

Syntenin-1 is recruited to the human immunodeficiency virus (HIV)-induced capping area but vanishes once the viral particles have entered the cell. Syntenin-1 limits HIV-1 infection. Moreover, syntenin-1 depletion specifically increases the HIV-1 entry step without affecting viral attachment to the cell surface. Silencing of syntenin-1 expression blocks actin polymerization triggered by HIV-1 contact and enhances phosphatidylinositol 4,5-bisphosphate production.
Abstract
Syntenin-1 is a cytosolic adaptor protein involved in several cellular processes requiring polarization. Human immunodeficiency virus type 1 (HIV-1) attachment to target CD4+ T-cells induces polarization of the viral receptor and coreceptor, CD4/CXCR4, and cellular structures toward the virus contact area, and triggers local actin polymerization and phosphatidylinositol 4,5-bisphosphate (PIP2) production, which are needed for successful HIV infection. We show that syntenin-1 is recruited to the plasma membrane during HIV-1 attachment and associates with CD4, the main HIV-1 receptor. Syntenin-1 overexpression inhibits HIV-1 production and HIV-mediated cell fusion, while syntenin depletion specifically increases HIV-1 entry. Down-regulation of syntenin-1 expression reduces F-actin polymerization in response to HIV-1. Moreover, HIV-induced PIP2 accumulation is increased in syntenin-1–depleted cells. Once the virus has entered the target cell, syntenin-1 polarization toward the viral nucleocapsid is lost, suggesting a spatiotemporal regulatory role of syntenin-1 in actin remodeling, PIP2 production, and the dynamics of HIV-1 entry.
INTRODUCTION
The adaptor protein syntenin-1 was originally described in relation to its association with the syndecan receptor and its recycling (Grootjans et al., 1997; Zimmermann et al., 2005). It was also termed melanoma differentiation-associated gene-9 (mda-9) for its up-regulated expression in several types of tumors, which correlates with high invasiveness (Sarkar et al., 2004, 2008). Structurally, syntenin-1 contains a regulatory N-terminal region and two tandem postsynaptic density protein, Drosophila disk large, and zonula occludens-1 (PDZ) domains. The N-terminal region is composed of two Tyr-based domains: one ITIM and one ITAM domain, the ITIM domain being important for CXCR4-mediated T-cell migration (Sala-Valdes et al., 2012). PDZ domains are involved in the formation of macromolecular complexes that often link transmembrane proteins to the actin cytoskeleton, mediating their clustering and polarized subcellular distribution (Fanning and Anderson, 1999; Brone and Eggermont, 2005; Hirbec et al., 2005; Ludford-Menting et al., 2005). PDZ domains interact with short amino acid sequences at the C-terminal end of plasma membrane proteins. Three consensus sequences bind PDZ domains: class I ([S/T]-X-Φ), class II (Φ-X-Φ), and class III ([D/E]-X-Φ), where X is any amino acid and Φ is a hydrophobic residue (Chimura et al., 2011). Through its PDZ domains, syntenin-1 binds to transmembrane proteins, such as CD6 (Gimferrer et al., 2005) and the tetraspanin CD63 (Latysheva et al., 2006); signaling proteins, such as the tyrosine kinase Src (Boukerche et al., 2008); and the actin-linker protein merlin (Jannatipour et al., 2001). Recruitment of syntenin-1 to the plasma membrane is also regulated through its PDZ-mediated interaction with the lipid second messenger phosphatidylinositol 4,5-bisphosphate (PIP2; Zimmermann, 2006).
Syntenin-1 has a cytosolic distribution, being enriched in vesicles and F-actin structures (Zimmermann et al., 2001). It is implicated in several actin-polarized processes, such as cell migration, immune synapse formation, intracellular trafficking, cell-surface targeting, axonal outgrowth, and synaptic transmission (Gimferrer et al., 2005; Hirbec et al., 2005; Zimmermann et al., 2005; Beekman and Coffer, 2008; Sarkar et al., 2008). We recently reported the involvement of syntenin-1 in CXCR4-mediated T-chemotaxis and T-APC antigen presentation, in which it regulates Rac-triggered actin polymerization (Sala-Valdes et al., 2012).
CD4/CXCR4-derived signaling and Rac-induced actin remodeling are also important for human immunodeficiency virus type 1 (HIV-1) infection (Pontow et al., 2004; Carter et al., 2009). Soon after HIV-1 contact with CD4+ T-cells, the viral envelope glycoprotein complex gp120/gp41 (Env) engages with CD4 and CXCR4, inducing several signaling pathways that trigger receptor clustering (Jimenez-Baranda et al., 2007; Barrero-Villar et al., 2009) in a structure called “cap” (Dianzani et al., 1995; Nguyen et al., 2005), local actin polymerization (Iyengar et al., 1998; Jolly et al., 2004), T-cell activation (Popik et al., 1998; Hiscott et al., 2001), and HIV-1 replication (Popik and Pitha, 2000; Mettling et al., 2008). These rearrangements enhance viral entry (Wu and Yoder, 2009; Juno and Fowke, 2010) by supplying an optimal density of CD4/CXCR4 complexes through actin-mediated surface reorganization (Iyengar et al., 1998; Doms, 2000; Jolly et al., 2004). Thus the actin-binding proteins moesin and filamin-A accumulate at the F-actin–enriched capping areas and influence CD4/CXCR4 clustering (Jimenez-Baranda et al., 2007; Barrero-Villar et al., 2009). Moreover, we previously showed that HIV-1 gp120, through its interaction with CD4, induces local production of PIP2 by activating PI4P5K-Iα (Barrero-Villar et al., 2008).
After viral attachment to the plasma membrane, the local F-actin accumulation can constitute a physical barrier for entry of the viral nucleocapsid (Vasiliver-Shamis et al., 2009). In this regard, CXCR4-gp120 interaction was recently shown to activate cofilin, which promotes actin clearance, facilitating nucleocapsid entry into the cytoplasm (Yoder et al., 2008).
In this study, we assessed the possible role of syntenin-1 as a mediator of CD4/CXCR4 clustering and actin remodeling during HIV-1 infection. Our data indicate that syntenin-1 associates with CD4 and negatively regulates HIV-1 infection by affecting the dynamic reorganization of actin cytoskeleton and PIP2 production triggered upon HIV-1 contact, specifically at the viral entry step.
RESULTS
Syntenin-1 is recruited to the plasma membrane by HIV-1 Env
PDZ proteins commonly determine the polarization of cellular events (Fanning and Anderson, 1999; Brone and Eggermont, 2005; Ludford-Menting et al., 2005). Because HIV-1 contact with target cells triggers the polarization of cellular components (Dianzani et al., 1995; Nguyen et al., 2005), we investigated whether the PDZ protein syntenin-1 was recruited upon HIV-1 contact with target CD4+ T-cells (CEM-T4). In the absence of HIV-1 virus, syntenin-1 displayed a diffuse cytosolic pattern with low clustering at the plasma membrane (Figure 1A). In contrast, after incubation of T-cells with free HIV-1 particles, syntenin-1 was clearly recruited toward the CD4/CXCR4 capping area (Figure 1, A and E). To confirm the specificity of syntenin-1 recruitment, we assessed CD45 distribution and observed that it did not accumulate at virus-induced capping areas (Figure 1, B and E). In addition, to assess that syntenin-1 recruitment was Env-mediated, target CEM-T4 cells were incubated with a Jurkat clone expressing the T-tropic HIV-1 envelope HxB (Env+ HxBc2 cells). Syntenin-1 was found at the Env-driven cell contacts, together with CD4 (Figure 1, C and E). We also cocultured target CEM-T4 cells with T-cells directly infected with HIV–green fluorescent protein (GFP) virus (MT4-HIV-GFP). In these cocultures, syntenin-1 accumulated at cell–cell contacts, areas in which viral synapses are established (Figure 1, D and E), but not at unspecific Env-independent cell–cell contacts (Figure1E, right-hand white bar; exemplified in Supplemental Figure S1). Quantification confirmed significant accumulation of syntenin-1 at contacts with HIV-1 viral particles and infected cells, in comparison with CD45 (Figure 1E).
