

Abstract
The results of computer simulations of the binding of etravirine (TMC125) and rilpivirine (TMC278) to HIV reverse transcriptase are reported. It is confirmed that consistent binding free energy estimates are obtained with or without the application of torsional restraints when the free energies of imposing the restraints are taken into account. The restraints have a smaller influence on the thermodynamics and apparent kinetics of binding of TMC125 compared to the more flexible TMC278 inhibitor. The concept of the reorganization free energy of binding is useful to understand and categorize these effects. Contrary to expectations, the use of conformational restraints did not consistently enhance convergence of binding free energy estimates due to suppression of binding/unbinding pathways and due to the influence of rotational degrees of freedom not directly controlled by the restraints. Physical insights concerning the thermodynamic driving forces for binding and the role of “jiggling” and “wiggling” motion of the ligands are discussed. Based on these insights we conclude that an ideal inhibitor, if chemically realizable, would possess the electrostatic charge distribution of TMC125, so as to form strong interactions with the receptor, and the larger and more flexible substituents of TMC278, so as to minimize reorganization free energy penalties and the effects of resistance mutations, suitably modified, as in TMC125, so as to disfavor the formation of non-binding competent extended conformations when free in solution.
1 Introduction
The strength of the association between a ligand molecule and its target receptor is measured by the binding constant or, equivalently, by the standard free energy of binding. From a medicinal perspective there is great interest in the development of computational models capable of predicting accurately protein-ligand binding free energies.[1, 2] A wide variety of methods have been developed to model the strength of protein-ligand association, spanning the field of QSAE knowledge-based approaches, to structure-based methodologies at various levels of theory ranging from empirical docking & scoring to quantum-mechanical descriptions.[2] This work is concerned a class of computational methodologies aimed at computing free energies of binding by employing high-level molecular mechanics descriptions of molecular interactions and classical descriptions of atomic motion.[3, 4, 5, 6, 7] Given a sufficiently accurate model of molecular interactions, these methods have the potential to incorporate details about the energetics and dynamics of the association equilibrium to address subtle aspects of drug development such as drug specificity and resistance.
Despite recent progress in phvsics-based binding free energy estimation methods, stemming from more accurate force field models, more extensive conformational sampling, improved free energy estimation methods, and faster computers, the conformational reorganization aspect of binding free energy calculations has received relatively little attention. The binding free energy is often the result of a large cancellation between the favorable work of forming receptor-ligand interactions and the unfavorable work to localize and reorganize the conformational ensembles of the ligand and receptor to their bound conformational states. The extent of this effect is likely widespread as for example it has been observed that that in protein-ligand complexes ligands assume energetically strained conformations.[8, 9] While drug design is often concerned with strengthening receptor-ligand interactions, the reorganization component can play a fundamental role in regulating binding specificity in cases where variations of binding energies are expected to be small. In such cases optimization of binding affinity can proceed by strategies aimed at preorganizing the ligand for binding, that is by minimizing the unfavorable reorganization. For example this strategy has been used to optimize the presentation of viral epitopes in HIV vaccine candidates.[10, 11] Examples exist of cases where optimization of a class of inhibitors was achieved by chemical rigidification of the ligands into their bound conformations.[12, 13, 14] Better correlation with experimental affinities have been reported when energetic scores are combined with ligand reorganization free energy estimates.[15, 16]
TMC125 (etravirine)[17] and TMC278 (rilpivirine)[18] (Fig. 2) are two of the newest and most effective drugs for antiviral AIDS therapy. Rilpivirine’s discovery has been announced in 2005[18] and has been approved for use by the Food and Drug Administration in May 2011. Both drugs act by inhibiting the function of the reverse transcriptase enzyme of the HIV virus (HIV-RT), which is essential for the initial transcription of viral RNA into DNA. TMC125 and TMC278 are members of the diarvlpyrimidine (DAPY) class of non-nucleoside inhibitors (NNRTI) of HIV-RT. These bind to an allosteric pocket in the so-called palm domain of HIV-RT (Fig. 1), causing a conformational change of the enzyme preventing it from properly processing the viral RNA.
Figure 2.
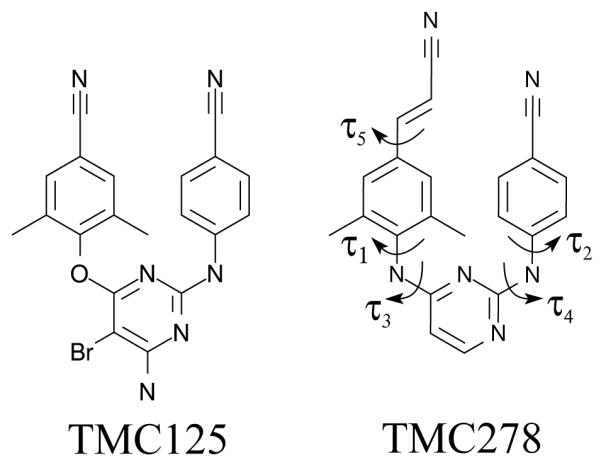
Chemical structures of TMC125 (etravirine) and TMC278 (rilpivirine). Hydrogen atoms are not shown. The nomenclature of the torsional angles are indicated for TMC278. The same nomenclature applies to TMC125, except for the τ5 angle which is specific to TMC278.
Figure 1.

Ribbon representation of portion of the reverse transcriptase (RT) enzyme of the HIV-1 virus. The palm, fingers, thumb, and connection domains of the p66 subunit are shown in red, blue, green, and yellow respectively. The p51 subunit is in dark purple. The crystal structure of HIV-RT in complex with TMC125 is shown (PDB id 3MEC[33]). TMC125 is shown in ball and stick representation within the non-nucleoside binding pocket. The circle represents schematically the region of the receptor that is included in the computational model.
The insurgence of drug-resistant HIV strains has been one of the most serious drawbacks of antiviral drug therapy, responsible for the growing inefficacy of the older drugs and causing the rebound of the viral load in patients after prolonged exposure to drugs. DAPY inhibitors have been developed specifically to target wild-type HIV-RT as well as common mutant forms that are known to be resistant to other NNRTI’s, so as to be more broadly applicable for more prolonged therapy regimens. Detailed structural studies[19, 20, 21] have characterized the molecular mechanisms of development of resistance and the properties of these drugs that help them evade resistance mutations better than older drugs. The main conclusion is that DAPY compounds possess specific flexibility that allows them to reposition (“wiggle”) into the binding site and to change shape (“jiggle”) to avoid clashes with mutated residues and form alternative contacts to replace those lost to mutations. While TMC125 and TMC278 have some of the most favorable properties among DAPY inhibitors in this respect, TMC278 has been shown to be superior to TMC125 in terms of adaptability to mutant forms of the HIV-RT enzyme partly due to its greater flexibility, and partly because of its unique cvanovinvl functionality which binds into an hydrophobic tunnel lined by conserved residues and provides an additional and reliable anchor in spite of mutations capable of disrupting the action of most other inhibitors.
The intrinsic flexibility of these DAPY compounds make them ideal proving ground for the study of conformational reorganization effects in binding free energy calculations. The conformational propensities of TMC278 have been previously characterized by both modeling and experimental techniques.[22, 21, 23, 24] The main conclusion is that in solution TMC278 preferentially assumes extended conformations very different from to the compact “U”-shaped conformation necessary for complexation with HIV-RT. Naturally, a conformational reorganization of TMC278 has to occur at a free energy cost disfavoring binding. In contrast, TMC125 is believed[25] to behave oppositely to TMC278 by incurring little conformational reorganization in going from the solution environment to the protein-bound state. This is because as it preferentially assumes the same “U”-shaped conformation in both conditions. We will show that the present modeling investigation confirms this hypothesis.
