

Abstract
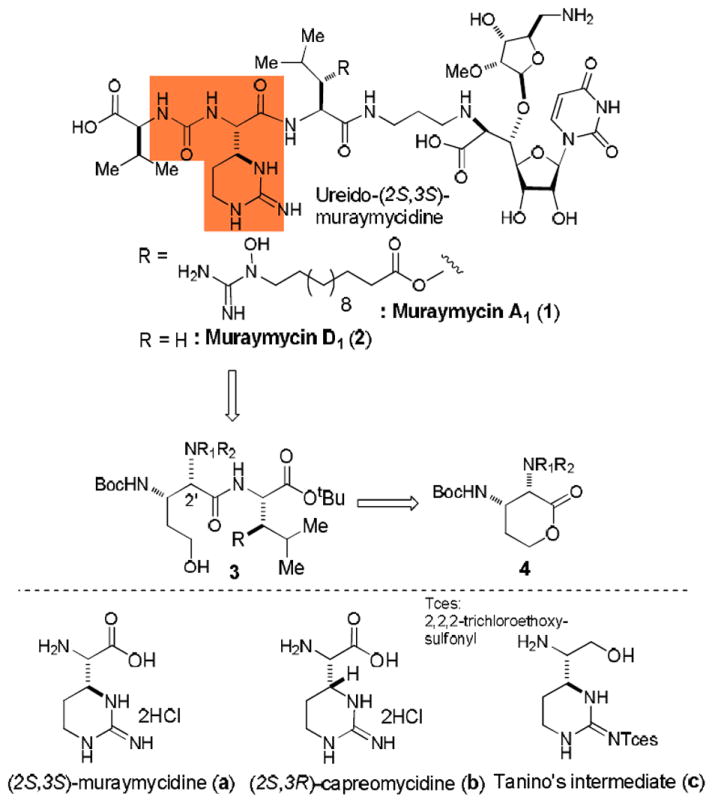
One of the key constituents of the muraymycins is the 6-membered cyclic guanidine, (2S,3S)-muraymycidine (or epi-capreomycidine). In order to diversify the structure of the oligo-peptide moiety of the muraymycins for thorough structure activity relationship studies, we have developed a highly stereoselective synthesis of ureido-muraymycidine derivatives with the lactone 4a.
INTRODUCTION
The increasing resistance among Gram-positive bacteria is concerning because they are responsible for one third of nosocomial infections.1 Multidrug resistance in Gram-positive cocci (i.e. staphylococci, pneumococci, and vancomycin resistance in enterococci) and mycobacteria has achieved great prominence in past 15 years.2 Over the last decade a few phase clinical drugs have been developed for Gram-positive bacterial infections.3 The ultimate goal of the development of the treatment of multidrug resistant strains is to find novel antibacterial agents which interfere with unexploited bacterial molecular targets.
Since peptidoglycan (PG) is an essential bacterial cell wall polymer, the machinery for PG biosynthesis provides a unique and selective target for antibiotic action. However, only a few enzymes in PG biosynthesis such as the penicillin binding proteins (PBPs) have been extensively studied.4 Thus, the enzymes associated with the early PG biosynthesis enzymes (i.e., MurA, B, C, D, E, and F, MraY, and MurG) are still considered to be a source of unexploited drug targets.5 Our interest in unexploited molecular targets related to PG biosynthesis is MraY,6 which catalyzes the transformation of UDP-N-acylmuramyl-l-alanyl-γ-d-glutamyl-meso-diaminopimelyl-d-alanyl-d-alanine (Park’s nucleotide) to prenylpyrophosphoryl-N-acylmuramyl-l-Ala-γ-d-glu-meso-DAP-d-Ala-d-Ala (lipid I).7 MraY is inhibited by nucleoside-based complex natural products such as muraymycin, liposidomycin, caprazamycin, and capuramycin. Muraymycins have been isolated from Streptmyces spp. and possess a common core structure of capuramycin, however, their structural diversity is observed in the ester moiety (R in Figure 1) and the appended C5’-ribose unit. Promising in vivo antibactericidal activity of muraymycin A1 (1) against S. aureus was highlighted by the Wyeth-Research groups.8 Thus, it is of our interest to validate the efficacy of 1 in vitro and in vivo against M. tuberculosis. In our effort on total synthesis of muraymycin A1 (1) and D1 (2), and their analogs for structure activity relationship studies against Gram-positive bacteria including M. tuberculosis, it is crucial to develop an efficient synthesis of (2S,3S)-2-amino-2-(2-iminohexahydropyrimidin-4-yl)acetic acid [(2S,3S)-muraymycidine (a in Figure 1)] derivative that can readily be incorporated in the syntheses of muraymycin analogs. The 6-membered cyclic guanidine moiety seems to be essential to exhibit strong antibactericidal activities for the muraymycidins.8b To date, several asymmetric syntheses of (2S,3R)-capreomycidine (b) have been reported for the total synthesis or biosynthetic studies of the capreomycins.9 On the other hand, very few synthetic efforts on (2S,3S)-muraymycidine derivative a have been reported.10 Recently, Tanino and co-workers reported a synthesis of the amino-alcohol possessing the cyclic guanidine c in which they accomplished the synthesis of c in 11 steps from an advanced intermediate with an overall yield of 7.9%.11 In the syntheses of the 6-membered cyclic guanidine containing α-amino acids reported to date, selectivities of the asymmetric induction to generate two consecutive chiral centers were moderate or very low and the synthetic schemes required multiple protecting-group manipulations. Herein, we report an efficient synthesis of the ureido-(2S,3S)-muraymycidine derivatives (highlighted in Figure 1) via the optically pure diamino lactone, (3S,4S)-3,4-diaminotetrahydro-2H-pyran-2-one derivative (4a).
Figure 1.
Retro-Synthesis of Muraymycins and Structures of Muraymycidine and Capreomycidine.
RESULTS AND DISCUSSION
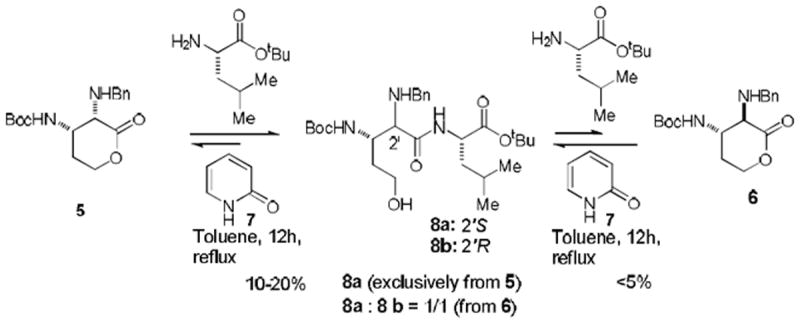
Our synthetic strategy to efficiently synthesize ureido-(2S,3S)-muraymycidine is illustrated in Figure 1. In our preliminary studies on the synthesis of the dipeptide intermediate 3 (Figure 1), we examined the efficiency of a strategy of lactone-opening of (2S,3S)-diamino-lactone 5 and (2R,3S)-diamino-lactone 6 with H-l-Leu-OtBu for the synthesis of 8 (Scheme 1).12 We observed that the lactone-opening of 6 with H-l-Leu-OtBu in the presence of 2(1H)-pyridinone (7) furnished a 1:1 mixture of the dipeptides 8a and 8b in very poor yield (< 5%). On the other hand, under the same conditions the lactone 5 yielded the desired 8a without contamination of 8b in 10-20% yield. These data clearly indicated that the lactone 6 was epimerized under the reaction conditions (2(1H)-pyridinone, toluene at reflux). Importantly, the stereochemistry of the lactone 5 was intact and the dipeptides 8a was not epimerized in the 2(1H)-pyridinone-catalyzed thermal lactone-opening reaction conditions. In addition, reactivity of the lactone 6 against H-l-Leu-OtBu was poorer than that of 5. Low conversion of the dipeptides 8 from the lactones in Scheme 1 can be attributed to the fact that δ-hydroxypentanoic acid derivatives tend to form δ-lactones even under weak acidic conditions.13 Indeed, the dipeptides 8a and 8b were relactonized to form 5 and 6, respectively during purification by a silica gel chromatography. In order to improve the conversion of 4 to 3 (Figure 1) and to realize epimerization of (2R,3S)-diamino-lactone derivatives (e.g. 6 in Scheme 1), we explored suitable N-protecting groups at the C2-position of lactone 4 (R1 and R2 in Figure 1) in which we expected that bulky N-protecting groups on (2R,3S)-diamino-lactone would prevent nucleophilic attack on the carbonyl group to form the undesired dipeptides possessing 2’R-configuration.
Scheme 1.
Preliminary Studies of Lactone-Opening Reactions.