FIGURE 1:

Syntenin-1 is recruited by HIV-1 envelope glycoproteins. (A) CEM-T4 cells incubated with or without HIV-1 particles for 30 min were fixed and immunostained for syntenin-1 and CD4 (HP2/6). Summative projections of confocal stack images are shown. Scale bars: 5 μm. Surface plots show fluorescence distribution and intensity for corresponding images. (B) CEM-T4 cells incubated with HIV-1 particles for 30 min were fixed and immunostained for CD4 (CD4v4-FITC) and CD45 (D3/9). Summative projections of confocal stack images are shown. Scale bar: 5 μm. Surface plots show fluorescence distribution and intensity for corresponding images. (C) Immunofluorescence microscopy images of target CEMT4 cells incubated for 2 h with Env+ HxBc2 cells (CMAC stained, blue) and stained for syntenin-1 and CD4 (HP2/6). Scale bars: 5 μm. (D) Target J77 cells were incubated for 2 h with HIV-1–infected T-cells (MT4-HIV-GFP) and immunostained for syntenin-1 and CD4 (HP2/6). Projections of confocal stack images are shown. Scale bar: 5 μm. (E) Quantification of syntenin-1 accumulation at HIV-1-induced capping areas (HIV-wt), contacts between target cells and HIV-infected cells (MT4-J77), or contacts between target cells and Env+ cells (HxB-J77). The control (white bars) for HIV wild-type and MT4-J77 represent the recruitment of CD45. The control (white bar) for HxB-J77 corresponds to the recruitment of syntenin at Env-independent cell–cell contacts (between two target J77 cells). The chart represents the mean ± SD of three independent experiments; more than 100 conjugates were counted in each condition.
Syntenin-1 associates with the cytoplasmic tail of CD4
PDZ domains usually bind to specific consensus sequences at the C-terminal ends of transmembrane proteins (Chimura et al., 2011). To determine whether syntenin-1 forms part of the CD4/CXCR4 complex at the capping areas, we first searched the CD4 and CXCR4 cytoplasmic domain sequences for potential PDZ-binding motifs. A class I PDZ-binding consensus sequence (Ser-Pro-Ile) was detected at the C-terminal end of CD4, suggesting potential direct interaction with syntenin-1 via its PDZ domains. To test this, we immobilized glutathione S-transferase (GST)-fused hemagglutinin (HA)- or Myc-tagged syntenin-1 on Sepharose beads, and incubated them with human primary lymphoblast lysates. GST-syntenin-HA and GST-syntenin-Myc, but not GST alone, precipitated CD4 (Figure 2A). Confirming this interaction, anti-CD4 antibody immunoprecipitated endogenous syntenin-1 from CEM T-cell lysates; syntenin-1 was detected with anti–syntenin-1 antibody as a double band of around 32 kDa (Figure 2B). To demonstrate the occurrence of CD4–syntenin-1 association in the context of HIV-1 infection, we performed reverse immunoprecipitation assays with Jurkat T-cells previously incubated with free viral particles. Immunoprecipitation of syntenin-1, followed by immunodetection of CD4, detected constitutive CD4–syntenin-1 interaction that was maintained during HIV-1 contact (Figure 2C). Together, these data suggest that syntenin-1 interacts with CD4 before and during HIV-1 infection.
FIGURE 2:
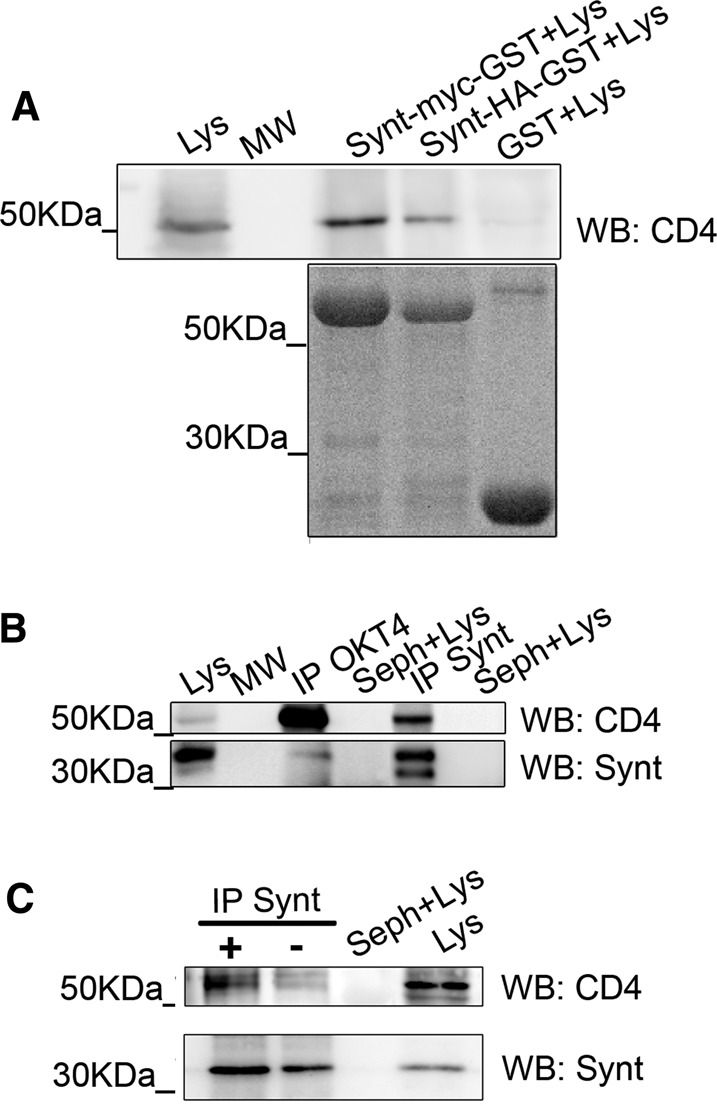
Syntenin-1 interacts with CD4 during HIV infection of T-cells. (A) T-lymphoblast cell lysates (Lys) were pulled down with GST, GST-syntenin-Myc, or GST-syntenin-HA, and blotted with anti-CD4 mAb MEM241. (B) CEM T-cell lysates were immunoprecipitated with anti-CD4 mAb OKT4 (IP-OKT4) and anti-syntenin pAb (IP-Synt), and then blotted for syntenin-1 (pAb) and CD4 (mAb MEM241). Total lysates (Lys) and Sepharose beads incubated with lysates (Seph+Lys) are shown. (C) Jurkat J77 T-cells incubated with or without HIV-1 particles were lysed, immunoprecipitated with anti–syntenin-1 pAb, and blotted with anti-CD4 mAb MEM241.
Syntenin-1 overexpression inhibits HIV-1–induced cell fusion and viral production
To assess the specific role of syntenin-1 during HIV-1 infection, target T-cells were transfected with GFP-tagged, wild-type syntenin-1 (syntenin-GFP) or a mutated protein, syntenin-Y1-GFP (Synt-Y1-GFP), which carries a Y4>F point mutation in the ITIM motif of the regulatory N-terminal region. Phosphorylation of endogenous syntenin-1 at this tyrosine residue has been shown to be essential for CXCR4-induced chemotaxis (Sala-Valdes et al., 2012). Syntenin-GFP and Synt-Y1-GFP showed a similar distribution to endogenous syntenin-1, being enriched at Env-driven cell–cell contacts (Figure 3A) and HIV-induced capping areas (Figure 3B and data not shown). Target J77 cells overexpressing these GFP-tagged proteins were infected with free HIV-1 particles, and viral production was measured 3 d later by p24 enzyme-linked immunosorbent assay (ELISA). Cells overexpressing syntenin-GFP produced lower viral titers than control cells (transfected with GFP alone). In contrast, overexpression of the mutated syntenin-1 had no effect on HIV-1 production (Figure 3C).
