaking the crystal in 0.4 M NaH2PO4/1.6 M K2HPO4. This latter condition was inspired by another successful crystallization condition for Δ1-KSTD1 (Fig. 3 ▶ h).
A three-wavelength MAD data set was collected from a single Pt- derivatized crystal (Table 1 ▶). The data could be processed to resolutions of 3.3, 3.5 and 3.7 Å for the peak, inflection-point and remote wavelengths, respectively, with basically the same unit-cell parameters as for the native crystal. The MAD data sets were collected sequentially at the peak, inflection-point and remote wavelengths; thus, the decreasing resolution is likely to be the result of radiation damage. All data sets were then limited to 3.7 Å resolution and used for phase calculation and density modification with autoSHARP (Vonrhein et al., 2007 ▶), which produced an electron-density map suitable for model building (Fig. 5 ▶). This map could be used for the manual placement of eight copies of a model of the F-domain of the 3-ketosteroid Δ4-(5α)-dehydrogenase structure (van Oosterwijk et al., unpublished work) and eight copies of its S-domain. After several cycles of manual model building, the resulting model was used to solve the native structure of Δ1-KSTD1 using Phaser (McCoy et al., 2007 ▶). Finally, automatic building using the program ARP/wARP (Langer et al., 2008 ▶) was performed to obtain the complete model for Δ1-KSTD1. Refinement and structure analysis are currently under way to unveil the structural basis of the substrate specificity and catalytic mechanism of Δ1-KSTD1.
Figure 5.

Electron-density map for Δ1-KSTD1 as obtained using autoSHARP (Vonrhein et al., 2007 ▶), showing a clear α-helix (arrow).
Acknowledgments
We thank Professor Lubbert Dijkhuizen, Dr Robert van der Geize and Dr Jan Knol of the Department of Microbiology, University of Groningen for providing the pET15b-kstD1 plasmid and initial Δ1-KSTD1 protein samples and for helpful discussions. We thank the beamline scientists at ID14-1 (ESRF) and PXI (SLS) for their assistance. AR was the recipient of a scholarship from the Directorate General of Higher Education (DIKTI), the Ministry of Education and Culture, Republic of Indonesia.
References
- Donova, M. V. (2007). Appl. Biochem. Microbiol. 43, 1–14.
- Drenth, J. (2007). Principles of Protein X-ray Crystallography, 3rd ed. New York: Springer.
- Ericsson, U. B., Hallberg, B. M., DeTitta, G. T., Dekker, N. & Nordlund, P. (2006). Anal. Biochem. 357, 289–298. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fernandes, P., Cruz, A., Angelova, B., Pinheiro, H. M. & Cabral, J. M. S. (2003). Enzyme Microb. Technol. 32, 688–705.
- Forneris, F., Orru, R., Bonivento, D., Chiarelli, L. R. & Mattevi, A. (2009). FEBS J. 276, 2833–2840. [DOI] [PubMed]
- Fujii, C., Morii, S., Kadode, M., Sawamoto, S., Iwami, M. & Itagaki, E. (1999). J. Biochem. 126, 662–667. [DOI] [PubMed]
- Geize, R. van der, Grommen, A. W., Hessels, G. I., Jacobs, A. A. & Dijkhuizen, L. (2011). PLoS Pathog. 7, e1002181. [DOI] [PMC free article] [PubMed]
- Geize, R. van der, Hessels, G. I. & Dijkhuizen, L. (2002). Microbiology, 148, 3285–3292. [DOI] [PubMed]
- Geize, R. van der, Hessels, G. I., van Gerwen, R., van der Meijden, P. & Dijkhuizen, L. (2001). FEMS Microbiol. Lett. 205, 197–202. [DOI] [PubMed]
- Geize, R. van der, Hessels, G. I., van Gerwen, R., Vrijbloed, J. W., van der Meijden, P. & Dijkhuizen, L. (2000). Appl. Environ. Microbiol. 66, 2029–2036. [DOI] [PMC free article] [PubMed]
- Geize, R. van der, Yam, K., Heuser, T., Wilbrink, M. H., Hara, H., Anderton, M. C., Sim, E., Dijkhuizen, L., Davies, J. E., Mohn, W. W. & Eltis, L. D. (2007). Proc. Natl Acad. Sci. USA, 104, 1947–1952. [DOI] [PMC free article] [PubMed]
- Horinouchi, M., Hayashi, T., Yamamoto, T. & Kudo, T. (2003). Appl. Environ. Microbiol. 69, 4421–4430. [DOI] [PMC free article] [PubMed]
- Itagaki, E., Matushita, H. & Hatta, T. (1990). J. Biochem. 108, 122–127. [DOI] [PubMed]
- Itagaki, E., Wakabayashi, T. & Hatta, T. (1990). Biochim. Biophys. Acta, 1038, 60–67. [DOI] [PubMed]
- Jaroszewski, L., Rychlewski, L., Li, Z., Li, W. & Godzik, A. (2005). Nucleic Acids Res. 33, W284–W288. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Knol, J., Bodewits, K., Hessels, G. I., Dijkhuizen, L. & van der Geize, R. (2008). Biochem. J. 410, 339–346. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Levy, H. R. & Talalay, P. (1959). J. Biol. Chem. 234, 2014–2021. [PubMed]
- Leys, D., Tsapin, A. S., Nealson, K. H., Meyer, T. E., Cusanovich, M. A. & Van Beeumen, J. J. (1999). Nature Struct. Biol. 6, 1113–1117. [DOI] [PubMed]
- Mahato, S. B. & Garai, S. (1997). Steroids, 62, 332–345. [DOI] [PubMed]
- Matsushita, H. & Itagaki, E. (1992). J. Biochem. 111, 594–599. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Pandey, A. K. & Sassetti, C. M. (2008). Proc. Natl Acad. Sci. USA, 105, 4376–4380. [DOI] [PMC free article] [PubMed]
- Ringold, H. J., Hayano, M. & Stefanovic, V. (1963). J. Biol. Chem. 238, 1960–1965. [PubMed]
- Sedlaczek, L. (1988). Crit. Rev. Biotechnol. 7, 187–236. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2007). Methods Mol. Biol. 364, 215–230. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.