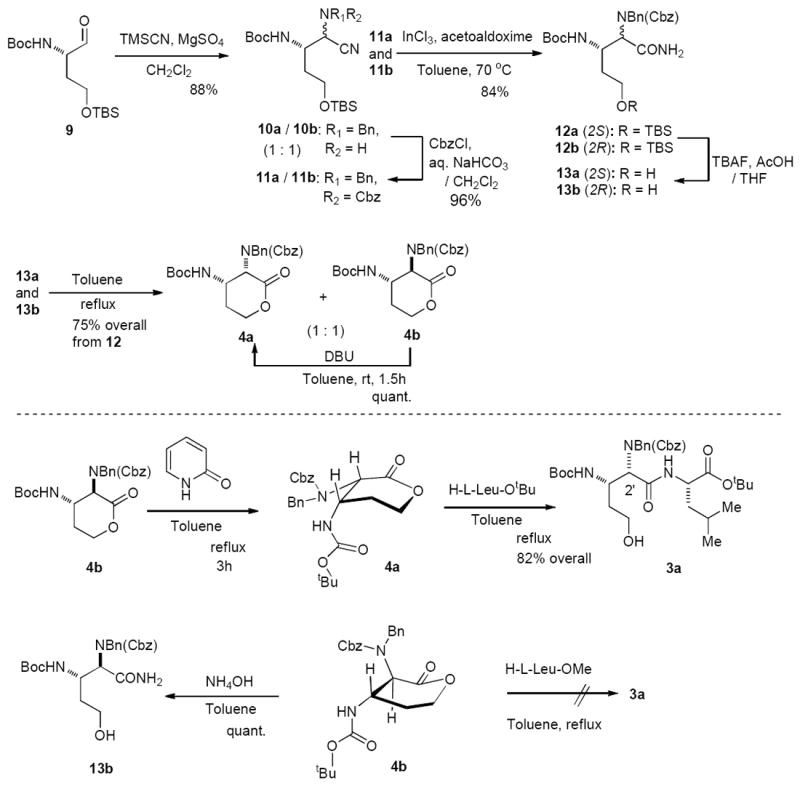
We first investigated chemical properties of N-benzyl-N-Cbz protected lactones 4a and 4b. The syntheses of 4a and 4b are illustrated in Scheme 2. The (2S)-aminobutanal derivative 9 was readily synthesized from (2S)-2-amino γ-butyrolactone according to the reported procedures.14 The aldehyde 9 was subjected to the Strecker reaction with benzyl amine and TMSCN to form a mixture of 2,3-diaminonitriles 10a and 10b.15 In our extensive reaction screening (9→10), the Strecker reaction conditions that provided 10 with greater than 80% yield are summarized in Table 1.
Scheme 2.
Syntheses of Lactones 4a and 4b, and Lactone-opening Reactions.
Table 1.
Strecker Reactions of the α-Amino-Aldedyde 9.
 | |||
|---|---|---|---|
| conditions | yield(%) | selectivity (10a : 10b) | |
| A |  |
88 | 1 : 3.5 |
| B | Ti(OiPr)4, HCO2H, H2O / CH2Cl2 | 85 | 1 : 1.5 |
| C | MgSO4 / CH2Cl2 | 88 | 1 : 1 |
The Strecker reactions with Lewis acids (e.g. ZnI2, Cu(OTf)2, Sn(OTf)2, La(OTf)3)16 provided the undesired product 10b as a major product with low yields (<30%) due probably to instability of the aldehyde 9 under strong Lewis acidic conditions. The reaction with the thiourea catalyst provided a 1 : 3.5 mixture of 10a and 10b in 88% yield (conditions A). A Ti-mediated Strecker reaction resulted in a 1 : 1.5 mixture of 10a and 10b (conditions B). It was found that the Stercker reaction of the benzyl imine 9 with TMSCN could be achieved via a convenient hydrating reagent, MgSO4 to furnish a 1 : 1 mixture of 10a and 10b in 88% yield (conditions C). The same Strecker reactions with the known chiral catalysts such as thioureas and salene-transition metal complexes resulted in the formation of a mixture of 10a and 10b in very poor yield (<30%) with low 10a/10b selectivity.17 The structures of 10b could unequivocally be determined by extensive 2D-NMR studies of 4b that were synthesized from 10b.18 With a 1 : 1 mixture of 10a and 10b in hand, we could establish the synthesis of 4a in 5 steps including epimerization of the C2-center (Scheme 2). Cbz protection of a mixture of the Strecker products was accomplished under buffered conditions in CH2Cl2 to afford 11a and 11b in 96% yield. Hydration of a mixture of the nitriles 11a and 11b was achieved by using InCl3 in the presence of acetaldoxime at 70 °C to afford a mixture of N-benzyl-N-Cbz protected primary amide 12a and 12b in 86% yield.19 Desilylation of 12 followed by a thermal lactonization of the resulting mixture of 13a and 13b in toluene at refluxing temperature provided 4a and 4b in 75% overall yield. The structure of (2R,3S)-diaminolactone 4b was established via extensive 2D-NMR techniques (vide supra). Gratifyingly, the undesired lactone 4b could be epimerized to the desired lactone 4a with DBU in quantitative yield. Epimerization of 4b to 4a was also observed under the conditions (2(1H)-pyridinone, toluene at reflux) used for opening of the lactone with H-l-Leu-OtBu (5→8a in Scheme 1). Under these conditions epimerization of 4b to 4a was completed in 3h. The synthesis of the desired dipeptide 3a could be accomplished via a one-pot operation in which H-l-Leu-OtBu was added into a solution of the completely epimerized lactone. We realized that the undesired lactone 4b could not be opened with H-l-Leu-OtBu even after prolonged reaction times. Among amine nucleophiles tested only NH3 could react with 4b at room temperature in the absence of 2(1H)-pyridinone to furnish 13b in quantitative yield. Therefore, the dipeptide 3a can be synthesized without contamination of the epimer of 3a from a 1: 1 mixture of 4a and 4b through epimerization.
In order to obtain more insight into epimerization followed by opening of the lactone 4b, we examined the lactone-opening reactions with a wide range of primary amines and α-amino acids (e.g. H-l-Leu-OtBu, H-l-Leu-OMe, Gly-OMe, H-l-Phe-OtBu, and others). Table 2 summarizes the selected examples of 2(1H)-pyridinone catalyzed lactone-opening reactions of a mixture of 4a and 4b (1 : 1). The lactone-opening reactions with the reactive amines (e.g. NH3, PhCH2NH2, C6H13NH2) were successfully achieved by addition of the amine nucleophiles after completion of epimerization (4b→4a) to afford the corresponding primary or secondary amides with 90-100% yield (conditions A). On the other hand, lactone-opening reactions with the α-amino acids did not require adding the nucleophiles after completion of the racemization of 4b. In all the reactions with α-amino acids summarized in Table 2, (2R,3S)-diaminolactone 4b did not react with salt free α-amino acid esters. Thus, the 2(1H)-pyridinone catalyzed epimerization of 4b to 4a could be completed in the presence of α-amino acid esters, and only (2S,3S)-diaminolactone 4a was smoothly reacted with α-amino acid esters. Lactone-opening reaction of a 1 : 1 mixture of 4a and 4b with H-l-Gly-OMe and 2(1H)-pyridinone in toluene at reflux for 5h furnished the desired 3c exclusively in 80% yield (conditions B).
Table 2.
Lactone-opening Reactions with Amines and α-Amino acid Esters.
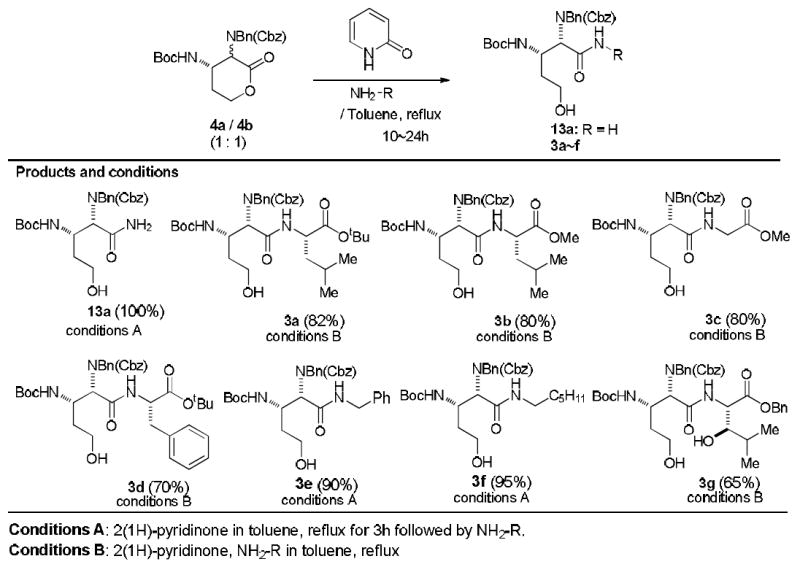 |
Under the same conditions (2S,3S)-benzyl-2-amino-3-hydroxy-4-methylpentanoate was reacted with a mixture of the lactones to furnish 3g in 65% yield (90% yield based on recovering 4a) without formation of the other diastereomers. The dipeptide 3g is a valuable intermediate for a total synthesis of muraymycin A1 (1). The dipeptides 3a-g in Table 2 were stable under weak acidic and basic conditions (pH 4.0-9.0) at room temperature; relactonizations of 3a-g to 4a were not observed. The plausible lowest-energy conformers of 4a and 4b are illustrated in Scheme 2. Those conformers were obtained via MM2 calculations20 and supported by the NOESY correlations.21 The syn-isomer 4a is significantly lower in energy than the anti-isomer 4b; the calculated free-energy difference was 6.86 Kcal/mol. Thus, we concluded that epimerization of 4b could readily be achieved by using 2(1H)-pyridinone in toluene at refluxing temperature. Due to the fact that the anti-isomer 4b exists as a pseudo-boat conformation, thus, the amino groups at the C2- and C3-positions hinder the nucleophilic additions of α-amino acids to the lactone carbonyl from both re- and si-faces.
Synthesis of the ureido-tripeptide 19 was achieved from the dipeptide 3a (Scheme 3). The primary alcohol of 3a was first protected as its acetate to afford 14 in quantitative yield. The N-Bn and N-Cbz groups of 14 were removed by hydrogenation to generate free amine which was subjected to the urea-forming reaction with the imidazolium salt 15 to furnish 16 in 70% overall yield.22 The Boc group of 16 was removed by using 50% TFA at 0 °C and the generated salt free amine was coupled with N,N’-di-tert-butoxycarbonyl-S-methyl isothiourea in the presence of Et3N, and HgCl2 to afford 17 in 62% yield.23 [tBu2Sn(OH)Cl]2 catalyzed deacetylation24 of 17 followed by an intramolecular Mitsunobu reaction with DIAD and PPh3 completed the synthesis of the fully-protected ureido-muraymycidine tripeptide 19 in 65% overall yield. The segment 19 possesses ideal protecting groups for a total synthesis of muraymycin D1 (2).
Scheme 3.