FIGURE 3:

Overexpression of wild-type syntenin-1, but not a phosphorylation-defective mutant, in target T-cells impairs HIV-1 infection. (A) CEM-T4 cells overexpressing syntenin-GFP or Synt-Y1-GFP (containing the Y4>F point mutation) were incubated with Env+ HxBc2 cells (tracked with CMAC, blue) for 2 h, fixed, and stained for CD4 (HP2/6 mAb). Summative projections of confocal stack images are shown. Scale bars: 5 μm. (B) CEM-T4 cells overexpressing wild-type syntenin-1 (syntenin-GFP) were incubated for 30 min with or without HIV-1 particles, fixed, and stained for CD4 (HP2/6 mAb). Projections of confocal stack images are shown. Scale bars: 5 μm. (C) HIV-1 infection is inhibited by syntenin-GFP but not by Synt-Y1-GFP. Infection was measured as viral production, quantified as p24 viral protein content in supernatants (ELISA). Data are the fold induction relative to control cells transfected with GFP alone (mean ± SD of four experiments performed in triplicate; p = 0.0058). (D) Cell fusion triggered by HIV-1 envelope is reduced in cells overexpressing syntenin-GFP. As a control, syncytia formation was blocked by incubation with the fusion inhibitor T20 (10 nM). Data are the fold induction relative to GFP-transfected cells (mean ± SD of four experiments performed in duplicate; p = 0.031). (E) Cells were transfected with GFP, syntenin-GFP, or Synt-Y1-GFP; immunoprecipitated with pAb anti-GFP; and blotted for CD4 (MEM241) and GFP (mAb).
To investigate the potential effect of syntenin-1 plasma membrane recruitment on HIV-1–induced cell fusion, we measured Env-mediated cell fusion by flow cytometry. Env+ HxBc2 cells were labeled with red intracellular tracker (CMTMR; 5-(and-6)(((4chloromethyl)benzoyl)amino) tetramethylrhodamine) and cocultured with target T-cells transfected with each GFP-tagged syntenin-1 construct. Env-driven cell fusion leads to syncytia formation, detected as double-fluorescence positive events of higher size and complexity. As a control of specificity, syncytia formation was blocked with the HIV-1 fusion inhibitor T-20 (Figure 3D). Quantification of these events showed that overexpression of wild-type syntenin-1 reduced Env-mediated cell fusion, whereas the Y4>F mutant had no significant effect (Figure 3D). These data suggest that syntenin-1 negatively regulates HIV-1–mediated membrane fusion through a mechanism requiring phosphorylation of tyrosine 4 in the ITIM motif. To assess whether the Y4>F point mutant was able to interact with CD4, cells were transfected with syntenin-GFP or Synt-Y1-GFP, immunoprecipitated with an anti-GFP, and blotted for CD4. As shown in Figure 3E, Synt-Y1-GFP also associated with CD4.
Knockdown of syntenin-1 expression increases HIV-1 cell fusion and viral entry
To further study syntenin-1 function during HIV-1 infection, we knocked down endogenous syntenin-1 expression using small interfering RNA (siRNA). Control cells were transfected with an siRNA sequence that does not hybridize with any eukaryotic mRNA. Syntenin-1 down-regulation was assessed for each experiment (Figure 4A and data not shown). Membrane expression levels of CD4 and CXCR4 were also routinely assessed to confirm that they were not affected by syntenin depletion (Figure 4B). To assess Env-driven cell fusion, silenced T-cells loaded with green cell tracker were cocultured with Env+ HxBc2 cells labeled with red cell tracker. Syntenin-1–depleted cells exhibited enhanced cell fusion with Env+ cells compared with control cells (Figure 4C). Furthermore, syntenin-1–depleted cells produced higher viral titers than control cells upon infection with free HIV-1 virions (Figure 4D). Importantly, this effect could be rescued by syntenin-GFP overexpression in silenced T-cells (Figure 4E). Taken together, these results confirm the inhibitory role of syntenin-1 in HIV-1 infection.
FIGURE 4:

Knockdown of syntenin-1 expression increases HIV-1 entry and syncytia formation. (A) Representative Western blot showing silencing of endogenous syntenin-1 expression using oligonucleotide J-008270-08 in T-cells 48 h after siRNA transfection. Numbers below the blot show band intensity ratios between cells transfected with control siRNA and syntenin-1 siRNA, expressed relative to tubulin. (B) Bar chart showing surface expression levels (mean fluorescence intensity) of CD4 and CXCR4 in control and syntenin-depleted cells measured by flow cytometry. (C) Syntenin-1 silencing in target T-cells favors syncytia formation induced by Env+ HxBc2 cells. Syncytia formation was quantified by flow cytometry detection of double-positive fluorescent cells and is presented as the fold induction relative to cells transfected with control siRNA (mean ± SD of four independent experiments performed in duplicate; p = 0.017). As a control, syncytia formation was blocked by incubation with the fusion inhibitor T20 (10 nM). (D) Effect of syntenin-1 silencing on HIV-1 infection. Infection was measured as viral production (p24 viral protein content in supernatants [ELISA]) and depicted as the mean fold induction relative to cells transfected with control siRNA (mean ± SD of four experiments performed in triplicate; p = 0.025). (E) Syntenin-1-GFP overexpression on cells knocked down for the endogenous syntenin-1 rescues the level of HIV-1 infection. Infection was measured as p24 viral protein content in supernatants (ELISA) and quantified as the mean fold induction relative to cells transfected with control siRNA + GFP (mean ± SD of two experiments performed in triplicate).
Syntenin-1 restricts HIV-1 entry
Syntenin-1 recruitment at the plasma membrane suggested its involvement during HIV-1 attachment or entry steps. To distinguish between these possibilities, we incubated syntenin-1–depleted cells with HIV-1 particles for 2 h at 4°C, a temperature that does not allow Env-induced membrane fusion, thus arresting the virus at the attachment step, or at 37°C, a permissive temperature for membrane fusion and consequently for viral entry. For both conditions a low infection rate was used to avoid superinfection and cooperative virion entry. The amount of p24 in the lysates was measured by ELISA (Figure 5A) and Western blot (data not shown); HIV-1 attachment was estimated from the p24 level at 4°C, and HIV entry was estimated from the difference between the levels at 37 and 4°C. Syntenin-1 silencing enhanced virus entry without affecting viral attachment (4°C), suggesting that syntenin-1 specifically impairs HIV entry (Figure 5A). To confirm these observations, we infected target cells with a one-cycle luciferase virus displaying a T-tropic envelope (X4-Luc), or the vesicular stomatitis virus (VSV) envelope (VSV-Luc) as a control. Higher luciferase levels were found in X4-Luc–infected syntenin-1–silenced cells, confirming that syntenin-1 knockdown specifically increases viral entry (Figure 5B). VSV infection was similar in silenced and control cells, demonstrating that the effect observed is HIV-1 Env-dependent (Figure S2). These results indicate that syntenin-1 negatively regulates HIV-1 entry without affecting viral attachment.