In this work we will address two main aspects of conformational reorganization. The first is understanding at a molecular level of how the opposite conformational propensities of the two drugs affect their affinity to HIV-RT. While direct measurements of binding constants are lacking, cell-based essays indicate that TMC278 has slightly higher affinity for wild-type HIV-RT than TMC125, despite that fact that reorganization effect are expected to favor the latter.[21] If so, the assumed larger reorganization free energy penalty for TMC278 must be somehow counterbalanced by stronger protein-ligand interactions so as to achieve higher or equivalent affinity than TMC125, for which reorganization is expected to be less significant.
The second aim of this work is to evaluate the role of conformational sampling[26, 27] and ligand conformational reorganization in protein-ligand binding free energy calculation practices. We have recently illustrated[28, 29] how an appropriate distribution of intermediate alchemical states is a necessary but not sufficient condition to ensure convergence of binding free energy calculations. Proper equilibration between unbound and bound conformational states, and between alternative binding modes, is also a critical requirement for obtaining reliable and reproducible results. The conformational sampling algorithm employed must be sufficiently powerful to allow frequent conformational transitions so as to rapidly explore conformational space with suitable frequency. While the Hamiltonian replica exchange (RE) algorithms we developed[30] are very useful to speed up equilibration along the alchemical thermodynamic path, the calculations reported here indicate that RE provides limited help in accelerating convergence when the kinetics is dominated by slow conformational reorganization processes. We will in particular examine the use of conformational restraints, devices that are often employed to speed up the convergence of binding free energy calculations.[31, 32, 6, 28] Confirmation that results are independent of the simulation setup, including the nature of imposed conformational restraints, is an important aspect of validation tests of binding free energy protocols. In addition we will investigate the conditions under which conformational restraints accelerate convergence and the molecular mechanisms at the basis of these effects.
2 Methods
2.1 Molecular systems
The chemical structures of the two DAPY NNRTI inhibitors of HIV-RT investigated are shown in Fig. 2. In this figure we indicate the main torsional degrees of freedom of the DAPY compounds based on established nomenclature.[21, 23] Non-nucleoside inhibitors bind to an allosteric pocket of HIV-RT in the so-called palm domain of the enzyme located between the thumb and fingers domains (see Fig. 1). HIV-RT is a heterodimer of the p66 and p51 subunits; while the non-nucleoside pocket is lined by residues of both subunits, residues of p66 accounts of most of the interactions with the inhibitors. The structures of HIV-RT complexes and the characteristics of the interactions with DAPY inhibitors has been thoroughly described.[?, 21, 33]
The initial structures of the complexes of HIV-RT in complex with TMC125 and TMC278 were obtained by the corresponding crystal structures (PDB id’s 3MEC[33] and 2ZD1[21], respectively), hydrogen atoms were added using the Maestro program (Schrödinger, Inc.), and protonation states were assigned based on neutral pH, Harmonic restraints with force constant of 1 kcal/mol/Å2 were imposed on all heavy atoms of the receptor, allowing for a range of motion of approximately 3 Å at room temperature. The structural models were then subjected to energy minimization before further processing. The calculations employed a reduced representation of the receptor which includes any residue of the receptor with atoms less than 12 Å from any atom of the ligand, resulting in a region surrounding the ligand of approximately 36 Å in diameter (Fig. 1). The reduced model was adopted only for reasons of computational convenience; the 12 Å cutoff on non-bonded interactions and the rigidity of the receptor, make model predictions independent from atomic interactions beyond the modelled region. The model of the receptor includes 114 residues (residues 88–112, 171–195, 220–243, 314–323, 347–350, and 378–385 of the p66 subunit and residues 132–142 of the p51 subunit) and 1,905 atoms. Residues at chain termini were capped using N-methvl-amine (NMA) and acetyl (ACE) groups, placed based on the coordinates of the backbone atoms of the corresponding deleted residues.
2.2 BEDAM Binding free energy computational protocol
Absolute binding free energies of the complexes with HIV-RT with TMC125 and TMC278 were computed using the Binding Energy Distribution Analysis Method method (BEDAM).[30] Briefly, BEDAM computes the standard binding free energy between a receptor R and a ligand L with implicit solvation by means of the expression
| (1) |
which follows, without approximations, from a well-established statistical mechanics theory of molecular association.[34] In Eq. (1) β = 1/kT, C° is the standard concentration of ligand molecules (set to C° = 1 M, or equivalently 1, 668 Å−3), Vsite is the volume of the binding site (see below), and ΔGb is the excess free energy of binding defined as the difference in free energy between the coupled state of the complex, in which the ligand and the receptor are fully interacting, and the decoupled state in which, while the ligand is confined in the binding site, the receptor and the ligand are interacting only with the solvent continuum and not with each other.
Central to the method is the concept of a binding energy function
| (2) |
defined for each conformation r = (rR, rL) of the complex as the difference between the effective potential energies U(rR, rL) and U(rR) + U(rL) of, respectively, the ligand-bound and ligand-separated conformations of the complex without internal conformational rear-rangements. BEDAM is based on a λ-dependent hybrid potential energy function of the form
| (3) |
where
| (4) |
is the potential energy of the uncoupled state of the complex. It is easy to see from Eqs. (2), (3), and (4) that Uλ=1 and Uλ=0 correspond to the effective potential energy functions of the coupled and decoupled states of the complex, respectively. The excess binding free energy ΔGb is by definition the difference in free energy between the λ = 1 and λ = 0 states. As described below, ΔGb is is obtained from statistical analysis of values of the binding energy [Eq. (2)] sampled from these states as well intermediate states with values of λ between these two extremes.
Rather than simulating each λ state independently, BEDAM employs a Hamiltonian replica exchange (HREM) λ-hopping strategy whereby simulation replicas periodically attempt to exchange λ values through Monte Carlo (MC) λ-swapping moves. λ-exchanges are accepted with the Metropolis probability min[1; exp(−βΔλΔu)][30] where Δλ is the difference in λ’s being exchanged and Δu is the difference in binding energies between the replicas exchanging them. Replica exchange strategies of this kind yield superior conformational sampling and more rapid convergence rates by allowing conformational transitions to occur at the value of λ at which they are most likely to occur and to be then propagated to other states.[26]
To improve convergence of the free energy near λ = 0, in this work we employ a modified “soft-core” binding energy function of the form
| (5) |
where umax is some large positive value (set in this work as 106 kcal/mol). This modified binding energy function, which is used in place of the actual binding energy function [Eq. (2)] wherever it appears, caps the maximum value of the binding energy while leaving unchanged the value of favorable binding energies.[35, 29]
In this work we employed multistate Bennett acceptance ratio estimator (MBAR)[36] to estimate the excess free energy of binding ΔGb, using as input binding energy samples obtained from the HREM simulations. Binding free energies are computed directly from the MBAR dimensionless free energies using the relationship
| (6) |
The MBAR dimensionless free energies, , are defined as the negative of the logarithm of the λ-dependent biased partition functions Zλ, In this case the dimensionless free energies are estimated by the self-consistent solution of the set of equations[36]
| (7) |
where ujn is the nth binding energy sample from replica j, K is the number of replicas and Nj is the total number of binding energy samples from replica j, For the MBAR analysis we employed the code provided by John Chodera and Michael Shirts (http://alchemistry.org). Statistical uncertainties were obtained from the standard deviation of the binding free energies from the last four 1 ns blocks of binding energy data (see below).
For later use, the reorganization free energy for binding is defined by the expression[37]
| (8) |
where ⟨u⟩1 is the average binding energy at λ = 1 and is the standard binding free energy from Eq. (1). The former is computed as described below and is computed by difference using Eq. (8).