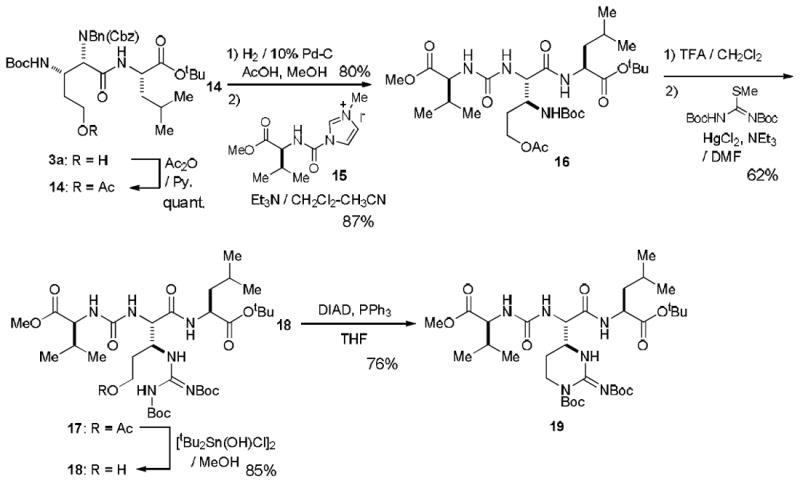
Synthesis of Ureido-Muraymycidine Tripeptide 19.
CONCLUSIONS
In summary, we present a highly stereoselctive synthesis of ureido-muraymycidine tripeptide 19 from a 1:1 mixture of the lactones 4a and 4b. δ-Lactones have not been widely utilized for functionalization of alcohols and amines due mainly to undesired reversible reactions.25 We realized that (2R,3S)-diaminolactone 4b can readily be epimerized to the stereoelectronically favored 4a with 2(1H)-pyridinone. In addition, the lactone 4b was not susceptible to lactone-opening reactions with α-amino acid derivatives. Thus, epimerization followed by selective lactone-opening reactions of a mixture of 4a and 4b with α-amino acids can be achieved in the presence of 2(1H)-pyridinone to furnish the corresponding dipeptides as a single diastereomer. Relactonizations of the δ-hydroxy dipeptides synthesized in this program were not observed under mild acidic and basic conditions, thus, high-yield dipeptide formations from the lactones 4a and 4b were achieved.26 The ureido-muraymycidine moiety of the muraymycins is an important functionality to show strong antibacterial activities.27 Thus, the ureido-(2S,3S)-muraymycidin (highlighted in Figure 1) should be retained as the intact stereochemistry for SAR studies of the muraymycins. As illustrated in Table 2, we will diversify the structure of muraymycin A1 and D1 for a thorough SAR study via the lactone-opening reactions of a 1:1 mixture of 4a and 4b, which could be synthesized from the known aldehyde 9 in over 50% overall yield. Total synthesis of muraymycins A1 and D1, and preliminary SAR of the muraymycins will be reported elsewhere.
EXPERIMENTAL SECTION
All reagents and solvents were of commercial grade and were used as received without further purification unless otherwise noted. Tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled from sodium benzophenone ketyl under an argon atmosphere prior to use. Methylene Chloride (CH2Cl2), acetonitrile (CH3CN), benzene, toluene and triethylamine (Et3N) were distilled from calcium hydride under an Argon atmosphere. Flash chromatography was performed with Whatman silica gel (Purasil 60 Å, 230-400 Mesh). Analytical thin-layer chromatography was performed with 0.25 mm coated commercial silica gel plates (EMD, Silica Gel 60F254) visualizing at 254 nm, or developed with ceric ammonium molybdate or anisaldehyde solutions by heating on a hot plate. 1H-NMR spectral data were obtained using 300, 400, and 500 MHz instruments. 13C NMR spectral data were obtained using 100 and125 MHz instruments. For all NMR spectra, δ values are given in ppm and J values in Hz.
(2S)-tert-Butyl (4-((tert-butyldimethylsilyl)oxy)-1-oxobutan-2-yl)carbamate (9)
MeNHOMe•HCl (1.89 g, 19.4 mmol) was suspended in CH2Cl2 (97 mL) and cooled to 0 °C. Me2AlCl (1M in hexanes, 19.4 mmol, 19.4 mL) was added dropwise and the reaction mixture was warmed to rt. After 1h, the reaction was cooled to 0 °C and a solution of 2S-[(tert-butyloxycarbonyl)amino]-4- butyrolactone (purchased from Aldrich, 1.95 g, 9.69 mmol) in CH2Cl2 (49 mL) was added via syringe pump over 15 min. The reaction mixture was stirred for 6h, and quenched with pH 8 phosphate buffer solution. The heterogeneous mixture was filtered and the filtrate was extracted with CH2Cl2. The generated Weinreb amide was used in the next step without purification. A stirred solution of the Weinreb amide (3.60 g, 13.70 mmol) and 2,6-lutidine (0.92 mL, 27.40 mmol) in CH2Cl2 (55 mL) was cooled to 0 °C. TBSOTf (3.46 mL, 15.10 mmol) was added and the reaction mixture was stirred for 30 min. The reaction was quenched with water and extracted with EtOAc. The extract was washed with 1N HCl, brine, dried over Na2SO4, and concentrated in vacuo. Purification by silica gel column chromatography gave (2S)-tert-butyl-(3,9,9,10,10-pentamethyl-4-oxo-2,8-dioxa-3-aza-9-silaundecan-5-yl)carbamate (4.71 g, 12.50 mmol, 91%) as an amorphous solid: TLC (hexanes:EtOAc 25:75): Rf = 0.7; [α]22D +0.4 (c = 0.9, CHCl3); IR (thin film) υmax = 3323 (br), 2930, 2858, 1716, 1669, 1500, 1390, 1366, 1253, 1173, 1101, 941, 836, 778 cm-1; 1H NMR (CDCl3, 400 MHz) δ 5.45 (d, J = 7.7 Hz, 1H), 4.74 (br s, 1H), 3.20 (s, 3H), 1.96 (dd, J = 4.7, 9.0 Hz, 1H), 1.79-1.61 (m, 1H), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 155.7, 79.4, 61.7, 59.9, 48.9, 34.9, 32.3, 28.5, 26.0, 18.3, -5.4; HRMS (ESI+): m/z calcd for C17H37N2O5Si [M+H], 377.2472; found 377.2472.
LiAlH4 (1M in THF, 15.90 mmol, 15.90 mL) was slowly added to a THF solution (40 mL) of (2S)-tert-butyl-(3,9,9,10,10-pentamethyl-4-oxo-2,8-dioxa-3-aza-9-silaundecan-5-yl)carbamate (3.00 g, 7.97 mmol) at 0 °C. After 1.5h, the reaction mixture was diluted with Et2O, and quenched with brine. The precipitates were filtered. The combined organic solution was dried over MgSO4, and evaporated. This was used for the next reaction without purification.
General Procedure of Strecker Reaction: Synthesis of a Mixture of 10a, 10b
A CH2Cl2 (72 mL) solution of aldehyde 9 (2.30 g, 7.24 mmol), benzylamine (0.87 mL, 7.97 mmol), and an excess of MgSO4 were stirred at room temparature for 2h. The solids were then filtered off and the mixture was concentrated in vacuo to give an intermediate imine. The imine was dissolved in CH2Cl2 (72 mL) and TMSCN (1.93 mL, 14.50 mmol) was then added. The reaction was stirred for 1h then poured into saturated NaHCO3 (aq.). The aqueous layer was extracted with CH2Cl2 (3×) and the combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 100:0 to 80:20) gave a mixture of 10a and 10b (2.77 g, 6.38 mmol, 88%) as an oil; IR (thin film) υmax = 3332 (br), 3065, 3031, 2932, 2228, 1714, 1505, 1367, 1255, 1172, 837 cm-1; 1H NMR (CDCl3, 400 MHz) δ 7.51-7.17 (m, 5H), 5.42 (d, J = 6.6 Hz, 0.5H), 5.21 (d, J = 8.4 Hz, 0.5H), 2.12-1.70 (m, 3H), 1.46 (s, 9H), 0.93-0.90 (m, 9H), 0.10-0.05 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ 155.7, 138.2, 128.6, 128.4, 128.4, 127.6, 119.0, 118.6, 80.0, 60.1, 59.8, 54.3, 52.1, 51.5, 51.3, 50.9, 34.1, 32.3, 28.5, 28.4, 26.1, 25.9, 25.9, 18.2, 18.2, -5.5; HRMS (ESI+): m/z calcd for C23H40N3O3Si [M+H], 434.2839; found 434.2839.
tert-Butyl ((3S,4S)-3-(benzylamino)-2-oxotetrahydro-2H-pyran-4-yl)carbamate (5)
tert-Butyl ((2S,3S)-1-amino-2-(benzylamino)-5-hydroxy-1-oxopentan-3-yl)carbamate (40 mg, 0.12 mmol) was dissolved in toluene (2 mL). The reaction mixture was stirred at reflux for 24h and cooled to rt. All volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 90:10 to 50:50) yielded product 5 as an amorphous white solid (25 mg, 0.08 mmol, 63%). Data for 5: TLC (hexanes:EtOAc 50:50): Rf = 0.4, [α]22D +36 (c = 0.85, CHCl3); IR (thin film) υmax = 3351 (br), 2979, 2929, 1693, 1524, 1459, 1418, 1364, 1259, 1170, 1075, 994, 873, 773, 739, 702 cm-1; 1H NMR (CDCl3, 500 MHz) δ 7.39-7.28 (m, 5H), 5.45 (s, 1H), 4.94 (s, 1H), 4.42 (m, 1H), 4.28 (m, 1H), 4.02 (d, J = 12.5 Hz, 1H), 3.91-3.77 (ddd, J = 13.5, 12.5, 14.0 Hz, 1H), 3.66 (m, 1H), 3.46-3.38 (dd, J = 7.5, 10.5 Hz, 1H), 2.51-2.49 (m, 1H), 1.98-1.94 (m, 2H), 1.48 (s, 9H); 13C NMR (CDCl3, 125 MHz) δ 172.9, 171.4, 156.2, 155.7, 139.5, 138.7, 128.7, 128.5, 128.1, 127.4, 79.9, 65.8, 65.5, 60.6, 57.9, 51.8, 50.5, 49.6, 45.0, 30.4, 28.4; HRMS (ESI+): m/z calcd for C17H24N2O4Na [M+Na], 343.1634; found 343.1637.