FIGURE 5:

Syntenin-1 specifically inhibits viral entry and coclusters with fluorescent VLPs at the plasma membrane. (A) Effect of syntenin-1 silencing on HIV-1 attachment (4°C) and entry (37–4°C) into target T-cells. Data present the fold induction relative to control cells (mean ± SD of three experiments performed in triplicate; p = 0.016). (B) HIV-1 entry in syntenin-silenced cells measured by infection with one-cycle viral particles carrying a luciferase reporter gene. Data present the fold induction of luciferase-HIV-1-envelope virus relative to control cells and normalized to values obtained with luciferase-VSV-envelope virus (mean ± SD from four independent experiments performed in triplicate; p = 0.038). (C) Confocal images showing the recruitment of endogenous syntenin-1 toward attached VLPs (Gag-GFP) at 4°C. Images are projections of the planes in which each individual VLP is observed. Scale bar: 5 μm. Syntenin-1 staining is shown in pseudocolor intensity-coding format next to the confocal image. The small panels are zoomed views of VLP contacts from pseudocolor syntenin-1 and merged immunostaining images (×3). Note that the maximum intensity of syntenin-1 staining (white on the pseudocolor scale images) overlaps the VLP attachment site. (D) Confocal images showing endogenous syntenin-1 distribution in target T-cells with internalized VLPs (37°C). Two VLPs are shown: one with low syntenin-1 clustering and another with no clustering. Scale bar: 5 μm. Small panels as in (C). (E) Confocal images showing endogenous syntenin-1 distribution in target T-cells forming cell–cell contacts with cells infected with VLPs. Contacts are productive sites for viral transfer and also recruit syntenin-1. Scale bar: 5 μm. Small panels as in (C). (F) Quantification of the number of VLPs that cluster syntenin-1 at the surface of target T-cells (4°C, attached particles), in the cytoplasm (37°C, intracellular particles) or at cell–cell contacts (37°C). Results are from at least three independent experiments and more than 100 VLPs were quantified for each condition.
To study the behavior of syntenin-1 during viral attachment and entry in more detail, we incubated Gag-GFP fluorescent virus-like particles (VLPs) with T-cells at 4°C or 37°C. At 4°C, VLPs attached preferentially to one pole of the target cell (Figure S3; see Supplemental Movie S1 for three-dimensional reconstruction), which suggests that the capping area behaves as the main site for viral entry, as has been proposed (Doms, 2000; Nguyen et al., 2005). At 37°C, VLPs could be observed inside target cells, usually accumulated at one pole of the cytoplasm (Figure S3; see Movie S2 for three-dimensional reconstruction).
We next examined the localization of endogenous syntenin-1 by confocal immunostaining of T-cells exposed to VLPs. To avoid false positives due to fortuitous superposition, we only analyzed planes in which VLPs were detected (usually one to two planes distanced by 0.25 μm). Syntenin-1 was enriched at VLP attachment sites in cells kept at 4°C (Figure 5, C and F), whereas barely any syntenin-1 localized at the internalized VLPs in cells incubated at 37°C (Figure 5, D and F). However, syntenin-1 did accumulate near VLPs positioned at cell–cell contacts at 37°C (Figure 5, E and F). Quantification of syntenin-1 accumulation at VLPs at each temperature and location confirmed that association of syntenin-1 with VLPs occurs only on the plasma membrane, and not once the particles are already internalized (Figure 5F).
Thus syntenin-1 is recruited to the plasma membrane by HIV-1 Env during the attachment and entry steps but affects only the effectiveness of viral entry, and it is released from the viral complex once the nucleocapsid has entered the cytoplasm.
Syntenin-1 regulates actin polymerization and PIP2 accumulation upon HIV-1 contact
Syntenin-1 was recently found to regulate F-actin polymerization via Rac GTPase in T-cells during CXCR4-induced chemotaxis and superantigen presentation (Sala-Valdes et al., 2012). This syntenin-1–mediated activation of Rac depends on phosphorylation of tyrosine 4 (Sala-Valdes et al., 2012). Moreover, Rac-mediated actin reorganization is known to be essential for HIV-1 entry (Iyengar et al., 1998; Pontow et al., 2004; Harmon et al., 2010). To analyze the relation between syntenin-1 and F-actin during HIV-1 infection, we incubated target cells with VLPs, stained them for syntenin-1 and F-actin, and analyzed them with confocal microscopy. Syntenin-1 clusters at the plasma membrane corresponded with the VLP contact area and partially overlapped with F-actin accumulation (Figure 6A; see Figure S4 for F-actin staining in noninfected target cells). Given that HIV-1 attachment to CD4 and CXCR4 triggers local actin polymerization (Jolly et al., 2004), we analyzed this process in syntenin-1–depleted cells. A similar percentage of VLPs colocalized with F-actin in control and syntenin-1–depleted cells (Figure 6A). However, when actin polymerization after HIV-1 contact was monitored at different time points, control cells showed a marked increase in F-actin content 30 min after HIV-1 contact, whereas F-actin polymerization was markedly lower in syntenin-1–depleted cells (Figure 6B). These data indicate that syntenin-1 is required for HIV-1–triggered F-actin polymerization at early stages, when most virus particles are observed at the plasma membrane. Moreover, F-actin accumulation at cell–cell contacts between target and infected MT4 cells was reduced in cells knocked down for syntenin in comparison with control cells (Figure 6C).
FIGURE 6:

Actin polymerization is reduced in syntenin-1–depleted T-cells. (A) J77 T-cells were incubated with fluorescent VLPs (Gag-GFP) for 30 min, fixed, and stained for syntenin-1 and F-actin. The panels show projections of selected confocal planes in which a VLP was observed, with corresponding pseudocolor images below. Scale bar: 5 μm. The zoomed image shows the particle site (boxed region in merged image) together with orthogonal sections. Quantification of VLPs that colocalized with F-actin in cells silenced or not for syntenin-1 is shown at the right. No significant difference was observed. (B) Time profile of F-actin content upon contact with HIV-1 particles, measured by flow cytometry in control or syntenin-depleted cells. Data are means ± SD of four independent experiments. (C) F-actin accumulation at contacts between infected MT4-HIV-GFP cells and silenced or control target cells was quantified using the synapse-measure plug-in of Image J. The medians and interquartile ranges are shown. More than 100 conjugates were counted for each condition in two independent experiments (p = 4.08 × 10−31). (D) MT4-HIV-GFP infected T-cells were incubated with control or syntenin-depleted target T-cells for 2 h, fixed, and stained for PIP2 with anti-PIP2 mAb. Quantified PIP2 labeling at Env-mediated cell contacts is presented as the percentage of PIP2 accumulated at cell contacts. More than 100 conjugates were counted for each condition (p = 1.1 × 10−5). (E) Env+ HxBc2 T-cells were incubated with control or syntenin-depleted target T-cells for 2 h and then fixed, and the conjugates were stained for PIP2 with anti-PIP2 mAb. Quantified PIP2 labeling at cell contacts is presented as the percentage of PIP2 accumulated at heterotypic Env-driven cell–cell contacts and at homotypic Env-independent contacts (between two target T-cells). More than 100 conjugates were counted for each condition (p = 2.1 × 10−4). (F) Control or syntenin-depleted cells expressing PLCδ-PH-GFP were incubated with fluorescent VLPs (Gag-Cherry) for 30 min, fixed, and analyzed by confocal microscopy. The bar chart shows quantification of PIP2 enrichment at VLP contacts, presented as the percentage of PIP2 recruited to VLP–cell contacts. More than 100 VLP contacts were counted for each condition (p = 0.038). The right panel shows a sample confocal image in which several VLPs are attached to one pole of the target T-cell (arrows). Scale bar: 5 μm.
