

Mass spectrometry identified multiple phosphorylation sites on the kinesin-13 protein Kif2b, some of which are acutely sensitive to inhibition of Polo-like kinase 1 (Plk1). Data demonstrate that Plk1 regulates the localization and activity of Kif2b during mitosis to promote the correction of kinetochore-microtubule attachment errors to ensure mitotic fidelity.
Abstract
Solid tumors are frequently aneuploid, and many display high rates of ongoing chromosome missegregation in a phenomenon called chromosomal instability (CIN). The most common cause of CIN is the persistence of aberrant kinetochore-microtubule (k-MT) attachments, which manifest as lagging chromosomes in anaphase. k-MT attachment errors form during prometaphase due to stochastic interactions between kinetochores and microtubules. The kinesin-13 protein Kif2b promotes the correction of k-MT attachment errors in prometaphase, but the mechanism restricting this activity to prometaphase remains unknown. Using mass spectrometry, we identified multiple phosphorylation sites on Kif2b, some of which are acutely sensitive to inhibition of Polo-like kinase 1 (Plk1). We show that Plk1 directly phosphorylates Kif2b at threonine 125 (T125) and serine 204 (S204), and that these two sites differentially regulate Kif2b function. Phosphorylation of S204 is required for the kinetochore localization and activity of Kif2b in prometaphase, and phosphorylation of T125 is required for Kif2b activity in the correction of k-MT attachment errors. These data demonstrate that Plk1 regulates both the localization and activity of Kif2b during mitosis to promote the correction of k-MT attachment errors to ensure mitotic fidelity.
INTRODUCTION
Faithful chromosome segregation is required for cell and organism viability. Chromosome missegregation leads to the state of aneuploidy, which is virtually irreversible (Lengauer et al., 1998). Normal cells display potent cell cycle arrest upon chromosome missegregation through a p53-dependent pathway that prevents the accumulation of aneuploid cells arising from spontaneous chromosome missegregation (Thompson and Compton, 2010). However, solid tumors circumvent this pathway and frequently propagate with aneuploid karyotypes. Moreover, most solid tumors have lost mitotic fidelity and show very high rates of ongoing chromosome missegregation in a phenomenon called chromosomal instability (CIN; Lengauer et al., 1997; Thompson et al., 2010).
Direct observation of aneuploid cancer cells with CIN has shown that the most common cause of chromosome segregation defects is the persistence of errors in the attachment of microtubules to kinetochores (Thompson and Compton, 2008; Bakhoum et al., 2009b). These errors arise in early mitosis due to the stochastic nature of kinetochore-microtubule (k-MT) interactions (Cimini et al., 2001). Normal cells efficiently correct these errors to ensure faithful chromosome segregation at anaphase. In contrast, the prevalence of k-MT attachment errors is elevated in aneuploid tumor cells because the rate of error formation is increased and the rate of error correction is decreased (Salmon et al., 2005; Bakhoum et al., 2009b; Ganem et al., 2009; Manning et al., 2010; Thompson et al., 2010). The rate-limiting step in the process of correction of these errors is the release of microtubules from kinetochores (Nicklas and Ward, 1994). This underscores the importance of the dynamics through which microtubules attach to and detach from kinetochores for the correction of k-MT attachment errors. Indeed, it has been shown that increasing k-MT turnover in aneuploid cancer cells with CIN by mildly destabilizing k-MT attachments is sufficient to restore faithful chromosome segregation (Bakhoum et al., 2009a).