tert-Butyl ((3R,4S)-3-(benzylamino)-2-oxotetrahydro-2H-pyran-4-yl)carbamate (6)
tert-Butyl ((2R,3S)-1-amino-2-(benzylamino)-5-hydroxy-1-oxopentan-3-yl)carbamate (30 mg, 0.089 mmol) was dissolved in toluene (2 mL). The reaction mixture was stirred at reflux for 24h and cooled to rt. All volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 90:10 to 50:50) yielded product 6 as an amorphous white solid (15mg, 0.048 mmol, 53%). Data for 6: TLC (hexanes:EtOAc 50:50): Rf = 0.4, [α]22D -0.6 (c = 0.75, CHCl3); IR (thin film) υmax = 3351 (br), 2979, 2929, 1693, 1524, 1459, 1418, 1364, 1259, 1170, 1075, 994, 873, 773, 739, 702 cm-1; 1H NMR (CDCl3, 500 MHz) δ 7.29-7.19 (m, 5H), 5.36 (s, 1H), 4.83 (s, 1H), 4.39-4.32 (m, 1H), 4.23-4.18 (m, 1H), 3.92 (d, J = 12.0 Hz, 1H), 3.82-3.65 (ddd, J = 14.0, 12.5, 13.5 Hz, 1H), 3.55 (m, 1H), 3.37-3.28 (dd, J = 5.0, 10.5 Hz, 1H), 2.43-2.34 (m, 1H), 2.07-2.02 (m, 1H), 1.39 (s, 9H); 13C NMR (CDCl3, 125 MHz) δ 172.9, 171.4, 156.2, 155.7, 139.6, 138.8, 128.7, 128.5, 128.1, 127.5, 79.9, 65.5, 60.6, 57.9, 51.8, 50.5, 49.6, 45.0, 30.36, 28.4, 27.5; HRMS (ESI+): m/z calcd for C17H24N2O4Na [M+Na], 343.1634; found 343.1636.
Benzyl benzyl((2S)-2-((tert-butoxycarbonyl)amino)-4-((tert-butyldimethylsilyl)oxy)-1-cyanobutyl)carbamate (12)
To a stirred solution of a 1:1 mixture of 10a and 10b (342 mg, 0.79 mmol) in CH2Cl2 (4 mL) were added aq. sat. NaHCO3 (4 mL), and CbzCl (0.23 mL, 1.58 mmol). This reaction mixture was stirred for 1h at rt. Upon completion, the aqueous layer was extracted with CH2Cl2 (3×), and the combined organic extract was dried over Na2SO4, and concentrated in vacuo. The crude mixture was purified by silica gel column chromatography (hexanes:EtOAc 90:10 to 80:20) to yield a 1:1 mixture of the Cbz-protected products 11a and 11b (430 mg, 0.76 mmol, 96%) as an oil: TLC (hexanes:EtOAc 75:25): Rf = 0.65; IR (thin film) υmax = 3362 (br), 3034, 2956, 2858, 2247, 1716, 1498, 1471, 1367, 1254, 1171, 1102, 1003, 837, 777, 698 cm-1; 1H NMR (CDCl3, 400 MHz) δ 7.46-7.14 (m, 10H), 5.31 (br s, 1H), 5.17 (br s, 2H), 4.87-4.69 (m, 2H), 4.23-4.15 (m, 1H), 4.20 (d, J = 3.9 Hz, 1H), 3.83-3.54 (m, 2H), 1.74 (br s, 1H), 1.57 (br s, 1H), 1.45 (s, 9H), 0.92-0.89 (m, 9H), 0.10-0.03 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ 155.3, 137.0, 135.6, 128.8, 128.6, 128.4, 128.2, 127.7, 127.7, 127.1, 116.4, 80.1, 77.2, 68.6, 65.4, 59.7, 53.1, 50.3, 32.9, 28.6, 28.5, 28.5, 28.4, 28.4, 26.1, 26.1, 26.0, 26.0, 25.9, 18.2, -5.3, -5.4, -5.5; HRMS (ESI+): m/z calcd for C31H45N3O5SiNa [M+Na], 590.3026; found 590.3017.
To a stirred solution of 11a and 11b (a 1:1 mixture, 2.34 g, 4.12 mmol) in toluene (27 mL) were added InCl3 (137.0 mg, 0.62 mmol) and acetaldoxime (1.26 mL, 20.6 mmol). The reaction mixture was heated at 70 °C for 4h. Upon completion, the reaction was cooled to rt and all volatiles were removed. Purified by silica gel column chromatography (hexanes:EtOAc 90:10 to 50:50) to yield 12a and 12b (2.03 g, 3.47 mmol, 84%) as an amorphous white solid. Data for 12a: TLC (hexanes:EtOAc 50:50): Rf = 0.5; [α]22D -0.4 (c = 3.1, CHCl3); IR (thin film) υmax = 3350 (br), 2956, 2930, 2857, 2556, 2490, 2406, 1682, 1454, 1412, 1366, 1255, 1169, 1094, 1030, 991, 837, 775, 735, 697 cm-1; 1H NMR (CD3OD, 400 MHz) δ 7.47-7.02 (m, 10H), 6.37 (br s, 1 H), 5.07 (br s, 2H), 4.69 (d, J = 16.0 Hz, 1H), 4.65-4.39 (m, 2H), 4.21 (br s, 1H), 3.55 (br s, 2H), 1.64 (br s, 1H), 1.49 (br s, 1H), 1.39 (s, 9H), 0.86 (s, 9H), 0.00 (br s, 6H); 13C NMR (CD3OD, 100 MHz) δ173.6, 158.3, 157.7, 157.6, 139.7, 137.5, 129.4, 129.3, 129.1, 128.4, 128.0, 80.2, 68.9, 63.4, 61.1, 36.0, 28.8, 26.5, 19.1, -5.2, -5.2; HRMS (ESI+): m/z calcd for C31H47N3O6SiNa [M+Na], 608.3132; found 608.3128. Data for 12b: TLC (hexanes:EtOAc 50:50): Rf = 0.55; [α]22D +0.6 (c = 1.3, CHCl3); IR (thin film) υmax = 3339 (br), 2956, 2930, 2857, 2541, 2474, 2406, 1683, 1499, 1463, 1407, 1366, 1254, 1172, 1098, 1030, 837, 776, 735, 697, 665 cm-1; 1H NMR (CD3OD, 400 MHz) δ 7.50-6.90 (m, 9H), 6.28 (d, J = 9.8 Hz, 1H), 5.18 (br s, 1H), 5.13-4.94 (m, 2H), 4.75-4.56 (m, 2H), 4.50(d, J = 16.0 Hz, 1H), 4.34-4.17 (m, 1H), 3.73 (d, J = 6.3 Hz, 2H), 1.77 (br s, 1H), 1.60 (br s, 1H), 1.40 (s, 9H), 0.91 (s, 9H), 0.06 (s, 6H); 13C NMR (CD3OD, 100 MHz) δ 178.1, 173.3, 157.7, 129.3, 129.2, 128.9, 128.9, 127.8, 127.6, 80.1, 68.8, 63.6, 61.3, 35.9, 28.8, 26.5, 19.1, -5.3; HRMS (ESI+): m/z calcd for C31H48N3O6Si [M+H], 586.3312; found 586.3306. A mixture of 12a and 12b was used for the next reaction.
Benzyl ((3S)-1-amino-3-((tert-butoxycarbonyl)amino)-5-hydroxy-1-oxopentan-2-yl)(benzyl)carbamate (13)
To a stirred solution of 12a and 12b (1:1, 2.03 g, 3.47 mmol) and HOAc (0.01 mL, 1.74 mmol) in THF (18 mL) was added TBAF (1M in THF, 6.93 mL, 6.93 mmol). After 1h at rt, all volatiles were concentrated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 50:50 to 0:100) gave a mixture of 13a and 13b (1.48 g, 3.13 mmol, 90%). Data for 13a: TLC (hexanes:EtOAc 25:75): Rf = 0.15; [α]22D -0.3 (c 2.1, CHCl3); IR (thin film) υmax = 3340 (br), 3200 (br), 2963, 2932, 1683, 1498, 1454, 1406, 1367, 1255, 1169, 1123, 1054, 1028, 1005, 771, 739, 698 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 7.48-7.14 (m, 10H), 7.00 (br s, 1H), 6.62-6.47 (m, 1H), 5.03 (br s, 2H), 4.64 (br s, 2H), 4.48 (d, J = 16.0 Hz, 1H), 4.30 (br s, 1H), 3.98 (br s, 1H), 3.32 (br s, 2H), 1.57-1.43 (m, 2H), 1.37 (s, 9H); 13C NMR (DMSO-d6, 100 MHz) δ 171.8, 170.5, 155.3, 128.7, 128.1, 128.0, 127.7, 127.6, 127.1, 126.4, 77.9, 66.6, 60.6, 57.8, 47.6, 35.0, 31.2, 28.4, 28.2, 22.1, 13.9; HRMS (ESI+): m/z calcd for C25H33N3O6Na [M+Na], 494.2267; found 494.2268. Data for 13b: TLC (hexanes:EtOAc 25:75): Rf = 0.05; [α]22D + 0.2 (c = 1.1, CHCl3); IR (thin film) υmax = 3346 (br), 3201 (br), 2976, 2933, 1684, 1513, 1499, 1453, 1404, 1366, 1345, 1258, 1170, 1052, 1029, 768, 737, 698 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 7.82 (br s, 1H), 7.48-7.04 (m, 10H), 6.90 (d, J = 5.5 Hz, 1H), 6.53 (d, J = 9.8 Hz, 1H), 5.05-4.95 (m, 1H), 4.91 (br s, 1H), 4.63 (d, J = 16.8 Hz, 1H), 4.54 (d, J = 10.2 Hz, 1H), 4.44-4.30 (m, 2H), 4.07-3.89 (m,1 H), 3.54-3.35 (m, 2H), 1.54 (br s, 2H), 1.34 (s, 9H); 13C NMR (DMSO-d6, 100 MHz) δ 175.4, 170.6, 155.4, 128.0, 127.8, 127.4, 127.0, 126.2, 126.0, 77.5, 66.4, 61.5, 58.1, 46.9, 34.2, 31.2, 28.3, 28.2, 22.1, 13.9; HRMS (ESI+): m/z calcd for C25H33N3O6Na [M+Na], 494.2267; found 494.2263. A mixture of these alcohols was used for the next reaction.