Interaction of the HIV-1 envelope with CD4 triggers PIP2 production (Barrero-Villar et al., 2008), and this lipid has been found to regulate not only syntenin-1 recruitment to the plasma membrane (Zimmermann et al., 2005) but also many other actin-related proteins (Saarikangas et al., 2010). We therefore assessed whether syntenin-1–dependent actin polymerization upon HIV-1 contact was related to local production of PIP2. Control or syntenin-1–depleted cells were incubated with MT4-HIV-GFP cells for 2 h and then fixed and stained for PIP2 with anti-PIP2 mAb. Syntenin-1–depleted T-cells showed increased accumulation of PIP2 at contacts with HIV-1–infected T-cells (Figure 6D). To confirm that this effect was related to HIV-1 Env-dependent contacts, we quantified PIP2 accumulation in target cells cocultured with Env+ HxBc2 cells (heterotypic, Env-driven contacts) or cultured alone (homotypic, Env-independent contacts between target cells; Figure 6E and see Figure S5 for an example image). Syntenin-1 knockdown specifically increased PIP2 accumulation at Env-driven contacts, while having no effect on Env-independent contacts (Figure 6E).
To assess whether syntenin-1 depletion increases PIP2 accumulation in virus–cell contacts, we transfected control and syntenin-1–depleted cells with PLCδ-PH-GFP (pleckstrin homology domain of PLC-delta tagged to GFP), a biosensor that binds to PIP2-enriched membranes (Varnai and Balla, 1998), and incubated them with Cherry-fluorescent VLPs expressing HxB (T-tropic) envelope. Patches of PLCδ-PH-GFP were frequently observed on the target cell surface at sites of VLP attachment (a representative image is shown in Figure 6F). The frequency of these events was higher in syntenin-1–depleted cells (Figure 6F). Taken together, these data indicate that syntenin-1 depletion increases PIP2 accumulation at Env-driven contacts both of cell–cell and of virus–cell interactions, correlating with lower F-actin polymerization and higher viral entry.
DISCUSSION
In this study we show that syntenin-1 is recruited toward HIV-1 Env-driven virus–cell and cell–cell contacts, associates with CD4, limits HIV-1–induced cell fusion and viral entry, and modulates Env-triggered F-actin remodeling and PIP2 production.
HIV-1 binding to CD4/CXCR4 complexes at the T-cell surface activates several signaling pathways and triggers local actin polymerization (Iyengar et al., 1998; Jolly et al., 2004), which is essential for receptor clustering at the plasma membrane (Iyengar et al., 1998; Jolly et al., 2004, 2007; Pontow et al., 2004). The actin-binding proteins filamin-A and ERMs are known to favor CD4/CXCR4 clustering during HIV-1 attachment (Jimenez-Baranda et al., 2007; Barrero-Villar et al., 2009). Filamin-A is needed for a transient cofilin inactivation through a RhoA-ROCK–dependent mechanism that stabilizes F-actin during HIV-1 attachment (Jimenez-Baranda et al., 2007). ERMs are activated by phosphorylation upon HIV-1 contact and are essential for F-actin polarization and CD4/CXCR4 clustering (Barrero-Villar et al., 2009). In contrast with these actin-binding proteins, syntenin-1 does not affect HIV-1 attachment and therefore does not seem to be involved in HIV-1–induced CD4/CXCR4 clustering.
Instead, syntenin-1 binding to CD4 might block Lck activation or other CD4-derived pathways, thus hampering HIV-1 entry. However, since syntenin-1 depletion reduces actin polymerization soon after HIV-1 contact, the most plausible explanation for its inhibitory effect is that it induces an actin accumulation at the viral attachment site that hinders viral nucleocapsid entry at the cell surface. Localized actin clearance during HIV-1 entry has been proposed as a mechanism for overcoming this physical restriction (Yoder et al., 2008; Liu et al., 2009; Vasiliver-Shamis et al., 2009), and cofilin activation and its actin-severing function have been linked to enhanced HIV entry (Yoder et al., 2008). Thus a reduced F-actin structure after HIV-1 attachment renders cells more susceptible to subsequent HIV-1 nucleocapsid entry, as occurs in syntenin-depleted cells.
Higher HIV-1–mediated cell fusion and viral entry in syntenin-1–depleted target T-cells also correlates with increased PIP2 production at Env-mediated cell–cell contacts. PIP2 is an important second messenger that targets proteins to the plasma membrane and regulates many actin-related proteins, such as ERMs, filamin, cofilin, and α-actinin (Saarikangas et al., 2010). Generation of PIP2 triggers local actin cytoskeleton changes by activating ERMs (Nakamura et al., 1999), recruiting cofilin (van Rheenen et al., 2009), and inhibiting α-actinin bundling activity (Corgan et al., 2004). Overall, these pathways might favor HIV-1 membrane fusion by facilitating receptor clustering and subsequent actin clearance (Liu et al., 2009). Remarkably, although PIP2-associated cofilin is inactive in tumor cells, cofilin activation in leukocytes is mainly regulated by dephosphorylation (van Rheenen et al., 2009), which is in fact induced upon HIV-1 contact through CXCR4-derived signaling (Yoder et al., 2008). Thus PIP2 might switch the association of membrane receptors and their cytosolic partners (Zimmermann et al., 2005; Saarikangas et al., 2010), modulating the links between transmembrane proteins and the subcortical actin cytoskeleton, and subsequently altering the association of proteins belonging to the same signaling pathway. In this regard, in a preliminary attempt to assess syntenin-1 partners, we have observed its association with ERMs and PI4P5K-I (Gordon-Alonso et al., unpublished data). One possibility is that syntenin-1 may be able to prevent ERMs from fulfilling their role of in facilitating HIV-1 entry but without affecting CD4/CXCR4 clustering. A second option is that syntenin-1 is limiting PI4P5K-I PIP2 production, and therefore, inhibiting HIV-1 entry (Barrero-Villar et al., 2008). The complexity of syntenin-1 relationships deserves further investigation. Alternatively, depletion of syntenin-1 might simply allow a higher pool of free PIP2 to accumulate at the plasma membrane after HIV-1 attachment, where it could mediate the recruitment of proteins, such as, ERMs, that facilitate HIV entry (Barrero-Villar et al., 2008, 2009).
The fact that syntenin-1 clustering around the virus-like particle vanishes after viral entry is consistent with participation of syntenin-1 at the plasma membrane during HIV-1 entry, probably as a consequence of its association with CD4 or its recruitment through PIP2. We therefore propose a dynamic model in which syntenin-1 is first recruited to the viral attachment site through CD4 and/or PIP2. At this location, syntenin-1 would trigger F-actin polymerization via Rac GTPase, as has also been suggested for other processes mediated by this GTPase (Sala-Valdes et al., 2012). This is consistent with the fact that Rac is the Rho GTPase specifically activated during HIV-1 entry (Pontow et al., 2004; Harmon et al., 2010). Because syntenin-1 regulates Rac-triggered F-actin polymerization through the adaptor protein M-Rip during T chemotaxis (Sala-Valdes et al., 2012), it is conceivable that this or a similar signaling axis could be involved during HIV-1 entry. Indeed, syntenin-1 Tyr-4 is essential for Rac-mediated T-cell chemotaxis and superantigen presentation (Sala-Valdes et al., 2012) and seems also to be crucial for syntenin-mediated inhibition of HIV infection. Subsequently, F-actin must be locally depolymerized to facilitate the nucleocapsid entry (Yoder et al., 2008; Vasiliver-Shamis et al., 2009), which is favored (F-actin depolymerization) in the absence of syntenin-1. In this context, PIP2 induction by HIV-1 might be the signal that shuts down local F-actin polymerization while activating F-actin clearance.
In summary, our results have unveiled a critical regulatory role for syntenin-1 in controlling HIV-1 entry into target T-cells through altering actin polymerization, most likely in a PIP2-regulated manner.