A key component of the machinery responsible for the correction of k-MT attachment errors is the kinesin-13 family of microtubule-depolymerizing enzymes (Ems-McClung and Walczak, 2010). Mammalian cells possess three genes in this subgroup of the kinesin gene family, and each fulfills different cellular functions (Manning et al., 2007; Ohi et al., 2007). MCAK and Kif2b have both been demonstrated to execute the correction of k-MT attachment errors (Maney et al., 1998; Andrews et al., 2004; Kline-Smith et al., 2004; Lan et al., 2004; Knowlton et al., 2006; Manning et al., 2007; Bakhoum et al., 2009b). Perturbation of MCAK or Kif2b function leads to significant increases in lagging chromosomes in anaphase cells (Maney et al., 1998; Lan et al., 2004; Kline-Smith et al., 2004; Bakhoum et al., 2009b), which is the consequence of the persistence of attachment of single kinetochores to microtubules oriented toward both spindle poles (i.e., merotely). Although lagging chromosomes rarely missegregate, they are symptomatic of the persistence of k-MT attachment defects that cause chromosome missegregation (Thompson and Compton, 2011), and they are prone to breakages that can contribute to chromosome translocations (Janssen et al., 2011). Of interest, the activities of MCAK and Kif2b are not strictly redundant, reflecting the need for a robust error correction machinery at all stages of mitosis before anaphase. MCAK localizes to centromeres and inner kinetochores and is most active in correcting k-MT attachment errors in metaphase (Maney et al., 1998; Bakhoum et al., 2009b). On the other hand, Kif2b localizes to outer kinetochores and is most active in correcting k-MT attachment errors in prometaphase (Manning et al., 2007; Bakhoum et al., 2009b). These differences in spatial and temporal activity suggest a tight cell cycle–dependent regulation. Indeed, it has been shown that Aurora B kinase regulates the activity of MCAK (Andrews et al., 2004; Lan et al., 2004; Knowlton et al., 2006). However, it remains unknown how Kif2b is regulated to ensure optimal activity during prometaphase, when numerous k-MT attachment errors must be corrected. Here we investigate the mechanism of cell cycle regulation of Kif2b through perturbation of mitotic kinases and analysis of the effect of Kif2b phosphorylation state on the correction of k-MT attachment errors.
RESULTS
The kinesin-13 Kif2b localizes to outer kinetochores during prometaphase and has been shown to be required for the correction of k-MT attachment errors during that stage of mitosis (Manning et al., 2007; Bakhoum et al., 2009b). To explore the mechanism of this tight cell cycle–dependent activity, we tested the sensitivity of the localization of GFP-Kif2b to inhibitors of Aurora B and Plk1 kinases (Figure 1), two kinases that have been shown to be required for proper k-MT attachments (Andrews et al., 2004; Lan et al., 2004; Knowlton et al., 2006; Pinsky et al., 2006; Petronczki et al., 2008; Li et al., 2010; Bader et al., 2011; Foley et al., 2011; Zhang et al., 2011). GFP-Kif2b serves as an appropriate surrogate for the localization of endogenous Kif2b (Manning et al., 2010) and localizes to numerous kinetochores in a majority (∼75%) of prometaphase cells induced by treatment with either monastrol or nocodazole but not in metaphase cells arrested with MG-132 (Figure 1, A and B, and Supplemental Figure S1, A and B). In the presence of the Aurora kinase inhibitor hesperadin (Hauf et al., 2003), only 10–20% of monastrol-treated cells display GFP-Kif2b localization at kinetochores (Figure 1, A and B), consistent with our previous data (Bakhoum et al., 2009b). In the presence of the Plk1 inhibitor BI-2536 (Lénárt et al., 2007) the percentage of cells with green fluorescent protein (GFP)–Kif2b kinetochore localization is reduced to ∼15–20%, a reduction roughly equivalent to that seen in the presence of hesperadin (Figure 1, A and B). GFP-Kif2b localizes to many kinetochores in nocodazole-treated cells, and this is abolished in the presence of either hesperadin or BI-2536 as well (Supplemental Figure S1A), demonstrating that the localization is independent of microtubule attachment. Cells arrested in metaphase with MG-132 rarely display Kif2b at kinetochores, and there is no significant change in the presence of either hesperadin or BI-2536 (Supplemental Figure S1B). These data demonstrate that both Aurora and Plk1 kinase activities are required to localize Kif2b to kinetochores specifically in prometaphase.