(3S,4S)- and (3S,4R)-3,4-Diaminotetrahydro-2H-pyran-2-one (4a and 4b)
A mixture of benzyl 1-amino-3-((tert-butoxycarbonyl)amino)-5-hydroxy-1-oxopentan-2-yl)(benzyl)carbamates (117 mg, 0.248 mmol) was dissolved in toluene (5 mL). The reaction mixture was stirred at reflux for 24h and cooled to rt. All volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 90:10 to 50:50) yielded 4a and 4b as an amorphous white solid (94 mg, 0.21 mmol, 83%). Data for 4a: TLC (hexanes:EtOAc 50:50): Rf = 0.4, (benzene:acetone 80:20): Rf = 0.75; [α]22D+46 (c = 0.75, CHCl3); IR (thin film) υmax = 3353 (br), 2978, 2932, 1699, 1519, 1454, 1420, 1366, 1261, 1171, 1075, 993, 871, 771, 737, 700 cm-1; 1H NMR (benzene-d6, 400 MHz) δ 7.37-7.21 (m, 2H), 7.21-7.08 (m, 3H), 7.04 (br s, 5H), 6.62 (d, J = 7.7 Hz, 1H), 5.08-5.05 (d, J = 12.4 Hz, 1H), 4.94-4.91 (d, J = 12.4 Hz, 1H), 4.49-4.45 (d, J = 16.0 Hz, 1H), 4.39-4.35 (d, J = 15.6 Hz, 1H), 4.11 (br s, 1H), 4.02-3.97 (dd, J = 10.5 Hz, 1H), 3.42-3.39 (d, J = 11.2 Hz, 1H), 3.33-3.31(d, J = 6.8 Hz, 1H), 1.57-1.54 (d, J = 12.0 Hz, 1H), 1.38 (s, 9H), 1.02-0.97 (dd, J = 11.2, 13.8 Hz, 1H); 13C NMR (benzene-d6, 100 MHz) δ 165.6, 157.7, 155.1, 136.7, 136.1, 128.3, 128.1, 127.8, 127.5, 127.3, 127.2, 126.7, 126.5, 78.6, 67.7, 63.8, 59.2, 52.7, 47.9, 27.9, 27.7; HRMS (ESI+): m/z calcd for C25H30N2O6Na [M+Na], 477.2002; found 477.1999. Data for 4b: TLC (hexanes:EtOAc 50:50): Rf = 0.4, (benzene:acetone 80:20): Rf = 0.8; [α]22D -0.8 (c 2.5, CHCl3); IR (thin film) υmax = 3368 (br), 2977, 2361, 1745, 1712, 1500, 1474, 1455, 1426, 1392, 1366, 1250, 1169, 1079, 992, 911, 865, 737, 699 cm-1; 1H NMR (CDCl3, 400 MHz) δ 7.40-7.18 (m, 10H), 5.12 (d, J = 14.5 Hz, 2H), 4.54 (d, J = 16.0 Hz, 2H), 4.31-4.10 (m, 2H), 4.01 (br s, 1H), 3.71-3.47 (m, 1H), 2.23-1.92 (m, 1H), 1.92-1.61 (m, 1H), 1.37 (s, 9H); 13C NMR (CDCl3, 100 MHz) δ 169.0, 154.9, 136.0, 135.3, 129.0, 128.8, 128.7, 128.7, 128.5, 128.3, 128.1, 127.8, 127.7, 79.9, 77.2, 68.7, 67.9, 66.5, 66.2, 62.1, 61.4, 54.0, 53.1, 49.7, 49.0, 30.2, 28.4; HRMS (ESI+): m/z calcd for C25H30N2O6Na [M+Na], 477.2002; found 477.1999.
Epimerization of the lactone 4b to 4a
To a stirred solution of the lactone 4b (20 mg, 0.044 mmol) in toluene (2 ml) was added DBU (14 mg, 0.088 mmol). After 1.5h at rt, all volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 90:10 to 50:50) gave the lactone 4a as a single diastereomer.
General procedure for Lactone-Opening Reaction
To a stirred solution of a 1:1 mixture of the lactones 4a and 4b (1 eq.) in toluene (0.4 M) were added 2(1H)-pyridinone (1-2 eq.), and α-amino acid (2-3 eq.). The reaction mixture was heated at reflux for 5h and cooled to rt. All volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 65:35 to 50:50) yielded the desired product (procedure A). To a stirred solution of a 1:1 mixture of the lactone 4a and 4b (1 eq.) in toluene (0.4 M) was added 2(1H)-pyridinone (1-2 eq.). After 3h at 130 °C, free amine (2-3 eq.) was added. The reaction mixture was heated at 130 °C for an additional 5h and cooled to rt. All volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 65:35 to 50:50) yielded the desired product (procedure B).
(S)-Methyl-2-((2S,3S)-2-(benzyl((benzyloxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)-5-hydroxypentanamido)-4-methylpentanoate (3b)
The dipeptide 3b was synthesized using general procedure A; 3b (22 mg, 0.036 mmol, 80%). A colorless oil: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.9 (c = 0.6, CHCl3); IR (thin film) υmax = 3330 (br), 2969, 2956, 1730, 1634, 1487, 1458, 1415, 1172, 1110, 1052, 1021, 773, 741, 692 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 9.15-8.92 (m, 1H), 7.61-7.05 (m, 8H), 6.90 (d, J = 6.7 Hz, 2H), 6.75-6.52 (m, 1H), 5.03 (br s, 1H), 4.95 (br s, 1H), 4.73 (d, J = 5.9 Hz, 2H), 4.49-4.37 (m, 2H), 4.37-4.24 (m, 1H), 4.12-3.99 (m, 1H), 3.69-3.59 (s, 3H), 3.46 (d, J = 7.4 Hz, 2H), 1.73-1.47 (m, 5H), 1.41 (s, 9H), 0.92 (d, J = 5.9 Hz, 3H), 0.86 (d, J = 6.3 Hz, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 185.6, 172.6, 168.6, 156.4, 155.4, 139.9, 136.5, 127.9, 127.7, 127.2, 127.0, 126.8, 126.2, 125.9, 77.7, 66.5, 61.6, 57.9, 51.9, 47.6, 47.2, 33.7, 28.1, 24.2, 22.9, 21.0; HRMS (ESI+): m/z calcd for C32H46N3O8 [M+H], 600.3285; found 600.3288.
(2S,3S)-Methyl-2-((2S,3S)-2-(benzyl((benzyloxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)-5-hydroxypentanamido)-3-hydroxy-4-methylpentanoate (3g)
The dipeptide 3g was synthesized using general procedure A; 3g (20 mg, 0.029 mmol, 65%). A colorless oil: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.6 (c = 0.8, CHCl3); IR (thin film) υmax = 3350 (br), 3015, 2975, 2962, 1728, 1510, 1464, 1412, 1182, 1109, 1063, 1035, 769, 735, 687 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 7.42-7.28 (m, 6H), 7.28-6.86 (m, 9H), 5.17-4.85 (m, 5H), 4.76-4.53 (m, 2H), 4.44-4.26 (m, 2H), 4.01 (d, J = 5.9 Hz, 1H), 3.36 (m, 2H), 1.54 (br s, 2H), 1.34 (s, 9H), 1.24, (br s, 1H), 0.94-0.71 (m, 6H); 13C NMR (DMSO-d6, 100 MHz) δ 169.3, 135.8, 128.5, 128.2, 127.9, 127.8, 127.7, 127.2, 127.1, 126.9, 125.9, 66.5, 66.4, 65.9, 58.0, 54.9, 31.3, 28.2, 28.1, 22.2, 13.9 ; HRMS (ESI+): m/z calcd for C38H50N3O9 [M+H], 692.3547; found 692.3543.
(S)-tert-Butyl-2-((2S,3S)-2-(benzyl((benzyloxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)-5-hydroxypentanamido)-3-phenylpropanoate (3d)
The dipeptide 3d was synthesized using general procedure A; 3d (21 mg, 0.031 mmol, 70%). A clear oil: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.8 (c = 0.7, CHCl3); IR (thin film) υmax = 3340 (br), 3004, 2969, 2964, 1732, 1642, 1630, 1485, 1474, 1412, 1171, 1118, 1063, 1037, 781, 755, 691 cm-1;1H NMR (DMSO-d6, 400 MHz) δ 8.92-8.83 (m, 1H), 7.17 (dd, J = 7.0, 15.7 Hz, 13H), 6.94-6.79 (m, 2H), 6.54-6.43 (m, 1H), 5.11-4.82 (m, 3H), 4.71-4.55 (m, 2H), 4.42-4.20 (m, 3H), 4.04-3.87 (m, 2H), 3.46-3.37 (m, 1H), 3.30-3.25 (m, 1H), 3.03-2.92 (m, 1H), 2.87-2.80 (m, 1H), 1.60-1.50 (m, 1H), 1.43-1.18 (m, 18H), 1.13-1.02 (m, 1H); 13C NMR (DMSO-d6, 100 MHz) δ 185.6, 170.4, 170.3, 170.1, 168.3, 156.4, 155.4, 155.3, 137.1, 136.1, 127.9, 127.7, 127.4, 127.1, 126.9, 126.5, 126.2, 125.8, 80.9, 80.8, 77.6, 66.4, 61.6, 57.6, 47.7, 46.9, 37.1, 31.3, 27.6, 22.1, 13.9; HRMS (ESI+): m/z calcd for C38H50N3O8 [M+H], 676.3598; found 676.3597.