MATERIALS AND METHODS
Cell lines and reagents
The Jurkat-derived human T-cell line J77 Vβ8+ J77cl20 was grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Cambrex Bioscience, Charles City, IA). The CEM-T4 human T-lymphoblast–like cell line (courtesy of Paul Clapman), and the HxBc2 Jurkat-derived T-cell line (courtesy of Joseph Sodrowski) were obtained from the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program and were cultured according to NIH instructions. The chronically infected MT4-NL4.3-GFP cell line (MT4-HIV-GFP) was generated by Nuria Izquierdo-Useros at the laboratory of Martinez-Picado (IrsiCaixa, Barcelona, Spain).
The biotinylated monoclonal anti-CXCR4 antibody was from BD PharMingen. Mouse monoclonal anti–α-tubulin was from Sigma-Aldrich (Saint Louis, MO). Anti-CD4 monoclonal antibodies used were HP2/6 (Gordon-Alonso et al., 2006), OKT4 (eBioscience), RPA-T4 (eBioscience, San Diego, CA), CD4v4-FITC (BD PharMingen, Franklin Lakes, NJ), and MEM 241 (Abcam, Cambridge, UK). Rabbit polyclonal anti–syntenin-1 was from Synaptic Systems (Göttingen, Germany). The anti-CD45 mAb was clone D3/9, previously described (Gordon-Alonso et al., 2006). Anti-PIP2 mAb was from Santa Cruz Biotechnology (clone 2C11; Santa Cruz, CA). Anti-GFP antibodies (pAb for IP and mAb for WB) were from Living colors (Clontech, Mountain View, CA). HRP-conjugated secondary antibodies were from Pierce (Rockford, IL).
The intracellular fluorescent trackers CMAC, Calcein-AM, and CM-TMR and all fluorochrome-conjugated secondary antibodies and phalloidins were from Molecular Probes (Camarillo, CA). The HIV-1-specific fusion inhibitor T20 (Enfuvirtide) was from Roche Diagnostics.
Cell transfection, DNA and siRNA
J77 cells (2 × 107) were electroporated in cold Optimem (Life Technologies-BRL, Camarillo, CA) with DNA (20 mg) or with siRNA (1.25 mM) using a Bio-Rad GenePulser II electroporator (240 V; 950°F). Fluorescent protein expression and siRNA knockdown were tested respectively by flow cytometry (24 h) and Western blot analysis (48 h).
The syntenin-1 GFP fusion proteins Psrα-HAGFP-syntenin-1 and syntenin-1-Y1-GFP have been described previously (Sala-Valdes et al., 2012). PIP2-enriched plasma membranes were monitored during Env-mediated membrane fusion or VLP attachment by transfecting cells with the biosensor PLCδ-PH-GFP (Varnai and Balla, 1998). GST-syntenin-1-HA and GST-syntenin-1-Myc were kindly donated by F. Lozano (Hospital Clinic, Barcelona, Spain).
Negative control siRNA was from Eurogentec (Serain, Belgium) and the specific siRNAs against syntenin-1 were from Thermo Scientific Dharmacon (oligonucleotides J-008270-08 and J-008270-06).
Preparation and production of HIV-1 and virus attachment and entry assay
Preparation of HIV-1 NL4.3 and measurement of viral replication were as previously described (Valenzuela-Fernandez et al., 2005). Gag-GFP and Gag-Cherry fluorescent VLPs were produced as previously described (Izquierdo-Useros et al., 2009) by cotransfecting 293T cells with the HIV Gag-eGFP/Cherry plasmids and the envelope plasmid pHXB2.
For p24 production, T-cells were infected with 100 ng of HIV-1 NL4.3 per million cells for 2 h at 37°C (which is equivalent to 1–2 multiplicity of infection or viral particles per cell), and then extensively washed with medium to remove nonattached viral particles. Infected T-cells were kept in culture for 3 d, at which time supernatants were harvested and p24 concentration measured by ELISA (Innotest HIV-1 antigen mAb; Innogenetic, Ghent, Belgium).
For HIV attachment and entry measurements, T-cells were infected with 20 ng of HIV-1 NL4.3 per million cells for 2 h at 4°C (attachment) or 37°C (attachment + entry), and then extensively washed to remove viral input and lysed with RIPA buffer (50 mM Tris HCl, pH 8, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). p24 was measured by ELISA (Innotest HIV-1antigen mAb; Innogenetic).
Luciferase virus assay
Replication-deficient luciferase-HIV-1 particles were kindly provided by Suryaram Gummuluru (Boston University, Boston, MA). Replication-deficient viral particles were produced by cotransfecting 293T cells with the luciferase-expressing reporter virus, HIV/Δnef/Δenv/luc+, which contains the luciferase gene inserted into the nef open reading frame and does not express env glycoprotein (Yamashita and Emerman, 2004), and a CXCR4-tropic (Lai) env glycoprotein or non–T-tropic VSV glycoprotein. Virus stocks were generated by PolyFect transient cotransfection of HEK293T cells (Gummuluru et al., 2002). Two days after transfection, cell-free virus-containing supernatants were clarified of cell debris and concentrated by centrifugation (16,000 × g, 1 h at 4°C) and stored at −80°C until required. HIV-1 virus preparations were titrated by p24-ELISA. T-cells were infected with 200 ng of luciferase-based virus with X4-tropic HIV-1 envelope or VSV envelope per million cells for 2 h. Cells were washed extensively and luciferase activity was measured at 48 h after infection with a luciferase assay kit (Promega Corporation, Madison, WI) on a 1450 Microbeta Luminescence Counter (Walax, Trilux).
Env-induced cell fusion assay
A dual-fluorescence cell-fusion assay was performed as described previously (Gordon-Alonso et al., 2006). Briefly, CMTMR-loaded Env+ Jurkat-Hxbc2 cells were mixed with calcein-AM–loaded parental or transfected CEM-T4 cells. Fluorescence events were detected 14 h later by flow cytometry. Fusion was quantified as the ratio of double-positive events to single-positive target cells.
Immunoprecipitation
T lymphoblasts and CEM T-cells were lysed in 1% NP-40, 1 mM MgCl2, 1 mM CaCl2 in Tris-buffered saline (TBS) supplemented with protease and phosphatase inhibitor cocktails. J77 T-cells (4 × 107) were lysed in 1% NP-40 in TBS supplemented with a protease inhibitor cocktail. Lysates were centrifuged at 11,000 rpm for 10 min at 4°C, and cell lysate supernatants were incubated for 2 h at 4°C with cyanogen bromide (CNBr) beads (Amersham, Uppsala, Sweden) blocked with bovine serum albumin (BSA). Supernatants were then incubated with the indicated antibody covalently coupled to CNBr beads for 2 h at 4°C. Pellets were washed with lysis buffer, resuspended in reducing Laemmli buffer, and processed for Western blotting with the indicated antibodies.
Immunofluorescence and confocal microscopy
T-cells incubated with viral particles, Env+ cells (HxBc2), or HIV-1 infected cells (MT4-HIV-GFP) were seeded on 50 μg/ml poly-l-lysine for 30 min and fixed with 3% paraformaldehyde. For staining of intracellular proteins (endogenous syntenin-1, F-actin, and PIP2), samples were permeabilized for 5 min with 0.5% Triton X-100 in TBS, stained and mounted in Prolong (Invitrogen, Camarillo, CA). Images were obtained with a photomicroscope (DMR; Leica, Germany) fitted with an HCX PL APO 63×/1.32-210.6 oil-immersion objective (Leica; Germany) and coupled to a COHU 4912–5010 charge-coupled camera.