2.3 Binding free energy calculations with conformational restraints
The concept of the reorganization free energy is useful to formalize situations in which, by imposing suitable conformational restraints, auxiliary states are introduced which do not match exactly with the end point states of the complex, [32, 37] Consider for example Fig. (3), in which the excess free energy of binding is first decomposed in the reorganization free energy ΔGrestrain of restraining the ensembles of conformations of the receptor and the ligand in solution to chosen macrostates R* and L*. For instance, in the application below the L* macrostate is defined as the ensemble of conformations of the ligand with torsional angles within a specified range. The free energy for this process is related to the population , defined as the probability of finding a conformation belonging to the specified macrostate, in absence of restraints and ligand-receptor interactions (λ = 0):
| (9) |
Following this step, we consider the binding free energy, ΔGb(Restr), between the R* and L* species, that is the excess binding free energy when the receptor and the ligand are limited to the chosen macrostates. In general the resulting state of the complex, denoted by (RL)* in Fig. 3, does not match the full complexed state RL because in the former the receptor and the ligand are limited to their respective macrostates. ΔGrelease, defined similarly to ΔGrestrain, is the free energy difference between these two states. In the present application we assume ΔGreiease = 0 because the restrained macrostate encompasses all of the conformational ensemble of the complex; that is the (RL)* and RL species are virtually equivalent.
Figure 3.

Thermodynamic cycle illustrating the restrain-and-release decomposition of the excess free energy of binding [Eq. (10)]. Although not indicated, the ligand here is assumed to be always sequestered in the binding site. R and L represent the free receptor and ligand, R* and L* represent the receptor and ligand restrained within a conformational macrostate, (RL)* represents the complex in which receptor and ligand are restrained within their macrostates, and RL represents the free complex.
Based on the above we have
| (10) |
Eq. (10) expresses the notion that a binding free energy calculation can be conducted with or without conformational restraints, and, if the the binding free energy, ΔGb(Restr), obtained with restraints is properly combined with the free energy, ΔGrestrain, of imposing the restraints, either approach should return the same binding free energy. Hence the use of restraints is purely a matter of efficiency; restraints should be considered whenever they can lead to faster convergence of binding free energy estimates.
2.4 Computational details
We employed the OPLS-AA/AGBNP2 effective potential in which the effect of the solvent is represented implicitly by means of the AGBNP2 implicit solvent model[38, 39] together with the OPLS-AA[40, 41] force field for covalent and non-bonded interatomic interactions. Parallel molecular dynamics simulations were conducted with the IMPACT program.[42] λ = 0, 10−6, 5 × 10−6, 10−5, 5 × 10−5, 10−4, 5 × 10−4, 10−3, 5 × 10−3, 0.01, 0.05, 0.075, 0.1, 0.175, 0.25, 0.3125, 0.375, 0.4375, 0.5, 0.58, 0.67, 0.75, 0.875 and 1. The binding site volume was defined as any conformation in which the middle nitrogen atom (the one directly in between the two sidechains) of the central pyridine ring of the ligand is within 2 Å of the center of mass of the Cα atoms of residues 97, 102, 179, and 181 of the p66 subunit of HIV-RT. The guest was sequestered within this binding site volume by means of a flat-bottom harmonic potential. Based on this definition, the volume of the binding site, Vsite, is calculated as approximately 33.3 Å3 and −kTln C°Vsite in Eq. (1) is 2.32 kcal/mol. BEDAM parallel λ-hopping calculations were performed for 8 ns of molecular dynamics per replica (192 ns of molecular dynamics for each of four binding free energy calculations with 24 replicas each, resulting in a total of 768 ns of simulations). The second half of the trajectory (4 ns per replica) was used for data analysis. Binding energies were sampled with a frequency of 1 ps. Statistical uncertainties are evaluated as the standard error of the distributions of estimates obtained from each 1 ns section of the trajectories.
Probability distributions and averages are computed by weighted averages over the trajectories of all of the replicas.[36] Briefly, each sample n from replica j is assigned a λ-dependent thermodynamic weight given by
| (11) |
where the fλi are the solution of the MBAR equation (7) and the other symbols are as for Eq. (7). Thermodynamic averages are formulated as weighted averages over the observations using the weights defined by Eq. (11). For example the average binding energy at λ = 1 is formulated as
| (12) |
and the probability density, pλ(ϕ), of a structural quantitv ϕ = ϕ(r) (a torsional angle for instance) at λ is expressed as
| (13) |
where ϕjn is the value of the structural quantity of sample jn and δϕ(ϕjn) is a function defined as 1 if the argument falls within the bin centered at ϕ with width Δϕ and zero otherwise.
BEDAM binding free energy calculations were conducted for TMC125 and TMC278 with and without torsional angle restraining potentials designed to restrict the ligand within the bound ligand macrostate, defined as any conformation with τ3 and τ4 torsional angles within a 120° interval around τ = 0° (see Figs. 2 and 4), Restraining potentials were implemented as fiat-bottom harmonic restraints on the τ3 and τ4 torsional angles centered on τ = 0 with a tolerance of 60°on either side. The population of the bound macrostate at λ = 0, required to compute ΔGrestrainfrom Eq. (9), was evaluated by numerical integration of the joint probability density, p0(τ3,τ4), of the τ3 and τ4 angles at λ = 0 within the region −60° < τ3,τ4 < 60°. p0(τ3,τ4) probability densities are displayed in terms of the corresponding potential of mean force (PMF)
| (14) |
taking the smallest value of the PMF as the zero of energies.
Figure 4.

The four main conformational states of the DAPY compounds based on the τ3 and τ4 torsional angles, illustrated for TMC278 . The U conformational state is centered around the “U”-shaped conformation with τ3 = τ4 = 0°. The U state corresponds to the bound conformation. The L1 and L2 conformational states are centered around the “L”-shaped conformations at (τ3,τ4) = (180°, 0) and (τ3,τ4) = (0°, 180°), respectively. Finally, the E or extended conformational state is centered around (τ3,τ4) = (180°, 180°)
3 Results
3.1 Conformational analysis of free and bound TMC125 and TMC278
Since they result in the largest conformational variations (see Fig. 4), we will focus first on the τ3 and τ4 torsional degrees of freedom of the ligands as defined in Fig. 2, The computed conformational distributions for TMC125 and TMC278 free in solution with respect to these torsional angles (see Methods) are shown in Figs. 5 and 6, respectively. As it can be seen from Fig. 5, TMC125 assumes predominantly U conformations in solution with a minor L1 component. Remarkably, TMC278 displays the opposite behavior by occupying mostly extended (E) and L2 conformations (Fig. 6), which are virtually never observed for TMC125. Of the two ligands, TMC278 can be considered the more flexible compound in solution since, although less frequently than TMC125, it also visits U and L1 conformations; so overall TMC278 is capable of adopting all of the main conformations available to DAPY compounds.
Figure 5.
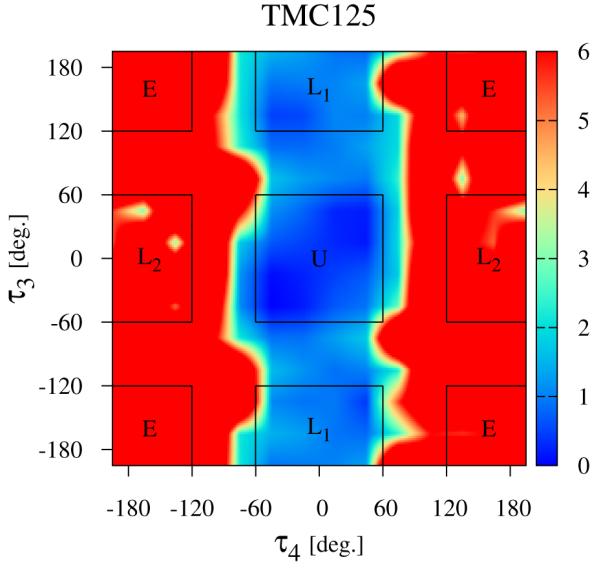
The distribution of conformations of free TMC125 in solution (λ = 0) expressed in terms of the potential of mean force (in kcal/mol) with respect to the τ3 and τ4 torsional angles (see Fig. 2), The regions corresponding to the main conformational states (see Fig. 4) are indicated.
Figure 6.