FIGURE 1:
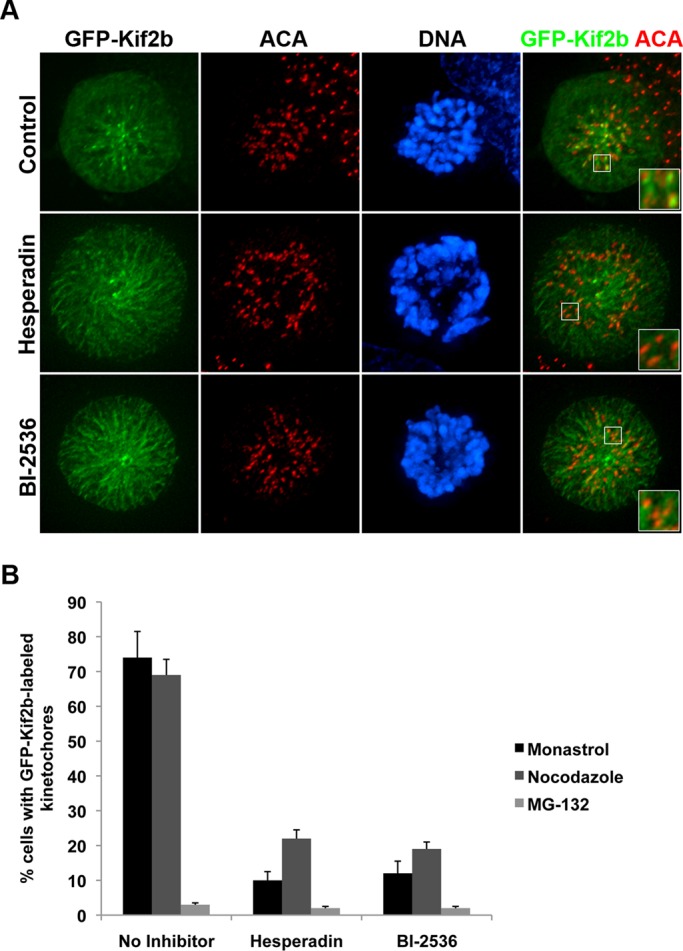
GFP-Kif2b kinetochore localization is sensitive to chemical inhibition of Aurora kinase and Plk1 kinase. (A) Representative images of monastrol-treated control, Aurora-inhibited (hesperadin), and Plk1-inhibited (BI-2536) human U2OS cells stably expressing GFP-Kif2b (green) and stained for DNA (blue) and centromeres (red). Scale bar, 5 μm. (B) Percentage of cells with GFP-Kif2b localized to kinetochores when arrested with monastrol, nocodazole, or MG-132 and treated with no inhibitor, hesperadin, or BI-2536. Error bars represent mean ± SEM, n = 300 cells, three experiments.
Mapping phosphorylation sites on Kif2b
To map specific sites of phosphorylation, we immunoprecipitated GFP-Kif2b from mitotically arrested cells and performed mass spectrometry (Figure 2). GFP-Kif2b localizes equivalently to endogenous Kif2b (Manning et al., 2010), making GFP-Kif2b a suitable surrogate for these experiments because the mechanisms regulating GFP-Kif2b are synonymous to those regulating endogenous Kif2b. We isolated the immunoprecipitated GFP-Kif2b using SDS–PAGE (Figure 2A), digested the protein with trypsin, and analyzed the resulting peptides for phosphorylation by high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS; Baker et al., 2009). In total, 78.6% of the Kif2b protein amino acid sequence was covered in this analysis, and 19 phosphorylation sites on Kif2b were identified (Figure 2B and Supplemental Table S1). The phosphorylation sites were primarily confined to two regions of Kif2b directly N- and C-terminal to the motor domain, although we also identified one phosphorylation site within the motor domain (Figure 2B). The sites within the N-terminal region, particularly around the neck domain, are of interest because the equivalent region of MCAK has been shown to be the site for regulation by Aurora B kinase (Andrews et al., 2004; Lan et al., 2004; Knowlton et al., 2006). In a few cases, insufficient information was obtained in the MS/MS spectra of some phosphopeptides to definitively assign a phosphorylation locus to a specific amino acid in the peptide sequence. For example, we were unable to distinguish between the phosphoacceptor residues serine 200 (S200) and serine 201 (S201) in the phosphopeptide HLDp(SS)KISVLEPPQEHR, although the peptide sequence with a phosphate was identified with high confidence.
FIGURE 2:

Mapping of phosphorylation sites on Kif2b. (A) GFP-Kif2b protein from mitotic U2OS cells was immunoprecipitated with anti-Kif2b (αKif2b) or control preimmunization (PreImm) serum. Total protein extracts (Input), unbound proteins (Unbound), and immunoprecipitations (IP) were resolved by SDS–PAGE and detected by immunoblot with anti-Kif2b (left) and Colloidal Blue stain (right). (B) Schematic of Kif2b denoting approximate locations of all phosphorylation sites identified by LC-MS/MS (not to scale). Asterisks denote phosphorylation sites identified on endogenous Kif2b. (C) Quantification of phosphorylated peptides in the presence of kinase inhibitors vs. untreated control cells. (D) Alignment of kinesin-13 protein sequences surrounding key phosphorylation sites.