Confocal images were obtained with a Leica TCS-SP5 confocal scanning laser microscope fitted with either an HCX PL APO lambda blue 63×/1.4 or an HCX PL APO lambda blue 100×/1.4 oil-immersion objective and analyzed with Image J. For clustering quantification, staining intensities were transformed with the pseudocolor-intensity look-up-table (LUT). Yellow-white patches at VLP locations were considered positive for clustering. For analyzing F-actin accumulation at cell–cell contacts, Image J software was used as previously described (Calabia-Linares et al., 2011).
Western blotting
J77 T-cells were lysed in 1% NP-40 in TBS supplemented with a cocktail of protease inhibitors and phosphatase inhibitors. Lysates were centrifuged at 11,000 rpm for 10 min at 4°C, and supernatants were mixed with reducing Laemmli buffer and boiled for 5 min. Lysates were separated by SDS–PAGE and immunoblotted with specific antibodies. Protein bands were analyzed using the LAS-1000 CCD system and Image Gauge 3.4 software (Fuji Photo Film, Tokyo, Japan).
Flow cytometry
Cells were incubated with TBS 5% BSA for 30 min at 4°C and then with primary monoclonal antibody (30 min at 4°C). After being washed, cells were incubated with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody and analyzed in a FACScalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Data were analyzed with Cell-Quest Pro (Becton Dickinson).
F-actin quantification
Cells were stimulated with HIV NL4.3 free virions (100 ng/million cells), fixed with 4% formaldehyde at different time points, permeabilized with TBS 0.5% Triton X-100 (5 min), and stained with Phalloidin-Alexa647 (Molecular Probes, Camarillo, CA). Mean fluorescence intensity of F-actin staining was analyzed on a FACScalibur cytometer (BD Biosciences, Franklin Lakes, NJ).
Statistical analysis
Statistical significance was calculated using Student's t test or the parametric one-way analysis of variance with Bonferroni's post hoc multiple-comparison test. Significant differences are labeled (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Supplementary Material
Acknowledgments
The authors thank José Román Cabrero for helpful biochemical advice, Manuel Perez-Martinez for confocal assistance, and Miguel Vicente-Manzanares for critical reading of the manuscript. Simon Barlett (CNIC) provided English editing. This work was supported by SAF2008-02635, SAF2011-25834 (from Ministerio de Economía y Competitividad), INSINET-0159/2006 (from the Comunidad de Madrid), and FIPSE 36658/07 (from the Spanish Society against AIDS), all to F.S.-M. M.G.-A. was funded by RECAVA and V.R.-P. by the CNIC.
Abbreviations used:
- APC
antigen presenting cell
- BSA
bovine serum albumin
- CD4
cluster of differentation-4
- CMTMR
5-(and-6)(((4chloromethyl)benzoyl)amino) tetramethylrhodamine
- CNBr
cyanogen bromide
- CXCR4
C-X-C chemokine receptor type-4
- ELISA
enzyme-linked immunosorbent assay
- ENV
viral envelope glycoprotein complex gp120/gp41
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HA
hemagglutinin
- HIV-1
human immunodeficiency virus type 1
- ITAM
immunoreceptor tyrosine-based activating motif
- ITIM
immunoreceptor tyrosine-based inhibitory motif
- mda-9
melanoma differentiation-associated gene-9
- PDZ
postsynaptic density protein, Drosophila disk large, and zonula occludens-1
- PI4P5K-Ia
phosphatidylinositol-4-phosphate-5 kinase type I-alpha
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC-delta-PH-GFP
pleckstrin homology domain of phospholipase C-delta tagged to GFP protein
- siRNA
small interfering RNA
- TBS
Tris-buffered saline
- VLP
virus-like particle
- VSV
vesicular stomatitis virus
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-1003) on April 25, 2012.
REFERENCES
- Barrero-Villar M, Barroso-Gonzalez J, Cabrero JR, Gordon-Alonso M, Alvarez-Losada S, Munoz-Fernandez MA, Sanchez-Madrid F, Valenzuela-Fernandez A. PI4P5-kinase Iα is required for efficient HIV-1 entry and infection of T cells. J Immunol. 2008;181:6882–6888. doi: 10.4049/jimmunol.181.10.6882. [DOI] [PubMed] [Google Scholar]
- Barrero-Villar M, Cabrero JR, Gordon-Alonso M, Barroso-Gonzalez J, Alvarez-Losada S, Munoz-Fernandez MA, Sanchez-Madrid F, Valenzuela-Fernandez A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J Cell Sci. 2009;122:103–113. doi: 10.1242/jcs.035873. [DOI] [PubMed] [Google Scholar]
- Beekman JM, Coffer PJ. The ins and outs of syntenin, a multifunctional intracellular adaptor protein. J Cell Sci. 2008;121:1349–1355. doi: 10.1242/jcs.026401. [DOI] [PubMed] [Google Scholar]
- Boukerche H, Su ZZ, Prevot C, Sarkar D, Fisher PB. mda-9/syntenin promotes metastasis in human melanoma cells by activating c-Src. Proc Natl Acad Sci USA. 2008;105:15914–15919. doi: 10.1073/pnas.0808171105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brone B, Eggermont J. PDZ proteins retain and regulate membrane transporters in polarized epithelial cell membranes. Am J Physiol Cell Physiol. 2005;288:C20–C29. doi: 10.1152/ajpcell.00368.2004. [DOI] [PubMed] [Google Scholar]
- Calabia-Linares C, et al. Endosomal clathrin drives actin accumulation at the immunological synapse. J Cell Sci. 2011;124:820–830. doi: 10.1242/jcs.078832. [DOI] [PubMed] [Google Scholar]
- Carter GC, Bernstone L, Baskaran D, James W. HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology. 2009;409:234–250. doi: 10.1016/j.virol.2010.10.018. [DOI] [PubMed] [Google Scholar]
- Chimura T, Launey T, Ito M. Evolutionarily conserved bias of amino-acid usage refines the definition of PDZ-binding motif. BMC Genomics. 2011;12:300. doi: 10.1186/1471-2164-12-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corgan AM, Singleton C, Santoso CB, Greenwood JA. Phosphoinositides differentially regulate α-actinin flexibility and function. Biochem J. 2004;378:1067–1072. doi: 10.1042/BJ20031124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianzani U, Bragardo M, Buonfiglio D, Redoglia V, Funaro A, Portoles P, Rojo J, Malavasi F, Pileri A. Modulation of CD4 lateral interaction with lymphocyte surface molecules induced by HIV-1 gp120. Eur J Immunol. 1995;25:1306–1311. doi: 10.1002/eji.1830250526. [DOI] [PubMed] [Google Scholar]
- Doms RW. Beyond receptor expression: the influence of receptor conformation, density, and affinity in HIV-1 infection. Virology. 2000;276:229–237. doi: 10.1006/viro.2000.0612. [DOI] [PubMed] [Google Scholar]
- Fanning AS, Anderson JM. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J Clin Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimferrer I, et al. The lymphocyte receptor CD6 interacts with syntenin-1, a scaffolding protein containing PDZ domains. J Immunol. 2005;175:1406–1414. doi: 10.4049/jimmunol.175.3.1406. [DOI] [PubMed] [Google Scholar]
- Gordon-Alonso M, Yanez-Mo M, Barreiro O, Alvarez S, Munoz-Fernandez MA, Valenzuela-Fernandez A, Sanchez-Madrid F. Tetraspanins CD9 and CD81 modulate HIV-1-induced membrane fusion. J Immunol. 2006;177:5129–5137. doi: 10.4049/jimmunol.177.8.5129. [DOI] [PubMed] [Google Scholar]