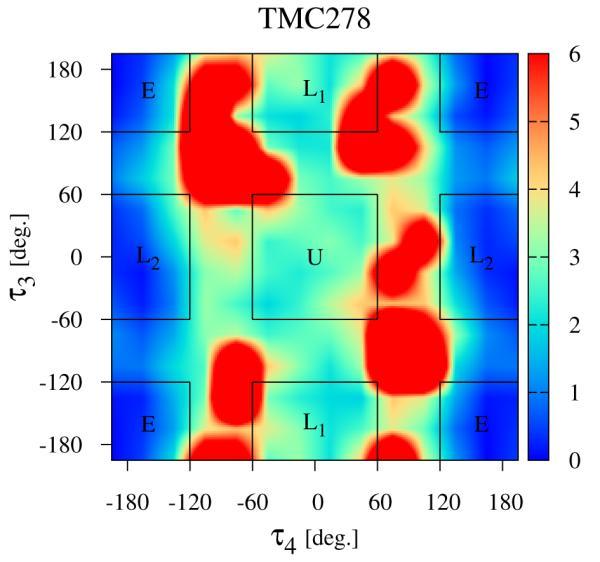
The distribution of conformations of free TMC278 in solution (λ = 0) expressed in terms of the potential of mean force (in kcal/mol) with respect to the τ3 and τ4 torsional angles (see Fig. 2), The regions corresponding to the main conformational states (see Fig. 4) are indicated.
By numerically integrating the conformational distributions in Figs. 5 and 6 within the U conformational state (indicated by the square central region of the diagrams), we obtain the restraining free energies ΔGrestrain defined by Eq. (9) and reported in Table 1, The restraining free energy for TMC125 (0.30 kcal/mol) is much smaller than the one for TMC278 (3.21 kcal/mol) reflecting the relative abundance of U conformations for the two molecules in solution, TMC125 has a natural propensity of forming U conformations so restraints that force it to adopt that state have little influence on its thermodynamics. Conversely, restraints have a large effect on the thermodynamics of TMC278 because in absence of restraints this preferentially adopts other conformations.
Table 1.
Experimental and computed standard binding free energies of the complexes of HIV-RT with the TMC125 and TMC278 ligands with and without torsional restraints.
Despite the large differences in conformational propensities when free in solution, the two compounds have similar conformational distributions in the bound state. The interactions with the protein receptor force each ligand to adopt conformations well contained within the U conformational macrostate. The thermodynamic analysis above indicates that TMC278 suffers a substantial reorganization free energy penalty associated with the reorganization of the τ3 τ4 degrees of freedom in order to adapt to the receptor. This is confirmed by the significantly smaller reorganization free energy (31.01 kcal/mol; Table 2, 5th column) obtained for TMC278 binding with torsional restraints on the τ3 τ4 angles, forcing it to remain withing the U state, compared to unrestrained conditions (34.55 kcal/mol). In contrast, the computed reorganization free energies for TMC125 with or without torsional restraints are very similar to each other (Table 2, 5th column) indicating minor reorganization of the distributions of τ3 τ4 angles.
Table 2.
Decomposition of computed binding free energies into average binding energy (⟨u⟩1) and reorganization free energy () components.
| Ligand | a,b | ⟨ u ⟩ 1a | a,c | a,d | |
|---|---|---|---|---|---|
| TMC125 | |||||
| Unrestrained | −13.32 ± 0.10 | −49.52 ± 0.06 | 36.20 ± 0.12 | 36.20 ± 0.12 | |
| Restrained | −13.16 ± 0.49 | −49.47 ± 0.18 | 36.31 ± 0.52 | 36.61 ± 0.53 | |
| TMC278 | |||||
| Unrestrained | −11.37 ± 0.67 | −45.92 ± 0.18 | 34.55 ± 0.69 | 34.55 ± 0.69 | |
| Restrained | −14.96 ± 0.60 | −45.97 ± 0.18 | 31.01 ± 0.63 | 34.22 ± 0.66 | |
3.2 Binding Free Energy Estimates
The computed binding free energies for the complexes of wild-type HIV-RT with the TMC125 and TMC278 inhibitors with and without torsional restraints are shown in Table 1, Based on Eqs. (1) and (9), the binding free energy values, , computed with torsional restraints (4th column) are combined with the free energies, ΔGrestrain, of imposing the restraints (5th column) to obtain the binding free energy estimates , which can be compared to the experimental measurements. The values obtained from the unrestrained simulations yield directly the actual binding free energy estimates without restraining corrections. The binding free energy estimates, , from the unrestrained and restrained simulations are in agreement within statistical uncertainty for both TMC125 and TMC278, thereby providing numerical validation to the theoretical requirement that binding free energy estimates should not depend on the calculation settings. Indeed, as discussed above, the binding free energy calculations conducted with conformational restraints, when appropriately combined with the free energy of imposing the restraints, should return equivalent results to those of binding free energy calculations without restraints.
The binding free energy estimates (6th column in Table 1) for TMC278 are in good agreement with the experimental measurement (2nd column). The computed binding free energy values for TMC125 are instead 1 to 2 kcal/mol more favorable to binding than the experimental value, causing the affinity ranking of the two compounds to be reversed relative to the experiments. Overall the agreement between calculations and experiments is reasonable considering the approximations in the potential energy model and the fact that we have not attempted to model the influence of receptor reorganization. The present model also neglects the influence of protonation and aggregation equilibria that are known to be important in these systems,[23] Furthermore, it should be noted that direct binding free energy measurements and inhibition constants are not available for these complexes. The experimental values listed in Table 1 have been derived from EC50 values from cell-based essays[43, 18] which are influenced by many characteristics of the drugs (propensity for aggregation, membrane transport, non-specific binding, degradation, etc.)[22] and represent only qualitatively their thermodynamic affinity for the HIV-RT enzyme, which is the only quantity modeled here.
4 Discussion
4.1 Use of conformational restraints in binding free energy calculations
Conformational restraints, often in the form of additional potential terms which restrict the position and conformation of the ligand and the receptor, are commonly used in binding free energy calculations. One use of restraints derives from the need to define a binding site volume which specifies the complexed state and introduces the effect of ligand concentration.[34] This issue and its influence on binding free energy estimates has been discussed.[44, 30, 37] Conformational restraints are, in addition, often used as computational devices to speed up the convergence of binding free energy calculations by reducing the size of conformational space of the complex that needs to be sampled to achieve convergence.[31, 6, 28] When used in the latter context, it is important that the inclusion of restraints affects only the efficiency of the calculations and not their outcomes. As discussed in Section 2, when appropriate corrections are applied representing the work corresponding to imposing and releasing the restraints, equivalent results should be obtained without restraints or with different restraining schemes.
4.1.1 Independence of binding free energy estimates
Protein-ligand binding free energy protocols are rarely subjected to validation tests with respect to variations of restraints. In this work we have numerically confirmed that the BEDAM protocol[30] combined with multi-state estimation of conformational free energies[24] returns consistent binding free energy estimates for two complexes of HIV-RT with DAPY inhibitors with or without the presence of ligand conformational restraints. Conformational restraints on key torsional degrees of freedom have been optionally used here to force the ligands to remain within the bound conformational macrostate even when free in solution, and all along the thermodynamic path. Restraints have very minor thermodynamic effects for TMC125, which preferentially adopts bound conformations in solution without the help of restraints. Restraints have instead a large effect on the affinity of TMC278 because this does not often visit bound conformations unless restraints are applied. The computed binding free energy of restrained TMC278 (−14.96 kcal/mol) is significantly larger in magnitude than the binding free energy of unrestrained TMC278 (−11.37 kcal/mol) because the latter includes a larger portion of the reorganization free energy penalty for reorganizing the ligand conformational ensemble into the bound macrostate. When, however, the work of imposing the restraints (estimated at 3.21 kcal/mol) is considered, the two approaches yield equivalent binding free energy estimates within statistical uncertainty.