To determine which of these phosphorylation sites may be responsible for the temporal and spatial control of Kif2b, we performed mass spectrometry on GFP-Kif2b isolated from mitotic cells treated with kinase inhibitors versus untreated mitotic cells. Using stable isotope labeling of amino acids in cell culture (SILAC; Ong, 2002; Baker et al., 2009; Kettenbach et al., 2011), we metabolically labeled the proteome of cells expressing GFP-Kif2b with analogues of lysine and arginine carrying heavy isotopes of carbon and nitrogen. These “heavy” cells were synchronized in mitosis with either nocodazole or MG-132 and then treated with either hesperadin or BI-2536 to inhibit Aurora or Plk1, respectively. In parallel, unlabeled (“light”) control cell populations were also synchronized in mitosis but were not treated with kinase inhibitors. Equal numbers of “heavy” and “light” cells were then combined, and mitotic extracts were prepared. GFP-Kif2b was isolated by immunoprecipitation, followed by SDS–PAGE (Supplemental Figure S2). We also used this approach to compare the phosphorylation status of GFP-Kif2b in cells arrested in prometaphase with nocodazole versus cells arrested in metaphase with MG-132. The difference in phosphorylation status of residues in these mixed extracts was then quantified by mass spectrometry (Figure 2C and Supplemental Table S2).
There were only slight changes in phosphorylation of residues in Kif2b between prometaphase (nocodazole treated) and metaphase (MG-132 treated). Surprisingly, no residues were found to be sensitive to chemical inhibition of Aurora (Figure 2C), despite evidence showing that GFP-Kif2b localization to kinetochores is perturbed at these concentrations of inhibitor (Figure 1; Bakhoum et al., 2009b). Of interest, we identified serine 147 as a phosphorylation site on Kif2b. This site is contained within a conserved Aurora B consensus sequence (Figure 2D; Kettenbach et al., 2011) and is homologous to the Aurora B phosphorylation site in MCAK (serine 192 in humans and serine 196 in Xenopus) that is required for inhibition of its activity (Andrews et al., 2004; Lan et al., 2004). Nonetheless this site in Kif2b was insensitive to inhibition of Aurora kinase activity.
Three phosphorylation sites (T125, S200/S201/S204, and S575) displayed a 5- to 50-fold decrease in phosphorylation in the presence of the Plk1 inhibitor BI-2536 compared with untreated cells (Figure 2C). Each of these sites is within a Plk1 consensus sequence (Figure 2D; Kettenbach et al., 2011). Two of these sites (T125 and S200/S201/S204) show moderate sequence conservation to other members of the kinesin-13 family, including MCAK. The other site (S575) shows sequence conservation with Kif2a but not MCAK (Figure 2D).
To verify the phosphorylation of these sites on the endogenous protein, we immunoprecipitated Kif2b from ∼108 mitotic U2OS cells. Phosphorylated peptides were enriched (Kettenbach and Gerber, 2011), and mass spectrometry identified phosphorylation of amino acids T125 and S204, among others (Figure 2B and Supplemental Table S3). Thus these are authentic phosphorylation sites on Kif2b during mitosis.
To demonstrate that Plk1 phosphorylates Kif2b directly at these sites, we performed in vitro phosphorylation assays with purified Plk1 and a segment of recombinant human Kif2b (Figure 3 and Supplemental Table S4). Full-length human Kif2b and a variety of truncated segments are insoluble when expressed in either bacteria or insect cells. We succeeded in getting small quantities of one fragment of Kif2b (Kif2bW115-N544) to remain soluble after renaturation after purification in the presence of urea. This protein was incubated with purified active Plk1 kinase, and the MS/MS spectra of phosphorylated peptides derived from this in vitro phosphorylation experiment were compared with the original MS/MS spectra from phosphorylated Kif2b immunoprecipitated from cells (Figure 3A). Phosphorylation spectra were similar between in vitro and in vivo conditions for peptides containing T125, S200, S201, and S204 (Figure 3B). Taken together with the sensitivity of phosphorylation of these sites to Plk1 inhibition, these data demonstrate that these sites on Kif2b are direct targets of phosphorylation by Plk1 during mitosis. The site S575 was not present in the fragment of Kif2b that we used in the in vitro phosphorylation assay and thus may be either a direct or indirect target of Plk1.