- Grootjans JJ, Zimmermann P, Reekmans G, Smets A, Degeest G, Durr J, David G. Syntenin, a PDZ protein that binds syndecan cytoplasmic domains. Proc Natl Acad Sci USA. 1997;94:13683–13688. doi: 10.1073/pnas.94.25.13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gummuluru S, KewalRamani VN, Emerman M. Dendritic cell-mediated viral transfer to T cells is required for human immunodeficiency virus type 1 persistence in the face of rapid cell turnover. J Virol. 2002;76:10692–10701. doi: 10.1128/JVI.76.21.10692-10701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon B, Campbell N, Ratner L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010;6:e1000956. doi: 10.1371/journal.ppat.1000956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbec H, Martin S, Henley JM. Syntenin is involved in the developmental regulation of neuronal membrane architecture. Mol Cell Neurosci. 2005;28:737–746. doi: 10.1016/j.mcn.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Hiscott J, Kwon H, Genin P. Hostile takeovers: viral appropriation of the NF-κB pathway. J Clin Invest. 2001;107:143–151. doi: 10.1172/JCI11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar S, Hildreth JE, Schwartz DH. Actin-dependent receptor colocalization required for human immunodeficiency virus entry into host cells. J Virol. 1998;72:5251–5255. doi: 10.1128/jvi.72.6.5251-5255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo-Useros N, et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood. 2009;113:2732–2741. doi: 10.1182/blood-2008-05-158642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jannatipour M, Dion P, Khan S, Jindal H, Fan X, Laganiere J, Chishti AH, Rouleau GA. Schwannomin isoform-1 interacts with syntenin via PDZ domains. J Biol Chem. 2001;276:33093–33100. doi: 10.1074/jbc.M105792200. [DOI] [PubMed] [Google Scholar]
- Jimenez-Baranda S, et al. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat Cell Biol. 2007;9:838–846. doi: 10.1038/ncb1610. [DOI] [PubMed] [Google Scholar]
- Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med. 2004;199:283–293. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Mitar I, Sattentau QJ. Requirement for an intact T-cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. J Virol. 2007;81:5547–5560. doi: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juno JA, Fowke KR. Clarifying the role of G protein signaling in HIV infection: new approaches to an old question. AIDS Rev. 2010;12:164–176. [PubMed] [Google Scholar]
- Latysheva N, Muratov G, Rajesh S, Padgett M, Hotchin NA, Overduin M, Berditchevski F. Syntenin-1 is a new component of tetraspanin-enriched microdomains: mechanisms and consequences of the interaction of syntenin-1 with CD63. Mol Cell Biol. 2006;26:7707–7718. doi: 10.1128/MCB.00849-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Belkina NV, Shaw S. HIV infection of T cells: actin-in and actin-out. Sci Signal. 2009;2:pe23. doi: 10.1126/scisignal.266pe23. [DOI] [PubMed] [Google Scholar]
- Ludford-Menting MJ, et al. A network of PDZ-containing proteins regulates T cell polarity and morphology during migration and immunological synapse formation. Immunity. 2005;22:737–748. doi: 10.1016/j.immuni.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Mettling C, Desmetz C, Fiser AL, Reant B, Corbeau P, Lin YL. Gαi protein-dependent extracellular signal-regulated kinase-1/2 activation is required for HIV-1 reverse transcription. AIDS. 2008;22:1569–1576. doi: 10.1097/QAD.0b013e32830abdaf. [DOI] [PubMed] [Google Scholar]
- Nakamura F, Huang L, Pestonjamasp K, Luna EJ, Furthmayr H. Regulation of F-actin binding to platelet moesin in vitro by both phosphorylation of threonine 558 and polyphosphatidylinositides. Mol Biol Cell. 1999;10:2669–2685. doi: 10.1091/mbc.10.8.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Giri B, Collins G, Taub DD. Dynamic reorganization of chemokine receptors, cholesterol, lipid rafts, and adhesion molecules to sites of CD4 engagement. Exp Cell Res. 2005;304:559–569. doi: 10.1016/j.yexcr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Pontow SE, Heyden NV, Wei S, Ratner L. Actin cytoskeletal reorganizations and coreceptor-mediated activation of rac during human immunodeficiency virus-induced cell fusion. J Virol. 2004;78:7138–7147. doi: 10.1128/JVI.78.13.7138-7147.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik W, Hesselgesser JE, Pitha PM. Binding of human immunodeficiency virus type 1 to CD4 and CXCR4 receptors differentially regulates expression of inflammatory genes and activates the MEK/ERK signaling pathway. J Virol. 1998;72:6406–6413. doi: 10.1128/jvi.72.8.6406-6413.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik W, Pitha PM. Inhibition of CD3/CD28-mediated activation of the MEK/ERK signaling pathway represses replication of X4 but not R5 human immunodeficiency virus type 1 in peripheral blood CD4(+) T lymphocytes. J Virol. 2000;74:2558–2566. doi: 10.1128/jvi.74.6.2558-2566.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikangas J, Zhao H, Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol Rev. 2010;90:259–289. doi: 10.1152/physrev.00036.2009. [DOI] [PubMed] [Google Scholar]
- Sala-Valdes M, Gordon-Alonso M, Tejera E, Ibanez A, Cabrero JR, Ursa A, Mittelbrunn M, Lozano F, Sanchez-Madrid F, Yanez-Mo M. Association of syntenin-1 with M-RIP polarizes Rac-1 activation during chemotaxis and immune interactions. J Cell Sci. 2012;125:1235–1246. doi: 10.1242/jcs.094912. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Boukerche H, Su ZZ, Fisher PB. mda-9/syntenin: recent insights into a novel cell signaling and metastasis-associated gene. Pharmacol Ther. 2004;104:101–115. doi: 10.1016/j.pharmthera.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Boukerche H, Su ZZ, Fisher PB. mda-9/syntenin: more than just a simple adapter protein when it comes to cancer metastasis. Cancer Res. 2008;68:3087–3093. doi: 10.1158/0008-5472.CAN-07-6210. [DOI] [PubMed] [Google Scholar]
- Valenzuela-Fernandez A, et al. Histone deacetylase 6 regulates human immunodeficiency virus type 1 infection. Mol Biol Cell. 2005;16:5445–5454. doi: 10.1091/mbc.E05-04-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci. 2009;122:305–311. doi: 10.1242/jcs.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliver-Shamis G, Cho MW, Hioe CE, Dustin ML. Human immunodeficiency virus type 1 envelope gp120-induced partial T-cell receptor signaling creates an F-actin-depleted zone in the virological synapse. J Virol. 2009;83:11341–11355. doi: 10.1128/JVI.01440-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Yoder A. Chemokine coreceptor signaling in HIV-1 infection and pathogenesis. PLoS Pathog. 2009;5:e1000520. doi: 10.1371/journal.ppat.1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Emerman M. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol. 2004;78:5670–5678. doi: 10.1128/JVI.78.11.5670-5678.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder A, et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P. The prevalence and significance of PDZ domain-phosphoinositide interactions. Biochim Biophys Acta. 2006;1761:947–956. doi: 10.1016/j.bbalip.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Zimmermann P, Tomatis D, Rosas M, Grootjans J, Leenaerts I, Degeest G, Reekmans G, Coomans C, David G. Characterization of syntenin, a syndecan-binding PDZ protein, as a component of cell adhesion sites and microfilaments. Mol Biol Cell. 2001;12:339–350. doi: 10.1091/mbc.12.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P, Zhang Z, Degeest G, Mortier E, Leenaerts I, Coomans C, Schulz J, N'Kuli F, Courtoy PJ, David G. Syndecan recycling [corrected] is controlled by syntenin-PIP2 interaction and Arf6. Dev Cell. 2005;9:377–388. doi: 10.1016/j.devcel.2005.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.