As it can be clearly seen from the data in Table 2, restraints have very little influence on average binding energy values, which measure the strength of ligand-receptor interactions. This is expected because in this case the bound conformational macrostate is well contained within the range allowed by the restraints. The influence of restraints is strictly limited to reorganization free energy penalties. The computed reorganization free energy of TMC278 is approximately 31 kcal/mol with restraints compared to 34:5 kcal/mol without them. The larger reorganization by 3:5 kcal/mol without restraints is clearly due to the reorganization free of the ligand, estimated at approximately 3:2 kcal/mol, due to the reorganization from the unrestrained ensemble in solution (Fig. 6) to the ensemble allowed by the restraints (the U macrostate in Fig. 6).
4.1.2 Effects of restraints on the rate of convergence
As discussed above increasing computational efficiency is the main motivation for using conformational restraints in binding free energy calculations. One measure of computational efficiency is the simulation time necessary to achieve a given statistical uncertainty of the free energy estimate or, equivalently, the magnitude of the statistical uncertainty for a given simulation time, smaller being better. One puzzling result of the calculations presented here is that the use of restraints did not lead to smaller statistical uncertainties (see Table 1). The estimated uncertainty for the binding free energy of TMC125 with restraints is significantly larger than for the unrestrained calculation (0:5 vs. 0:1 kcal/mol, respectively). The statistical uncertainties for the two calculations for TMC278 binding are very similar (both are approximately 0:65 kcal/mol) with the uncertainty (0:2 kcal/mol, see Table 1) of the restraining free energy, ΔGrestrain, contributing somewhat to the overall uncertainty of the free energy estimate with restraints.
Some insights on this issue can be gained by examining how binding free energy estimates vary as the simulation progresses (Fig. 7). In this figure we plot binding free energy estimates from data from 1 ns sections of the simulation trajectories. Note that these are not cumulative results. We see that for each calculation there is a relatively short equilibration time (the results presented in Table 1 are from the second half of the trajectory); furthermore the free energy estimates for TMC125 show significantly smaller variations than for TMC278. The unrestrained TMC125 calculation shows the smallest variations reflecting its small statistical uncertainty. The restrained TMC125 calculation displays larger variations. There are no obvious differences in the variations of the free energy estimates for TMC278 with or without restraints, however the variations for TMC278 are consistently greater than those for TMC125.
Figure 7.

Binding free energy estimates from 1 ns sections of trajectories as a function of simulation time. Note that these are not cumulative estimates.
In prior studies[28, 29] we have identified as one major cause of slow convergence the long timescales to achieve conformational transitions between unbound conformations with large binding energies to bound conformations with low binding energies. In Figs. 8 and 9 we show the time trajectories of the binding energies of some of the replicas of the HREM simulations of the complexes with TMC125 and TMC278 with and without torsional restraints. In general, a larger number of transitions from high binding energies to low binding energies and vice versa is indicative of better conformational sampling and, in turn, faster convergence.
Figure 8.

Time trajectories of the binding energies of some of the replicas of the BEDAM simulation of TMC125 in complex with HIV-RT without (A) and with (B) torsional restraints.
Figure 9.

Time trajectories of the binding energies of some of the replicas of the BEDAM simulation of TMC278 in complex with HIV-RT without (A) and with (B) torsional restraints. Fewer long-lived binding/unbinding transitions occur compared to TMC125 (Fig. 8). Note that without torsional restraints (A) the region of binding energies near −20 kcal/mol is rarely crossed. Also note that longer trajectories are shown for unrestrained TMC278 (A).
Based on this criterion, of the four calculations, the unrestrained TMC278 simulation appears to undergo the fewest conformational transitions (Fig. 9A); several of the replicas remain in either bound conformations or unbound conformation for the entire duration of the simulation and never undergo transitions, while few replicas undergo one or two transitions. The corresponding simulation for TMC278 with restraints (Fig. 9B) shows better behavior; all of the replicas display rapid transitions between low binding energies (around −50 kcal/mol) and high binding energies (103-106 kcal/mol). More rarely and similarly to the unrestrained simulation, long-lived transition to high binding energies occur (see for example bottom-center panels in Figs. 9A and 9B). The simulations for TMC125 binding (Fig. 8) display analogous behavior except that the kinetics of the unrestrained and restrained simulations are more similar to each other and undergo more long-lived transitions than the TMC278 simulations.
Structural analysis shows that low-to-high and high-to-low rapid transitions in binding energy space observed in the TMC125 simulations and the restrained TMC278 simulation (Figs. 8 and 9B) correspond to excursions in which replicas rapidly traverse λ-space from λ = 1 to λ ≃ 0 and concomitantly assume high binding energies, but with the ligand molecule remaining in the neighborhood of the bound orientation and getting quickly “recaptured” in the bound state at low binding energies. The infrequent long-lived transitions are cases in which instead during such excursions the ligand leaves the bound orientation and starts to freely diffuse in orientational space before eventually finding again the bound conformation. Compared to TMC125, the TMC278 ligand spends more time exploring orientations before finding an orientation suitable for binding. This is due in part of the presence of the extra orientational constraint due to the positioning of the eyanovinyl group in its receptor cavity and, for the unrestrained simulation, also because of the tendency of TMC278 to migrate to the E, L1, and L2 rotameric states, which are incompatible to the formation of strong interactions with the receptor.
It appears therefore that orientational degrees of freedom play an important role in the kinetics of binding and unbinding in these systems and that they strongly influence the rate of convergence. Our conclusion is that the convergence rate is for the most part dictated by slow orientational diffusion. An equivalent conclusion is that slow convergence is due to the difficulty of estimating precisely the size of the region of orientational space compatible with binding relative to the size of the overall orientational space. We predict therefore that imposing restraints on the ligand orientation would speed up convergence of binding free energy considerably.[28] On the other hand restraints must be designed based on prior structural knowledge so at to not interfere with the bound conformational state (or calculation of ΔGrelease must be be considered, see above), hence they are not easily applicable to situations in which structural information about the complex is uncertain or when multiple binding modes are possible.
4.1.3 Can restraints slow down convergence?
One puzzling result of this work is that the statistical uncertainty for the binding free energy of TMC125 estimated from the restrained simulation is five times larger than that from the unrestrained simulation (see Table 1). Restraints are believed to generally aid convergence of binding free energy calculations[6] rather than worsen it, as in this case. We found indications that the poorer convergence of the restrained simulation of TMC125 binding is caused by the reduced number of binding pathways available to the ligand compared to the unrestrained simulation.
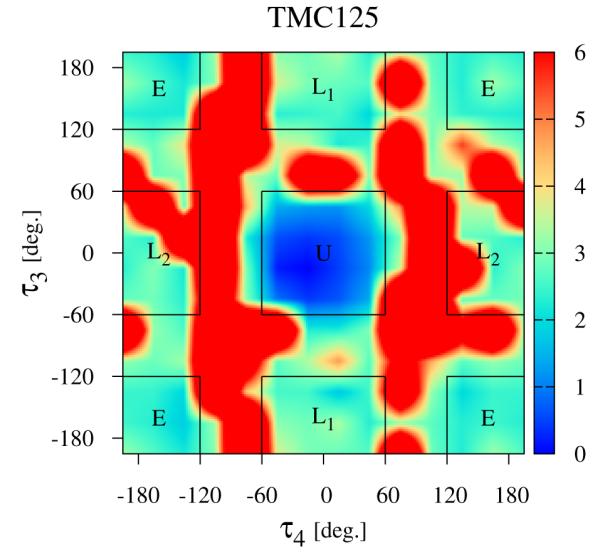
Indication of this phenomenon is apparent from the analysis of ligand conformational distributions at intermediate values of λ from the unrestrained simulation (Figs. 10 and 11). As shown in Fig. 5, TMC125 assumes mostly U and L1 conformations when dissociated from the receptor and exclusively U conformations when it is fully associated. However, the shift from U and L1 conformations to exclusively U conformations does not appear to occur as a direct population transfer from L1 to U. At intermediate values of λ the L2 and E conformational states become also significantly populated (Fig. 10). This occurs mainly by population transfer from the L1 macrostate to the L2 and E maerostates (Fig. 11); with increasing λ, the populations of the L2 and E states increase rapidly at the expense of the L1 population. At larger values of λ’s, population transfer from the L2 and E states to the U state then occurs (Fig. 11). This observation is clearly indicative of the existence of binding routes whereby TMC125, initially in a L1 conformation, reach the bound U state by going through E and/or L2 intermediate states. These binding mechanisms are absent in the restrained simulation in which, by construction, only U conformations are allowed.