FIGURE 3:

In vitro phosphorylation of Kif2b by Plk1. (A) Schematic diagram showing how spectra from mass spectrometry from in vitro and in vivo derived protein samples are compared. (B) Comparison of in vitro and in vivo spectra for two different peptides of Kif2b.
Functional evaluation of Kif2b phosphorylation
To determine whether the phosphorylation sites identified by mass spectrometry regulate Kif2b function, we tested the localization and error correction activity of GFP-Kif2b harboring mutations in these sites in human mitotic cells. We selected a subset of phosphorylation sites for this analysis. First, we included the sites that are direct targets of Plk1 (T125, S200, S201, S204), and the site that is sensitive to Plk1 inhibition (S575). We also included the site that is highly conserved and homologous to the Aurora B phosphorylation site in MCAK (S147). Finally, we included phosphorylation sites adjacent to the motor domain (S616, S617) for which there was no other functional data to serve as negative controls for mutations in the protein. We mutated each site alone, or in some cases in combination, to either a nonphosphorylatable alanine or a putative phoshomimic glutamic acid or aspartic acid. We then expressed each mutant protein in human U2OS cells using a Tet-inducible system. There was no detectable expression by fluorescence microscopy or immunoblotting in the absence of doxycycline (Supplemental Figure S3). Robust expression was induced by the addition of 1 μg/ml doxycycline (Supplemental Figure S3A). Each protein in this induction system displayed the correct molecular mass as judged by immunoblotting protein from two independent clones, and the level of induction was similar for all proteins (Supplemental Figure S3B).
First, we tested the cell cycle–dependent localization of GFP-Kif2b mutant proteins in mitosis. Wild-type Kif2b is recruited to kinetochores, the spindle, and the spindle poles in prometaphase, and it is removed from kinetochores in metaphase when chromosomes align (Manning et al., 2007). Under doxycycline-induction conditions, wild-type GFP-Kif2b localized appropriately to spindles and kinetochores in prometaphase in a majority of cells (Figure 4, A and B). All single mutants were able to localize to kinetochores normally except for one, the nonphosphorylatable GFP-Kif2bS204A mutant (Figure 4, A and B). GFP-Kif2bS204A localized to kinetochores in <10% of prometaphase cells, compared with an average of 70% of prometaphase cells expressing GFP-Kif2bWT. In contrast, the putative phosphomimic GFP-Kif2bS204D mutant localized normally to kinetochores in prometaphase and was displaced normally from kinetochores in metaphase (Supplemental Figure S4). This localization pattern is equivalent to wild type, indicating that although phosphorylation of S204 is required for Kif2b recruitment to kinetochores, its dephosphorylation is not required for its timely removal, assuming the aspartic acid mutation accurately reflects a phosphorylated state.
FIGURE 4:

A nonphosphorylatable GFP-Kif2bS204A mutant fails to localize to kinetochores in prometaphase. (A) Representative images of U2OS cells expressing wild-type or mutant GFP-Kif2b (green). Cells were fixed and stained for DNA (blue) and centromeres (red). Scale bar, 5 μm. (B) Percentage of prometaphase cells with GFP-Kif2b localized to kinetochores. Error bars represent mean ± SEM. *p < 0.05, chi-squared test, n = 400 cells from two clonal populations, two experiments each.
It was previously shown that the protein astrin competes with Kif2b for binding of Clasp1 at kinetochores and that recruitment of astrin to Clasp1 is required for the removal of Kif2b in metaphase and subsequent stabilization of k-MT attachments (Manning et al., 2010). Conversely, Kif2b occupancy of Clasp1 prevents astrin localization in prometaphase. To determine whether astrin localization is perturbed in the presence of Kif2b mutants, we induced GFP-Kif2b mutant expression and then stained for astrin protein (Supplemental Figure S5). In cells expressing GFP-Kif2bWT, astrin is present on the spindle and the spindle poles in prometaphase and is recruited to kinetochores in metaphase. None of the GFP-Kif2b mutants analyzed altered astrin localization in prometaphase or metaphase (Supplemental Figure S5, A and B). This result was expected, since none of the GFP-Kif2b mutants remains on kinetochores in metaphase.