Methyl 2-((2S,3S)-2-(benzyl((benzyloxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)-5-hydroxypentanamido)acetate (3c)
The dipeptide 3c was synthesized using general procedure A; 3c (20 mg, 0.036 mmol, 80%). A colorless oil: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.8 (c = 0.8, CHCl3); 1H NMR (DMSO-d6, 400 MHz) δ 8.55-8.38 (m, 1H), 7.45-7.01 (m, 10H), 6.52 (m, 1H), 5.05 (br s, 2H), 4.78-4.65 (m, 2H), 4.51-4.48 (m, 1H), 4.34 (s, 1H), 3.99 (d, J = 8.5 Hz, 1H), 3.70-3.66 (m, 2H), 3.61 (s, 3H), 3.36-3.25 (m, 2H), 1.48 (m, 2H), 1.36 (s, 9H); 13C NMR (CDCl3, 125 MHz) δ 169.6, 156.9, 137.7, 135.9, 128.6, 128.3, 127.3, 79.9, 68.3, 60.6, 58.4, 52.4, 48.7, 40.9, 36.5, 28.4; HRMS (ESI+): m/z calcd for C28H38N3O8 [M+H], 544.2659; found 544.2657.
Benzyl-benzyl((2S,3S)-1-(benzylamino)-3-((tert-butoxycarbonyl)amino)-5-hydroxy-1-oxopentan-2-yl)carbamate (3e)
The amide 3e was synthesized using general procedure B; 3e (23 mg, 0.04 mmol, 90%). A white foam: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.4 (c = 0.3, CHCl3); IR (thin film) υmax = 3345 (br), 3301, 2952, 2954, 1712, 1638, 1452, 1472, 1411, 1169, 1110, 1052, 1033, 774, 748, 691 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 8.97 (br s, 1H), 7.48-6.81 (m, 15H), 6.62-6.52 (m, 1H), 4.98 (m, 1H), 4.91 (m, 1H), 4.68-4.62 (m, 1H), 4.58-4.51 (m, 1H), 4.47-4.34 (m, 2H), 4.20-3.97 (m, 3H), 3.49-3.34 (m, 2H), 1.61-1.46 (m, 2H), 1.34 (s, 9H); 13C NMR (DMSO-d6, 100 MHz) δ 185.6, 168.4, 156.3, 155.4, 139.6, 138.8, 136.4, 128.2, 128.0, 127.8, 127.5, 127.4, 127.0, 126.8, 126.1, 126.0, 77.5, 66.4, 61.7, 58.0, 47.4, 46.9, 42.1, 34.0, 31.2, 28.3, 28.2, 22.1; HRMS (ESI+): m/z calcd for C32H40N3O6 [M+H], 562.2917; found 562.2917.
Benzyl ((2S,3S)-1-amino-3-((tert-butoxycarbonyl)amino)-5-hydroxy-1-oxopentan-2- yl)(benzyl)carbamate (13a)
The amide 13a was synthesized using general procedure B; 13a (21 mg, 0.044 mmol, 100%). A white foam: Data for 13a: TLC (hexanes:EtOAc 25:75): Rf = 0.15; [α]22D -0.3 (c 2.1, CHCl3); IR (thin film) υmax = 3340 (br), 3200 (br), 2963, 2932, 1683, 1498, 1454, 1406, 1367, 1255, 1169, 1123, 1054, 1028, 1005, 771, 739, 698 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 7.48-7.14 (m, 10H), 7.00 (br s, 1H), 6.62-6.47 (m, 1H), 5.03 (br s, 2H), 4.64 (br s, 2H), 4.48 (d, J = 16.0 Hz, 1H), 4.30 (br s, 1H), 3.98 (br s, 1H), 3.32 (br s, 2H), 1.57-1.43 (m, 2H), 1.37 (s, 9H) ; 13C NMR (DMSO-d6, 100 MHz) δ 171.8, 170.5, 155.3, 128.7, 128.1, 128.0, 127.7, 127.6, 127.1, 126.4, 77.9, 66.6, 60.6, 57.8, 47.6, 35.0, 31.2, 28.4, 28.2, 22.1, 13.9; HRMS (ESI+): m/z calcd for C25H33N3O6Na [M+Na], 494.2267; found 494.2268.
Benzyl-benzyl((2S,3S)-3-((tert-butoxycarbonyl)amino)-5-hydroxy-1-(octylamino)-1-oxopentan-2-yl)carbamate (3f)
The amide 3f was synthesized using general procedure B; 3f (24 mg, 0.042 mmol, 95%). A white foam: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.4 (c = 0.6, CHCl3); IR (thin film) υmax = 3355 (br), 2951, 2944, 1632, 1451, 1462, 1112, 1051, 1023, 772, 751, 695 cm-1; 1H NMR (DMSO-d6, 400 MHz) δ 8.45-8.34 (m, 1H), 7.53-7.28 (m, 2H), 7.19 (d, J = 7.0 Hz, 5H), 7.06 (d, J = 6.7 Hz, 2H), 6.92 (br s, 1H), 6.56-6.45 (m, 1H), 4.99 (br s, 1H), 4.93-4.85 (m, 1H), 4.66-4.57 (m, 1H), 4.51 (br s, 1H), 4.39 (d, J = 17.6 Hz, 2H), 4.07-3.95 (m, 1H), 3.39 (d, J = 5.9 Hz, 2H), 2.86 (br s, 2H), 1.56-1.42 (m, 2H), 1.34 (s, 9H), 1.30-1.16 (m, 12H), 0.88-0.83 (m, 3H); 13C NMR (DMSO-d6, 100 MHz) δ 185.7, 168.1, 156.3, 155.4, 139.8, 136.5, 128.1, 127.7, 127.5, 127.2, 127.1, 126.9, 126.4, 126.1, 125.9, 77.6, 66.4, 61.8, 58.1, 47.3, 46.9, 31.3, 28.5, 26.4, 22.1, 13.9; HRMS (ESI+): m/z calcd for C33H50N3O6 [M+H], 584.3700; found 584.3701.
(S)-tert-Butyl 2-((2S,3S)-2-(benzyl((benzyloxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)-5-hydroxypentanamido)-4-methylpentanoate (3a)
To a stirred solution of a 1:1 mixture of 4a and 4b (37.0 mg, 0.081 mmol) and 2(1H)-pyridinone (15.4 mg, 0.16 mmol) in toluene (0.4 mL) was added H-L-Leu-OtBu (60.0 mg, 0.413 mmol). The reaction mixture was stirred at 130 °C for 5h and cooled to rt. Purification by silica gel column chromatography (hexanes:EtOAc 65:35 to 50:50) provided 3a (43 mg, 0.067 mmol, 82%) as a colorless oil: TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D +0.8 (c = 0.5, CHCl3); IR (thin film) υmax = 3340 (br), 2965, 2954, 1732, 1634, 1498, 1464, 1410, 1172, 1112, 1057, 1031, 771, 745, 697 cm-1; 1H NMR (CDCl3, 500 MHz) δ 7.27-7.33 (m, 10H), 6.67 (s, 1H), 5.32 (s, 1H), 5.19 (m, 2H), 4.48-4.56 (m, 3H), 4.28 (m, 2H), 3.64 (br s, 2H), 1.75 (br s, 2H), 1.45 (s, 9H), 1.29 (m, 3H), 0.89 (br s, 6H); 13C NMR (CDCl3, 125 MHz) δ 171.5, 157.5, 156.3, 137.4, 135.9, 128.8, 128.6, 128.2, 127.9, 81.9, 79.8, 68.1, 64.7, 58.8, 51.5, 47.1, 41.4, 35.3, 28.4, 27.9, 24.9, 22.7, 22.1; HRMS (ESI+): m/z calcd for C35H52N3O8 [M+H], 642.3754; found 642.3756.
(S)-tert-Butyl 2-((2S,3S)-5-acetoxy-2-(benzyl((benzyloxy)carbonyl)amino)-3-((tert-butoxycarbonyl)amino)pentanamido)-4-methylpentanoate (14)
To a stirred solution of the dipeptide 3a (43 mg, 0.067 mmol) in pyridine (0.1 mL) was added acetic anhydride (0.1 mL). The reaction mixture was stirred for 6h at rt, and all volatiles were evaporated in vacuo. Purification by silica gel column chromatography (hexanes:EtOAc 80:20 to 50:50) gave 14 (44 mg, 0.064 mmol, 95%) as a white foam. TLC (hexanes:EtOAc 50:50): Rf = 0.7; [α]22D +0.8 (c = 0.75, CHCl3); IR (thin film) υmax = 3336, 2974, 1739, 1718, 1677, 1516, 1453, 1367, 1246, 1152, 1043, 751, 697 cm-1; 1H NMR (CDCl3, 500 MHz) δ 7.21-7.30 (m, 10H), 6.72 (s, 1H), 5.18 (s, 2H), 4.61 (d, J = 16.5 Hz, 1H), 4.48 (s, 1H), 4.42 (d, J = 11.0 Hz, 1H), 4.31(m, 1H), 4.19 (m, 2H), 4.12 (m, 2H), 2.05(s, 3H), 1.98 (m, 2H), 1.81 (m, 2H), 1.46 (m, 1H), 1.44 (s, 9H), 1.39 (s, 9H), 0.95 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 171.4, 171.1, 168.1, 157.5, 155.5, 137.8, 135.9, 128.5, 128.1, 127.9, 127.7, 127.3, 81.8, 79.5, 67.9, 64.3, 63.8, 61.4, 51.7, 51.4, 51.3, 50.4, 47.4, 42.1, 41.5, 30.8, 29.8, 28.4, 27.9, 24.9, 24.8, 22.8, 22.4, 22.2, 20.9; HRMS (ESI+): m/z calcd for C37H54N3O9 [M+H], 684.3860; found 684.3863.