Figure 10.
The distribution of conformations of TMC125 in complex with HIV-RT at λ = 0.005 expressed in terms of the potential of mean force (in kcal/mol) with respect to the τ3 and τ4 torsional angles (see Fig. 2). At this intermediate value of λ TMC125 visits all of the main rotamers (see Fig. 4).
Figure 11.

Conformational populations of TMC125 as a function of λ from the unrestrained binding simulation. At λ = 0 only the U and L1 conformations are significantly populated. As λ increases the L1 population decreases rapidly concomitantly with a rapid increase of the populations of the L2 and E populations. At even larger values of λ only the U (bound) conformation is significantly populated.
We therefore suggest that in this case conformational restraints oppose convergence rather than aid it because they block additional binding pathways and decreasing the chances of more rapid interconversion between unbound and bound conformations of TMC125. This conclusion goes contrary to the generally accepted wisdom that conformational restraints always speed up convergence by reducing the size of conformational space that needs to be sampled. This example shows that reducing the accessible conformational space may have the side effect of precluding some conformational transition pathways and slow down convergence.
4.1.4 Order-disorder pseudo-phase transition
It is apparent from Figs. 8 and 9 that the unrestrained simulation of TMC278 (Fig. 9A) behaves differently from the others in terms of binding/unbinding transitions. In this simulation a rarely crossed barrier exists in binding energy space separating unbound and bound conformations. The location of this barrier is clearly seen in the binding energy distributions pλ(u) of this system (Fig 12). For λ < 0.2 binding energies are predominantly above −20 kcal/mol, whereas for λ > 0.35 much lower values of binding energies are sampled. Values of binding energies near −20 kcal/mol are rarely visited at any λ and binding energy distributions straddling this region are bimodal (see distributions in Fig. 12 at λ = 0.25 and 0.3125).
Figure 12.

Probability densities of the binding energy, u, for the simulation of unrestrained TMC278 complexed with HIV-RT as a function of λ near the critical state. The distributions near the critical λ (λc ≃ 0.25) are bimodal and the intermediate region of binding energies around −20 kcal/mol is infrequently visited at any λ.
This behavior is the tell-tale indication of an order-disorder pseudo-phase transition;[45] a situation in which a macrostate at high energy and high entropy is in equilibrium with another macrostate at low energy and low entropy. In this specific case the two macrostates correspond to the L1, L2, and E rotamers of TMC278, which are not capable of binding strongly to HIV-RT, and the U rotamer that becomes predominant when interactions with the receptor are turned on. The switch to U conformations occurs rapidly as a function of λ (Fig. 13), a consequence of the large difference in average binding energies between bound and unbound conformations.[28] Pseudo-phase transitions of similar strength are not observed in the simulations of TMC125 and the simulation of TMC278 with restraints because of reduced conformational space and entropy of the unbound macrostate in these cases. Order-disorder transitions are also not observed for unrestrained TMC125 because of its relative rigidity compared to TMC278.
Figure 13.

Conformational populations of the U, L1, L2 and E rotamers of unrestrained TMC278 in complex with HIV-RT as a function of the coupling parameter λ. The transition from rotamer states prevalent near the decoupled state (λ = 0) to the U rotamer occurs in a relatively small region of λ. As we have previously shown,[28] the gradient of the population profile for the U state at 50% population is proportional to the difference in average binding energy between the bound and unbound macrostates of the complex.
As previously observed,[28] the rarely visited intermediate region of binding energies (Fig. 12) constitutes an obstacle to binding/unbinding transitions, thereby hampering convergence of binding free energy calculations. Advanced conformational sampling tools are being developed in this and other research fields to address conformational sampling challenges of this kind.[45, 46]
4.2 Insights on thermodynamic driving forces for binding
Our original expectation was that, due to its higher flexibility in solution, binding of TMC278 would be characterized by a larger reorganization free energy penalty than TMC125. The calculations however indicate the opposite. As it can be seen by the values in Table 2 the binding reorganization free energy of TMC125 is approximately 36 kcal/mol; 2 kcal/mol greater than the reorganization of TMC278. This is despite the fact that reorganization of the τ3 and τ4 torsional angles alone within the defined ranges (see above) opposes binding of TMC278 by approximately 3 kcal/mol (see ΔGrestrain in Table 1, 5th column). Clearly, collective reorganization of other degrees of freedom of TMC125 must be contributing more to the overall reorganization free energy than the τ3 and τ4 angles which account for only approximately 0:3 kcal/mol of . Hints concerning the additional possible sources of reorganization come from a comparative analysis of the bound conformational ensembles of TMC125 and TMC278.
The motion of the DAPY compounds within the non-nucleoside binding site of HIV-RT has been described as “wiggling”, that is internal conformational rearrangements such as rotations of sidechains, and “jiggling”, or overall translation and rotational motion of the molecule. As illustrated in Fig. 14, analysis of conformational ensembles of the complexes at λ = 1 reveals that, when bound to HIV-RT, TMC278 is significantly more mobile than TMC125 in both respects. Fig. 14A represents the distribution of τ1and τ3 torsional angles characterizing the “wiggling” motion of the left sidechain of the ligands in Fig. 2, which contains the oxy-dimethylbenzonitrile moiety of TMC125 or the amino-dimethylbenzocyanovinyl moiety of TMC278. This data clearly indicates that the sidechain of TMC278 undergoes a wider range of libration motion than TMC125, despite the fact that the cyanovinyl group specific to TMC278 is known to be anchored in what has been a described as a hydrophobic tunnel of the receptor.[21] In comparison (data not shown), we observed little difference in the distribution of τ2 and τ4 torsional angles of the amino-benzonitrile group, which is the sidechain common to both compounds (see Fig. 2).
Figure 14.

(A) Distribution of the τ1 and τ3 torsional angles of TMC125 (blue triangles) and TMC278 (red squares). The τ1 and τ3 torsional angles probe the motion of the ligand sidechain which contains the oxy-dimethylbenzonitrile moiety of TMC125 or the amino-dimethylbenzoeyanovinyl moiety of TMC278 (“wiggling” motion). (B) Probability density (in Å−1) of displacement δd of the N1 atom of the central pyrimidine ring (the nitrogen atom in between the two substituents) relative to the its position in the corresponding crystal structure (“jiggling” motion).
We have also observed a larger amount of “jiggling” motion of TMC278 relative to TMC125. As an illustration, Fig. 14B shows the distribution of distances δd of the position of the N1 atom of the central pyrimidine ring (the nitrogen atom in between the two substituents) relative to the its position in the corresponding crystal structure. The variance of the distance distribution for TMC278 is 60% greater than that for TMC125, underscoring greater translational motion, or “jiggling”, of TMC278. Collectively these observations indicate that within the bound conformational macrostate the motion of TMC125 is more restricted than TMC278 thereby explaining in part the greater reorganization free energy penalty for binding of the former. A large contribution to reorganization may also originate from librational and vibrational degrees of freedom that we have not analyzed.[47]
The larger unfavorable reorganization free energy for TMC125 is counterbalanced by a more favorable average binding energy; the latter is approximately 3.5 kcal/mol more negative than for TMC278 (see Table 2, 4th column), compared to a deficit of roughly 2 kcal/mol for the reorganization free energy component (Table 2, 6th column), resulting in a net ~ 1.5 kcal/mol advantage for TMC125 in terms of binding free energy. Among the energetic contributions to the binding energy (ligand-receptor electrostatic and van der Waals interactions, and desolvation free energy), stronger receptor-ligand electrostatic interactions accounts for the majority of the difference in average binding energy in favor of binding of TMC125.