We further quantified the kinetochore localization of Kif2b mutants as relative fluorescence intensity (Supplemental Figure S6). In cells expressing GFP-Kif2bWT, the GFP fluorescence intensity at kinetochores is approximately double the intensity of the background cytosolic GFP intensity. In contrast, the fluorescence intensity of GFP-Kif2bS204A at kinetochores is no greater than its intensity in the cytosol. This is equivalent to the reduction in GFP-Kif2b kinetochore intensity observed in cells treated with hesperadin or BI-2536. Conversely, the kinetochore intensity of GFP-Kif2bS204D was equivalent to that of GFP-Kif2bWT. Thus, among all the phosphorylation sites that we tested, only one site regulates the recruitment of Kif2b to kinetochores during prometaphase. This site is a substrate for Plk1, demonstrating that phosphorylation of Kif2b at S204 by Plk1 is required for targeting of Kif2b to kinetochores in early mitosis.
Next we tested whether phosphorylation of specific residues regulates Kif2b function in correcting defects in k-MT attachments that lead to lagging chromosomes in anaphase. Aneuploid and chromosomally unstable U2OS cells display at least one lagging chromosome in ∼30% of anaphases, indicating inherent defects that increase the prevalence of k-MT attachment errors (Figure 5, A and B). On induction of expression of GFP-Kif2bWT, the rate of defective anaphases is reduced to ∼15% (Figure 5B). This provides a functional readout for the biological activity of Kif2b in cells, as shown previously (Bakhoum et al., 2009b). All single phosphomutants of GFP-Kif2b analyzed successfully reduced the rate of defective anaphases to levels statistically equivalent to wild type, except for two. The GFP-Kif2bS204A nonphosphorylatable mutant failed to correct k-MT attachment defects, which was expected since it fails to localize to kinetochores in prometaphase, when Kif2b is responsible for k-MT attachment error correction. The other mutant that failed to correct k-MT attachment errors was GFP-Kif2bT125A. The proportion of cells displaying lagging chromosomes in anaphase in the presence of GFP-Kif2bT125A is equivalent to cells in which expression of exogenous GFP-Kif2b has not been induced, suggesting that this mutant is completely inactive in correcting k-MT attachment errors despite the fact that it localizes correctly to kinetochores in prometaphase. Conversely, the putative phosphomimic GFP-Kif2bT125E and GFP-Kif2bS204D mutants rescued k-MT attachment errors as efficiently as wild-type GFP-Kif2b. There is no significant difference in the mitotic indices in these cells, indicating that differences in error correction efficiency is not the result of differences in time spent in prometaphase and metaphase (Supplemental Figure S7, A and B). Thus, among all the phosphorylation sites tested, only two regulate the functional ability of Kif2b to correct k-MT attachment errors during prometaphase. These sites are direct substrates for Plk1, demonstrating that phosphorylation of Kif2b at T125 by Plk1 is required for the functional activity of Kif2b in correcting k-MT attachment errors in early mitosis, whereas phosphorylation of Kif2b at S204 is required for its timely kinetochore localization.
FIGURE 5:

Nonphosphorylatable GFP-Kif2bT125A and GFP-Kif2bS204A mutants fail to correct k-MT attachment errors. (A) Representative images of anaphase U2OS cells expressing wild-type or mutant GFP-Kif2b (green). Cells were fixed and stained for DNA (blue) and centromeres (red). Scale bar, 5 μm. (B) Percentage of anaphase cells with lagging chromosomes in cells expressing wild-type or mutant GFP-Kif2b compared with U2OS cells in which expression has not been induced. Error bars represent mean ± SEM. *p < 0.05, chi-squared test, n = 400 cells from two clones, two experiments each.