(S)-1-((1-Methoxy-3-methyl-1-oxobutan-2-yl)carbamoyl)-3-methyl-1H-imidazol-3-ium-iodide (15)
A stirred suspension of the HCl•H-L-Val-OH (500 mg, 2.99 mmol) in CH2Cl2 (0.3M) were added Et3N (0.92 ml, 6.58 mmol) and DMAP (37 mg, 0.3 mmol) and N,N-carbonyldiimidazole (534 mg, 3.29 mmol) was added at 0 °C. The reaction mixture was warmed to rt, and stirred for 2 h. The reaction mixture was diluted with CH2Cl2, and the combined organic phase was washed with H2O, brine, and dried over Na2SO4. The crude material was purified by basic alumina column chromatography to give (S)-methyl 2-(1H-imidazole-1-carboxamido)-3-methylbutanoate as colorless oil (587 mg, 2.61 mmol, 87%). TLC (CHCl3:MeOH 90:10): Rf = 0.25; 1H NMR (CDCl3, 500 MHz) δ 8.19 (s, 1H), 7.43 (s 1H), 6.75 (d, J = 8.0 Hz, 1H), 4.59 (m, 1H), 3-81 (s, 3H), 2.28 (m, 1H), 1.01 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 172.1, 148.9, 136.1, 130.7, 115.9, 58.8, 52.6, 31.4, 18.9, 17.9; HRMS (ESI+): m/z calcd for C10H15N3O3, 225.1113; found 225.1115.
To a stirred solution of (S)-methyl 2-(1H-imidazole-1-carboxamido)-3-methylbutanoate (587 mg, 2.61 mmol) in dry CH3CN (13 ml) were added Et3N (0.40 ml, 2.88 mmol) and MeI (0.18 ml, 2.88 mmol). The reaction mixture was stirred at rt for 18h. All volatiles were evaporated in vacuo. The resulting light yellow solid 15 (959 mg) was used in the following reactions without further purification.
(2R,6S,7S)-Methyl-7-(2-acetoxyethyl)-6-(((S)-1-(tert-butoxy)-4-methyl-1-oxopentan-2-yl)-carbamoyl)-2-isopropyl-11,11-dimethyl-4,9-dioxo-10-oxa-3,5,8-triazadodecan-1-oate (16)
To a stirred solution of 14 (100.0 mg, 0.15 mmol) in MeOH (30 mL) was added AcOH (20 μL) and Pd(OH)2/C (25 wt% 10 mg) under N2. H2 gas was introduced via double-folded balloon and the reaction mixture was stirred for 6h under H2. Upon completion, the solution was filtered through Celite. The crude mixture was dissolved in EtOAc, and washed with aq. sat. NaHCO3. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to yield the desired primary amine. To a stirred solution of the primary amine in CH2Cl2 (0.5 mL) was added a solution of imidazolium salt 15 (2.5 eq) in CH3CN (0.5 mL) at rt. After 12h, the reaction mixture was diluted with EtOAc, and washed with NaHCO3 (aq.), brine, and dried over Na2SO4. The crude material was purified by silica gel column chromatography to give 16 as white foam (79.0 mg, 0.13 mmol, 87%). TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D -2.5 (c = 0.5, CHCl3); IR (thin film) υmax = 3356, 2983, 1741, 1684, 1631, 1572, 1275, 1260, 764 cm-1; 1H NMR (CDCl3, 500 MHz) δ 6.82 (d, J = 8.0 Hz, 1H), 6.36 (s, 1H), 5.13 (d, J = 7.6 Hz, 1H), 5.01 (d, J = 8.0 Hz, 1H), 4.51 (m, 1H), 4.42 (m, 1H), 4.38 (m, 1H), 4.38 (m, 1H), 4.29 (m, 1H), 3.74 (s, 3H), 2.09 (m, 1H), 2.06 (s, 3H), 1.93 (d, J = 5.5 Hz, 1H), (m, 2H), 1.56-1.58 (m, 2H), 1.46 (s, 9H), 1.42 (s, 9H), 0.94 (m, 12H); 13C NMR (CDCl3, 125 MHz) δ 173.3, 171.8, 171.5, 170.2, 157.7, 81.8, 80.1, 61.5, 58.4, 57.3, 52.1, 51.5, 50.1, 41.6, 41.2, 31.4, 28.3, 28.2, 27.9, 24.9, 24.8, 22.8, 22.7, 22.1, 21.1, 19.1, 17.9; HRMS (ESI+): m/z calcd for C29H52N4O10, 616.3683; found 616.3686.
(8S,9S,13R)-Methyl-8-(2-acetoxyethyl)-9-(((S)-1-(tert-butoxy)-4-methyl-1-oxopentan-2-yl)carbamoyl)-6-((tert-butoxycarbonyl)amino)-13-isopropyl-2,2-dimethyl-4,11-dioxo-3-oxa-5,7,10,12-tetraazatetradec-5-en-14-oate (17)
To a stirred solution of 16 (20.0 mg, 0.033 mmol) was added cooled TFA (50% in CH2Cl2, 1 mL). The reaction mixture was stirred at 0 °C for 30 min, warmed to rt, diluted with CH2Cl2 (10 mL), and poured into NaHCO3 solution. The aqueous layer was extracted with CHCl3 (3×). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to provide the free amine as an oil: TLC (CH2Cl2:MeOH 90:10): Rf = 0.25. To a stirred solution of the free amine (15.0 mg, 0.028 mmol) in DMF (0.3 mL) were added N,N’-di-tert-butoxycarbonyl-S-methyl isothiourea (12.2 mg, 0.042 mmol), Et3N (8.5 mg, 0.084 mmol), and HgCl2 (11.4 mg, 0.042 mmol). The reaction mixture was stirred at rt for 14h. Upon completion, the reaction mixture was diluted with EtOAc, and filtered through celite. The combined organic phase was washed with brine (2×), dried over Na2SO4 and concentrated in vacuo. The crude material was purified by silica gel column chromatography (hexanes:EtOAc 50:50) to give 17 (13.0 mg, 0.018 mmol, 62%). TLC (hexanes:EtOAc 50:50): Rf = 0.25; [α]22D -3.16 (c = 0.3, CHCl3); IR (thin film) υmax = 3284, 2978, 1792, 1726, 1639, 1614, 1540, 1369, 1264, 1100, 1058, 737 cm-1; 1H NMR (CDCl3, 500 MHz) δ 11.34 (s, 1H,), 8.65 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H), 5.03 (d, J = 9.0 Hz, 1H), 4.61 (t, J = 7.0 Hz, 1H), 4.42 (m, 3H), 4.16 (m, 1H), 4.12 (m, 1H), 3.73 (s, 3H), 2.18 (s, 1H), 2.08 (m, 2H), 2.07 (s, 3H), 2.05 (m, 2H), 1.98 (m, 2H), 1.43-1.53 (m, 27H), 0.91 (m, 12H); 13C NMR (CDCl3, 125 MHz) δ 173.3, 171.5, 171.2, 170.1, 157.9, 156.7, 152.7, 83.6, 81.4, 79.9, 61.3, 60.4, 58.1, 57.8, 51.9, 51.5, 51.4, 41.4, 31.6, 29.1, 28.4, 28.1, 27.9, 24.9, 22.9, 21.9, 20.9, 19.2, 17.9; HRMS (ESI+): m/z calcd for C35H62N6O12, 758.4426; found 758.4428.
(S)-tert-Butyl-2-((tert-butoxycarbonyl)imino)-4-((4R,8S,11S)-11-isobutyl-4-isopropyl-14,14-dimethyl-3,6,9,12-tetraoxo-2,13-dioxa-5,7,10-triazapentadecan-8-yl)tetrahydropyrimidine-1(2H)-carboxylate (19)
To a stirred solution of 17 (12.5 mg, 0.016 mmol) in MeOH (0.5 mL) was added [tBu2Sn(OH)Cl]2 (0.0008 mmol). After 12h at rt, all volatiles were evaporated in vacuo. The crude product was passed through silica gel pad (hexanes:EtOAc 50:50) to provide the free alcohol 18 (10.0 mg, 0.014 mmol, 85%) as a white foam. TLC (hexanes:EtOAc 50:50): Rf = 0.20; [α]22D -2.13 (c = 0.5, CHCl3); IR (thin film) υmax = 3273 (br), 2929, 2927, 1732, 1645, 1556, 1430, 1369, 1264, 1210, 1155, 1050, 1075, 1020, 764, 669 cm-1; 1H NMR (CDCl3, 500 MHz) δ 11.39 (s, 1H), 8.82 (d, J = 9.0 Hz, 1H), 6.56 (d, J = 9.0 Hz, 1H), 6.25 (s, 1H), 5.14 (d, J = 9.0 Hz, 1H), 4.62 (m, 1H), 4.59 (m, 1H), 4.59 (m, 2H), 4.39 (m, 1H), 3.73 (s, 3H), 3.68 (m, 1H), 3.55 (t, 1H), 2.16 (m, 1H), 1.95 (m, 1H), 1.74 (s, 9H), 1.51 (s, 9H), 1.47 (s, 9H), 0.96 (m, 12H); 13C NMR (CDCl3, 100 MHz) δ 173.4, 172.0, 170.1, 162.6, 157.5, 156.6, 152.6, 83.6, 82.0, 79.6, 58.4, 57.9, 56.4, 52.1, 51.3, 51.1, 41.6, 34.9, 31.3, 29.7, 28.2, 28.1, 27.9, 24.8, 23.0, 21.6, 19.1, 17.9; HRMS (ESI+): m/z calcd for C33H61N6O11 [M+H], 717.4398; found 717.4399. The alcohol 18 (10.0 mg, 0.014 mmol) was dissolved in THF (0.3 mL), and PPh3 (36.7 mg, 0.14 mmol) and DIAD (28.3 mg, 0.14 mmol) were added. The reaction mixture was stirred at rt for 18h. Upon completion, the crude mixture was concentrated in vacuo and the crude product was purified by silica gel chromatography (hexanes:EtOAc 60:40) to yield 19 (7.0 mg, 0.011 mmol, 76%) as a colorless oil. TLC (hexanes:EtOAc 50:50): Rf = 0.30; [α]22D -2.15 (c = 0.1, CHCl3); IR (thin film) υmax = 3276, 2933, 1728, 1637, 1617, 1544, 1372, 1276, 1105, 1063, 739 cm-1; 1H NMR (CD3OD, 500 MHz) δ 4.63 (m, 1H), 4.55 (m, 2H), 4.33 (m, 1H), 4.21 (d, J = 5.0 Hz, 1H), 3.72 (s, 3H), 3.63 (m, 1H), 3.59 (m, 1H), 2.13 (m, 1H), 1.91 (m, 1H), 1.62 (m, 2H), 1.59 (m, 1H), 1.56 (s, 9H), 1.46 (s, 18H), 1.31 (m, 2H), 0.87-0.98 (m, 12H); 13C NMR (CD3OD, 100 MHz) δ 174.7, 173.2, 172.1, 164.1, 160.1, 158.3, 153.9, 84.8, 82.8, 80.6, 59.8, 58.9, 57.3, 52.9, 52.5, 41.5, 36.4, 32.2, 28.6, 28.3, 28.2, 25.9; HRMS (ESI+): m/z calcd for C33H58N6O10 [M+H], 699.4293; found 699.4291.