Given its weaker interactions with the receptor (see Table 2), the affinity of TMC278 for HIV-RT would have been even weaker relative to TMC125 if not for its smaller (and unexpected) reorganization free energy. The present results indicate that the influence of the cyanovinyl group on the thermodynamics of binding of TMC278 can be ascribed less to increased strength of receptor-ligand interactions and more to increases redundancy and flexibility of these interactions, so that they are maintained in a wider range of conformations. This hypothesis is supported by the less favorable average binding energy of TMC278 relative to TMC125 (Table 2) and the lower reorganization free energy penalty for binding of TMC278, due in part to the greater freedom of motion of the sidechain containing the cyanovinyl group (Fig. 14A). Based on structural studies of complexes of TMC278 with mutant forms of HIV-RT, Das at al,[21] have observed that the ability of the cyanovinyl group to reposition and switch contacts between receptor residues is key to the greater potency of TMC278 against a wide range of drug-resistant mutants of HIV-RT.
Structure-based drug optimization strategies are often aimed at increasing the number and strength of ligand-receptor interactions. This example shows that both greater affinity and improved resistance profiles can be achieved by focusing instead on increasing the redundancy and flexibility of ligand-receptor interactions so that they are maintained over a wider range of conformations of the complex.
5 Conclusions
We have investigated some aspects of the complexation equilibrium between HIV-RT and TMC125 (etravirine) and TMC278 (rilpivirine), two antiviral drugs recently approved for the treatment of AIDS. We have confirmed that the BEDAM computational protocol we employed yields consistent results regardless of applied conformational restraining potentials on torsional degrees of freedom. The presence of the restraints is shown however to have significant influence on the convergence rate of the simulations and the conformational transition mechanisms. Unexpectedly, torsional restraints do not enhance convergence for these systems. Torsional restraints on the ligands slow down convergence by precluding some pathways for binding/unbinding transitions and moreover equilibration along ligand orientational degrees of freedom, which are not limited by the applied restraints, seem to dominate the rate of binding/unbinding transitions and ultimately the rate of convergence of binding free energies. We have also shown that restraints have little influence on the ligand-receptor interaction energy driving force for binding, while they lower the reorganization free energy penalty.
Physical insights concerning the thermodynamic driving forces for or against binding have been obtained. Contrary to expectations TMC125 forms stronger interactions with the receptor than TMC278 and, despite the higher flexibility of the latter when dissociated by the receptor, TMC125 suffers a higher reorganization free energy penalty for binding than TMC278. This is rationalized by the wider range of conformations of the TMC278/HIV-RT complex partly due to the ability of the cyanovinyl substituent of TMC278 to form interactions with the receptor in multiple orientations. Mindful of the approximations in the computational model, and in particular to the lack of modelling of receptor reorganization effects, this computational study proposes ligand design principles to increase affinity to HIV-RT and possibly to achieve more favorable inhibition profiles across mutant forms of the receptor. An ideal inhibitor, if chemically realizable, would possess the electrostatic charge distribution of TMC125 to form strong interactions with the receptor and the large and flexible substituents of TMC278 to minimize reorganization free energy penalties and the effects of resistance mutations, suitably modified as in TMC125 so as to disfavor the formation of non-binding competent extended conformations (L1; L2 and E) in solution.
Acknowledgments
The author expresses sincere gratitude to Dr. Ronald Levy for his contributions to the development of the BEDAM method and stimulating collaborative work on the statistical thermodynamics of protein-ligand binding. This work has been supported by grants by the National Institute of Health (GM30580) and the National Science Foundation (CDI type II no. 1125332). The calculations reported in this work have been performed at the BioMaPS High Performance Computing Center at Rutgers University funded in part by the NIH shared instrumentation grants no. 1 S10 RR022375 and 1 S10 RR027444, and on the Trestles cluster at the San Diego supercomputing center under XSEDE National Science Foundation allocation grant no. TG-MCB100145.
References
- [1].Jorgensen William L. The many roles of computation in drug discovery. Science. 2004 Mar;303(5665):1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- [2].Guvench Olgun, MacKerell Alexander D. Computational evaluation of protein-small molecule binding. Curr. Opin. Struct. Biol. 2009 Feb;19(1):56–61. doi: 10.1016/j.sbi.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gilson Michael K, Zhou Huan-Xiang. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- [4].Shirts MR, Mobley DL, Chodera JD. Alchemical free energy calculations: ready for prime time? Ann. Rep. Comput. Chem. 2007;3:41–59. [Google Scholar]
- [5].Mobley David L, Dill Ken A. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure. 2009 Apr;17(4):489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Deng Yuqing, Roux Benoǐt. Computations of standard binding free energies with molecular dynamics simulations. J. Phys. Chem. B. 2009 Feb;113(8):2234–2246. doi: 10.1021/jp807701h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chodera John D., Mobley David L., Shirts Michael R., Dixon Richard W., Branson Kim, Pande Vijay S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011;21:150–160. doi: 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Perola Emanuele, Charifson Paul S. Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding. J Med Chem. 2004 May;47(10):2499–2510. doi: 10.1021/jm030563w. [DOI] [PubMed] [Google Scholar]
- [9].Liebeschuetz John, Hennemann Jana, Olsson Tjelvar, Groom Colin. The good, the bad and the twisted: a survey of ligand geometry in protein crystal structures. J. Comp. Aided Mol. Des. 2012 doi: 10.1007/s10822-011-9538-6. page Online First. 10.1007/s10822-011-9538-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lapelosa Mauro, Gallicchio Emilio, Arnold Gail Ferstandig, Arnold Eddy, Levy Ronald M. In silico vaccine design based on molecular simulations of rhinovirus chimeras presenting hiv-1 gp41 epitopes. J. Mol. Biol. 2009 Jan;385(2):675–691. doi: 10.1016/j.jmb.2008.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lapelosa Mauro, Arnold Gail Ferstandig, Gallicchio Emilio, Arnold Eddy, Levy Ronald M. Antigenic characteristics of rhinovirus chimeras designed in silico for enhanced presentation of HIV-1 gp41 epitopes. J .Mol. Biol. 2010 Apr;397(3):752–766. doi: 10.1016/j.jmb.2010.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].DeLorbe John E, Clements John H, Teresk Martin G, Benfield Aaron P, Plake Hilary R, Millspaugh Laura E, Martin Stephen F. Thermodynamic and structural effects of conformational constraints in protein-ligand interactions. entropic paradoxy associated with ligand preorganization. J. Am. Chem. Soc. 2009 Nov;131(46):16758–16770. doi: 10.1021/ja904698q. [DOI] [PubMed] [Google Scholar]
- [13].Bizzarri Marco, Marsili Simone, Procacci Piero. Intraligand hydrophobic interactions rationalize drug affinities for peptidyl-prolyl cis-trans isomerase protein. J. Phys. Chem. B. 2011 May;115(19):6193–6201. doi: 10.1021/jp110585p. [DOI] [PubMed] [Google Scholar]
- [14].Bizzarri Marco, Tenori Eleonora, Martina Maria Raffaella, Marsili Simone, Caminati Gabriella, Menichetti Stefano, Procacci Piero. New perspective on how and why immunophilin fk506-related ligands work. J. Phys. Chem. Lett. 2011 Oct;2(22):2834–2839. [Google Scholar]