As noted earlier, some of the phosphorylation sites identified are found in tight clusters of serine residues, so we tested the localization and function of proteins with multiple mutations within each cluster. All of the double (GFP-Kif2bS200A/S201A, GFP-Kif2bS616A/S617A) and triple (GFP-Kif2bS200A/S210A/S204A) nonphosphorylatable alanine mutant proteins failed to localize to kinetochores or the spindle and rarely to spindle poles, and yet they exacerbated the level of anaphase cells with lagging chromosomes (Supplemental Figure S8, A and B). Conversely, proteins with multiple aspartic acid mutations localized normally but failed to correct k-MT attachment errors (Supplemental Figure 8, A and B). Therefore the phenotypes demonstrated by the double and triple mutants are worse than those of the single mutants. This suggests that either the phosphoregulation of Kif2b is more complex than the single mutants indicate or they do not accurately reflect the physiological function of the protein (e.g., the multiple-alanine mutants may be dominant negative).
Kif2b corrects improper k-MT attachments through its microtubule depolymerization activity (Bakhoum et al., 2009b). To determine whether the observed decreases in error correction activity of GFP-Kif2bT125A and GFP-Kif2bS204A mutants are due to a reduction in their microtubule depolymerization activity, we measured microtubule density in vivo. GFP-Kif2b mutant constructs were transiently transfected into U2OS cells, and fluorescence intensities of total microtubule polymer in expressing versus nonexpressing mitotic cells was determined. In cells transiently transfected with GFP alone, the total microtubule polymer intensity was equivalent to the intensity in nonexpressing cells. In cells transfected with GFP-Kif2bWT, the microtubule intensity was significantly reduced (Figure 6), as previously described (Manning et al., 2007). This reduction was observed in mitotic cells with either monopolar (monastrol treated) or bipolar (MG-132 treated) spindles. As expected, in cells transfected with GFP-Kif2bT125A, the total microtubule populations were equivalent to untransfected cells, indicating that a T125A mutation completely inactivates Kif2b's microtubule depolymerization activity. Conversely, GFP-Kif2bT125E expression reduced microtubule populations as efficiently as wild type. Surprisingly, the S204A mutation, which prevents Kif2b localization to kinetochores, also completely abolished Kif2b's microtubule depolymerization activity (Figure 6). GFP-Kif2bS204D depolymerized microtubules as efficiently as wild type. Therefore Plk1 phosphorylation of Kif2b at T125 is required for Kif2b's microtubule depolymerization activity, whereas phosphorylation at S204 is required for both Kif2b localization and activity.
FIGURE 6:
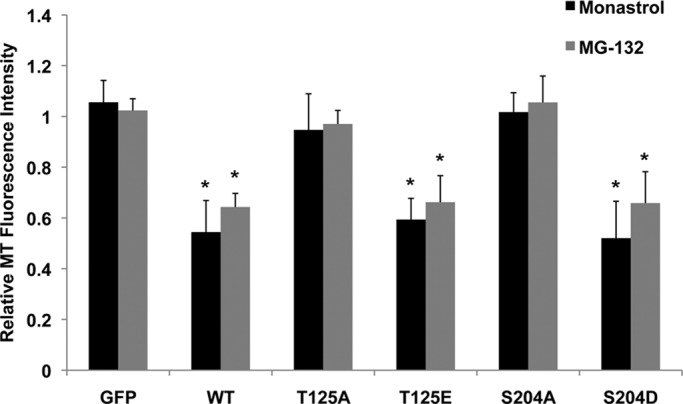
In vivo microtubule depolymerization activity of GFP-Kif2b mutants. U2OS cells were transiently transfected with GFP alone, wild-type GFP-Kif2b, or mutant GFP-Kif2b, arrested with monastrol or MG-132, and then fixed and stained for microtubules and DNA. Fluorescence intensity of monopolar (monastrol) and bipolar (MG-132) spindles is reported as the ratio of fluorescence intensity in transfected cells to untransfected cells in the same field of view. *p < 0.0001, t test, n = at least 30 cells per condition, three experiments.
Finally, it is noteworthy that mutations at the Aurora B consensus site, GFP-Kif2bS147A and GFP-Kif2bS147D, localized normally to kinetochores, the spindle, and the spindle poles in prometaphase and were absent from kinetochores in metaphase (Figure 4B). Both mutants were also as effective at correcting k-MT attachment errors as wild-type Kif2b, as judged by the percentage of anaphase cells with lagging chromosomes (Figure 5B). Thus, despite our evidence for phosphorylation of S147 and the apparent homology of this site to a key Aurora B regulation site on MCAK (Andrews et al., 2004; Lan et al., 2004), these data demonstrate that the phosphorylation state of this residue alone does not influence the normal function of Kif2b.