Supplementary Material
Acknowledgments
The National Institutes of Health is greatly acknowledged for financial support of this work (AI084411-02). We also thank University of Tennessee for generous financial support. NMR data were obtained on instruments supported by the NIH Shared Instrumentation Grant.
Footnotes
1H and 13C NMR spectra, and NOESY data. This material is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES AND NOTES
- 1.Grenet K, Guillemot D, Jarlier V, Moreau B, Dubourdieu S, Ruimy R, Armand-Lefevre L, Brau P, Andremont A. Emerg Infect Dis. 2004;10:1150. doi: 10.3201/eid1006.031015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaynes R, Edwards JR. Clin Infect Dis. 2005;41:848. doi: 10.1086/432803. [DOI] [PubMed] [Google Scholar]
- 3.Sekigichi J, Fujino T, Saruto K, Kawano F, Takami J, Miyazaki H, Kuratsuji T, Yoshikura H, Kirikae T. Jpn J Infect Dis. 2003;56:133. [PubMed] [Google Scholar]
- 4.Wright GD. Science. 2007;315:1373. doi: 10.1126/science.1140374. [DOI] [PubMed] [Google Scholar]
- 5.(a) Cudic P, Behenna DC, Yu MK, Kruger RG, Szwczuk LM, McCafferty DG. Bioorg Med Chem Lett. 2001;11:3107. doi: 10.1016/s0960-894x(01)00653-9. [DOI] [PubMed] [Google Scholar]; (b) Helm JS, Hu Y, Chen L, Gross B, Walker S. J Am Chem Soc. 2003;125:11168. doi: 10.1021/ja036494s. [DOI] [PubMed] [Google Scholar]; (c) Bachelier A, Mayer R, Klein CD. Bioorg Med Chem Lett. 2006;16:5605. doi: 10.1016/j.bmcl.2006.08.021. [DOI] [PubMed] [Google Scholar]; (d) Antane S, Caufield CE, Hu W, Keeney D, Labthavikul P, Morris K, Naughton SM, Petersen PJ, Rasmussenm BA, Singh G, Yang Y. Bioorg Med Chem. 2006;16:176. doi: 10.1016/j.bmcl.2005.09.021. [DOI] [PubMed] [Google Scholar]; (e) Taha MO, Atallah N, Al-Bakri AG, Pradis-Bleau C, Zalloum H, Yonis KS, Levesque PC. Bioorg Med Chem. 2008;16:1218. doi: 10.1016/j.bmc.2007.10.076. [DOI] [PubMed] [Google Scholar]; (f) Bryskier A, Dini AC. Antimicr Agents: Antibacter Antifung. 2005:377. [Google Scholar]; (g) Kotnik M, Humljan J, Contreras-Martel C, Oblak M, Kristan K, Hervé M, Blanot D, Urleb U, Gobec S, Dessen A, Solmajer T. J Mol Biol. 2007;370:107. doi: 10.1016/j.jmb.2007.04.048. [DOI] [PubMed] [Google Scholar]; (h) Perdih A, Kovac A, Wolber G, Blanot D, Gobec S, Solmajer T. Bioorg Med Chem Lett. 2009;19:2668. doi: 10.1016/j.bmcl.2009.03.141. [DOI] [PubMed] [Google Scholar]; (i) Humljan J, Kotnik M, Contreras-Martel C, Blanot D, Urleb U, Dessen A, Solmajer T, Gobec S. J Med Chem. 2008;51:7486. doi: 10.1021/jm800762u. [DOI] [PubMed] [Google Scholar]
- 6.Bugg TDH, Timothy DH, Lloyd AJ, Roper DI. Infect Disorders: Drug Targets. 2006;6:85. doi: 10.2174/187152606784112128. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kurosu M, Mahapatra S, Narayanasamy P, Crick DC. Tetrahedron Lett. 2007;48:799. [Google Scholar]; (b) Kurosu M, Narayanasamy P, Crick DC. Heterocycles. 2007;72:339. [Google Scholar]; (c) Kurosu M, Li K. J Org Chem. 2008;73:9767. doi: 10.1021/jo801408x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kurosu M, Li K, Crick DC. Org Lett. 2009;11:2393. doi: 10.1021/ol900458w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. J Am Chem Soc. 2002;124:10260. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]; (b) Lin YI, Li Z, Francisco GD, McDonald LA, Davis RA, Singh G, Yang Y, Mansour TS. Bioorg Med Chem Lett. 2002;12:2341. doi: 10.1016/s0960-894x(02)00469-9. [DOI] [PubMed] [Google Scholar]
- 9.(a) Johnson AW, Bycroft BW, Cameron D. J Chem Soc C. 1971:3040. [Google Scholar]; (b) Wakamiya T, Mizuno K, Ukita T, Teshima T, Shiba T. Bull Chem Soc Jpn. 1978;51:850. [Google Scholar]; (c) Shiba T, Ukita T, Mizuno K, Teshima T, Wakamiya T. Tetrahedron Lett. 1977;18:2681. [Google Scholar]; (d) Jackson MD, Gould SJ, Zabriskie TM. J Org Chem. 2002;67:2934. doi: 10.1021/jo016182c. [DOI] [PubMed] [Google Scholar]; (e) DeMong DE, Williams RM. J Am Chem Soc. 2003;125:8561. doi: 10.1021/ja0351241. [DOI] [PubMed] [Google Scholar]
- 10.Sarabia F, Martín-Ortiz L. Tetrahedron. 2005;61:11850. [Google Scholar]
- 11.(a) Tanino T, Ichikawa S, Shiro M, Matsuda A. J Org Chem. 2010;75:1366. doi: 10.1021/jo9027193. [DOI] [PubMed] [Google Scholar]; (b) Tanino T, Ichikawa S, Matsuda A. Org Lett. 2011;13:4028. doi: 10.1021/ol201527k. [DOI] [PubMed] [Google Scholar]
- 12.The optically pure lactones 5 and 6 were synthesized via the synthetic procedures summarized in Scheme 2 with a minor modification.
- 13.Barrett AGM, Bezuidenhoudt BCB, Dhanak D, Gasiecki AF, Howell AR, Lee AC, Russell MA. J Org Chem. 1989;54:3321. [Google Scholar]
- 14.(a) Shimizu T, Osako K, Nakata T. Tetrahedron Lett. 1997;38:2685. [Google Scholar]; (b) Ragains JR, Winkler JD. Org Lett. 2006;8:4437. doi: 10.1021/ol061577+. [DOI] [PubMed] [Google Scholar]
- 15.Herranz R, Suarez-Gea ML, Vinuesa S, Garcia-Lopez MT. J Org Chem. 1993;58:5186. [Google Scholar]
- 16.Merino P, Marqués-López E, Tejero T, Herrera RP. Tetrahedron. 2009;65:1219. [Google Scholar]
- 17.Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968. doi: 10.1038/nature08484.Vachal P, Jacobsen EN. Org Lett. 2000;2:867. doi: 10.1021/ol005636+.Martens J. Chem Cat Chem. 2010;2:379. and references cited therein.
- 18.The NOESY correlations of one of most lower-energy conformers of 4b are illustrated in Supporting Information
- 19.Kim ES, Lee HS, Kim SH, Kim JN. Tetrahedron Lett. 2010;51:1589. [Google Scholar]
- 20.MM2 calculations were performed using SPARTAN on a Silicon Graphics O2 work station.
- 21.See Supporting Information.
- 22.(a) Batey RA, Santhakumar V, Yoshina-Ishii C, Taylor SD. Tetrahedron Lett. 1998;39:6267. [Google Scholar]; (b) Maresca KP, Hillier SM, Femia FJ, Keith D, Barone C, Joyal JL, Zimmerman CN, Kozikowski AP, Barrett JA, Eckelman WC, Babich JW. J Med Chem. 2009;52:347. doi: 10.1021/jm800994j. [DOI] [PubMed] [Google Scholar]
- 23.Kim KS, Qian L. Tetrahedron Lett. 1993;34:7677. [Google Scholar]
- 24.Orita A, Hamada Y, Nakano T, Toyoshima S, Otera J. Chem Eur J. 2001;7:3321. doi: 10.1002/1521-3765(20010803)7:15<3321::aid-chem3321>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 25.A limited number of examples have been found in the literature: for example, see. Fernández MM, Diez A, Rubiralta M, Montenegro E, Casamitjana N. J Org Chem. 2002;67:7587. doi: 10.1021/jo025999i.
- 26.Openshaw HC, Whittaker N. J Chem Soc. 1969:89. doi: 10.1039/j39690000089. [DOI] [PubMed] [Google Scholar]
- 27.Yamashita A, Norton E, Petersen PJ, Rasmussen BA, Singh G, Yang Y, Mansour TS, Ho DM. Bioorg Med Chem Lett. 2003;13:3345. doi: 10.1016/s0960-894x(03)00671-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.