- [15].Yang Chao-Yie, Sun Haiying, Chen Jianyong, Nikolovska-Coleska Zaneta, Wang Shaomeng. Importance of ligand reorganization free energy in protein-ligand binding-affinity prediction. J. Am. Chem. Soc. 2009 Sep;131(38):13709–13721. doi: 10.1021/ja9039373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gao Cen, Park Min-Sun, Stern Harry A. Accounting for ligand conformational restriction in calculations of protein-ligand binding affinities. Biophys. J. 2010 Mar;98(5):901–910. doi: 10.1016/j.bpj.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Andries Koen, Azijn Hilde, Thielemans Theo, Ludovici Donald, Kukla Michael, Heeres Jan, Janssen Paul, De Corte Bart, Vingerhoets Johan, Pauwels Rudi, de Béthune Marie-Pierre. Tmc125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004 Dec;48(12):4680–4686. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bèthune M-P, Pawels R, Das K, Clark AD, Jr., Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. In search of a novel anti-hiv drug: Multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1e)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (r278474, rilpivirine) J. Med. Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- [19].Das Kalyan, Clark Arthur D, Lewi Paul J, Heeres Jan, De Jonge Marc R, Koymans H. Lucien M H, Vinkers Maarten, Daeyaert Frederik, Ludovici Donald W, Kukla Michael J, De Corte Bart, Kavash Robert W, Ho Chih Y, Ye Hong, Lichtenstein Mark A, Andries Koen, Pauwels Rudi, De Béthune Marie-Pierre, Boyer Paul L, Clark Patrick, Hughes Stephen H, Janssen Paul A J, Arnold Eddy. Roles of conformational and positional adaptability in structure-based design of tmc125-r165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant hiv-1 variants. J Med Chem. 2004 May;47(10):2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- [20].Das Kalyan, Lewi Paul J, Hughes Stephen H, Arnold Eddy. Crystallography and the design of anti-aids drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside hiv-1 reverse transcriptase inhibitors. Prog Biophys Mol Biol. 2005 Jun;88(2):209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- [21].Das Kalyan, Bauman Joseph D, Clark Arthur D, Frenkel Yulia V, Lewi Paul J, Shatkin Aaron J, Hughes Stephen H, Arnold Eddy. High-resolution structures of hiv-1 reverse transcriptase/tmc278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci U S A. 2008 Feb;105(5):1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Frenkel Yulia Volovik, Clark Arthur D., Das Kalyan, Wang Yuh-Hwa, Lewi Paul J., Janssen Paul A. J., Arnold Eddy. Concentration and ph dependent aggregation of hydrophobic drug molecules and relevance to oral bioavailability. J. Med. Chem. 2005 Feb;48(6):1974–1983. doi: 10.1021/jm049439i. [DOI] [PubMed] [Google Scholar]
- [23].Frenkel Yulia Volovik, Gallicchio Emilio, Das Kalyan, Levy Ronald M, Arnold Eddy. Molecular dynamics study of non-nucleoside reverse transcriptase inhibitor 4-[[4-[[4-[(e)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (tmc278/rilpivirine) aggregates: correlation between amphiphilic properties of the drug and oral bioavailability. J Med Chem. 2009 Oct;52(19):5896–5905. doi: 10.1021/jm900282z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Okumura Hisashi, Gallicchio Emilio, Levy Ronald M. Conformational populations of ligand-sized molecules by replica exchange molecular dynamics and temperature reweighting. J. Comput. Chem. 2010;31:1357–1367. doi: 10.1002/jcc.21419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Frenkel Yulia. PhD thesis. Rutgers University; 2009. The roles of structural variability and amphiphilicity of TMC278/rilpivirine in mechanisms of HIV drug resistance avoidance and enhanced oral bioavailability. [Google Scholar]
- [26].Gallicchio Emilio, Levy Ronald M. Advances in all atom sampling methods for modeling protein-ligand binding affinities. Curr. Opin. Struct. Biol. 2011 Apr;21(2):161–166. doi: 10.1016/j.sbi.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mobley David L. Let’s get honest about sampling. J Comput Aided Mol Des. 2012 Jan;26(1):93–95. doi: 10.1007/s10822-011-9497-y. [DOI] [PubMed] [Google Scholar]
- [28].Lapelosa Mauro, Gallicchio Emilio, Levy Ronald M. Conformational transitions and convergence of absolute binding free energy calculations. J. Chem. Theory Comput. 2012;8:47–60. doi: 10.1021/ct200684b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gallicchio E, Levy RM. Prediction of sampl3 host-guest affinities with the binding energy distribution analysis method (BEDAM) J Comp Aided Mol Design. 2012 doi: 10.1007/s10822-012-9552-3. page In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gallicchio Emilio, Lapelosa Mauro, Levy Ronald M. Binding energy distribution analysis method (BEDAM) for estimation of protein-ligand binding affinities. J. Chem. Theory Comput. 2010 Sep;6(9):2961–2977. doi: 10.1021/ct1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mobley David L, Chodera John D, Dill Ken A. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations. J. Chem. Phys. 2006 Aug;125(8):084902. doi: 10.1063/1.2221683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mobley David L, Chodera John D, Dill Ken A. The confine-and-release method: Obtaining correct binding free energies in the presence of protein conformational change. J. Chem. Theory Comput. 2007;3(4):1231–1235. doi: 10.1021/ct700032n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lansdon Eric B, Brendza Katherine M, Hung Magdeleine, Wang Ruth, Mukund Susmith, Jin Debi, Birkus Gabriel, Kutty Nilima, Liu Xiaohong. Crystal structures of hiv-1 reverse transcriptase with etravirine (tmc125) and rilpivirine (tmc278): implications for drug design. J Med Chem. 2010 May;53(10):4295–4299. doi: 10.1021/jm1002233. [DOI] [PubMed] [Google Scholar]
- [34].Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Buelens Floris P., Grubmüller Helmut. Linear-scaling soft-core scheme for alchemical free energy calculations. J. Comput. Chem. 2012;33(1):25–33. doi: 10.1002/jcc.21938. [DOI] [PubMed] [Google Scholar]
- [36].Shirts Michael R, Chodera John D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008 Sep;129(12):124105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gallicchio Emilio, Levy Ronald M. chapter Recent Theoretical and Computational Advances for Modeling Protein-Ligand Binding Affinities. vol. 85. Academic Press; 2011. Advances in Protein Chemistry and Structural Biology; pp. 27–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gallicchio E, Levy RM. AGBNP: an analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. J. Comput. Chem. 2004;25:479–499. doi: 10.1002/jcc.10400. [DOI] [PubMed] [Google Scholar]
- [39].Gallicchio Emilio, Paris Kristina, Levy Ronald M. The agbnp2 implicit solvation model. J. Chem. Theory Comput. 2009 Sep;5(9):2544–2564. doi: 10.1021/ct900234u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jorgensen WL, Maxwell DS, Tirado-Rives J. Developement and testing of the opls all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- [41].Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparameterization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- [42].Banks JL, Beard JS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, Maple JR, Murphy R, Philipp DM, Repasky MP, Zhang LY, Berne BJ, Friesner RA, Gallicchio E, Levy RM. Integrated modeling program, applied chemical theory (IMPACT) J. Comp. Chem. 2005;26:1752–1780. doi: 10.1002/jcc.20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, Desmyter J, De Clercq E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-hiv compounds. J Virol Methods. 1988 Aug;20(4):309–321. doi: 10.1016/0166-0934(88)90134-6. [DOI] [PubMed] [Google Scholar]
- [44].Mihailescu Mihail, Gilson Michael K. On the theory of noncovalent binding. Biophys. J. 2004 Jul;87(1):23–36. doi: 10.1529/biophysj.103.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kim Jaegil, Straub John E. Generalized simulated tempering for exploring strong phase transitions. J. Chem. Phys. 2010 Oct;133(15):154101. doi: 10.1063/1.3503503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Min Donghong, Chen Mengen, Zheng Lianqing, Jin Yonghao, Schwartz Martin A., Amy Sang Qing-Xiang, Yang Wei. Enhancing qm/mm molecular dynamics sampling in explicit environments via an orthogonal-space-random-walk-based strategy. J. Phys. Chem. B. 2011 Apr;115(14):3924–3935. doi: 10.1021/jp109454q. [DOI] [PubMed] [Google Scholar]
- [47].Chang Chia-en A, Chen Wei, Gilson Michael K. Ligand configurational entropy and protein binding. Proc. Natl. Acad. Sci. USA. 2007 Jan;104(5):1534–1539. doi: 10.1073/pnas.0610494104. [DOI] [PMC free article] [PubMed] [Google Scholar]