DISCUSSION
Faithful chromosome segregation relies on a robust machinery to correct k-MT attachment errors that frequently arise in early mitosis (Cimini et al., 2001). The microtubule depolymerase Kif2b has been shown to execute this error correction function in early mitosis (Bakhoum et al., 2009b), and we demonstrate that the temporal regulation of Kif2b is mediated by phosphorylation by Plk1 (Figure 7). Plk1 has been implicated in regulating other kinesin-13 family members in mammalian cells, including Kif2a (Jang et al., 2009) and MCAK (Zhang et al., 2011), although the sites of phosphorylation on Kif2a are unknown, and the conclusion that Plk1 regulates MCAK was based on the simultaneous mutation of six putative phosphorylation sites. Of note, one of these sites was homologous to S575 on Kif2b, a residue that was sensitive to Plk1 inhibition but whose phosphorylation state did not affect Kif2b function. Our data provide direct evidence that Plk1 phosphorylates Kif2b on two sites to regulate its cellular activity. Plk1 phosphorylates Kif2b at serine 204 to promote its recruitment to kinetochores in prometaphase. In addition, it phosphorylates Kif2b at threonine 125 and serine 204 to activate its function in promoting the correction of k-MT attachment errors to foster faithful chromosome segregation.
FIGURE 7:
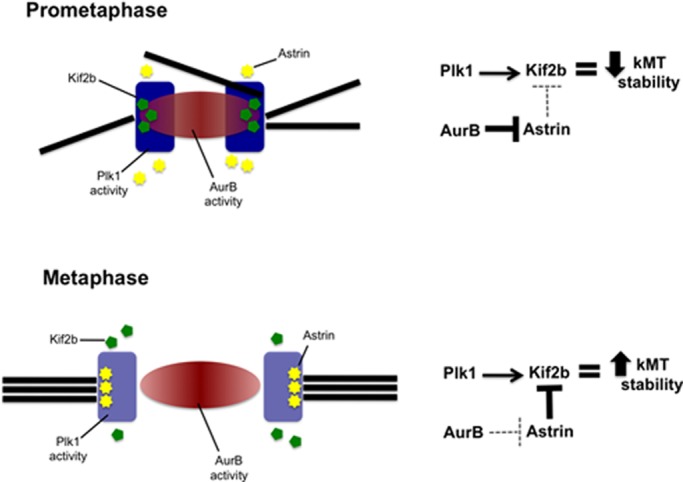
Model of Kif2b regulation at the kinetochore in prometaphase and metaphase. In prometaphase, Plk1 phosphorylates Kif2b to recruit it to kinetochores and activate it for the purpose of correcting attachment errors. Aurora B strongly prevents astrin from displacing Kif2b at this time (solid line). In metaphase, sister kinetochores separate. This weakens the influence of Aurora B on astrin (dashed line), which subsequently blocks Kif2b from localizing to kinetochores, resulting in stabilization of kinetochore-microtubule attachments.
Plk1 levels are high on kinetochores of unaligned chromosomes in prometaphase (Ahonen et al., 2005; Lénárt et al., 2007), positioning it at the right place and time to regulate Kif2b activity. Moreover, inactivation studies have shown that Plk1 is an important regulator of the initial formation of k-MT attachments (Sumara et al., 2004; Lénárt et al., 2007) and may regulate the conversion of MT sidewall attachments into end-on attachments at kinetochores (Lénárt et al., 2007). Other evidence suggests that Plk1 activity is modulated by the competing activity of phosphatase PP2A to support appropriate k-MT attachment stability during mitosis (Foley et al., 2011). However, those studies largely relied on the complete inactivation of Plk1 function, making it difficult to determine the relative contributions of specific Plk1 substrates to regulating k-MT attachment stability. For example, BubR1 is required for stable k-MT attachment (Lampson and Kapoor, 2005), and the ability of BubR1 to stabilize k-MT attachments is dependent on Plk1 phosphorylation (Elowe et al., 2007). Our work demonstrates that Plk1 also activates Kif2b to destabilize k-MT attachments. Thus the mechanism through which Plk1 fine-tunes k-MT attachments during mitosis is complex and involves the regulation of multiple substrates, including both k-MT attachment stabilizers (BubR1) and destabilizers (Kif2b).
Kif2b localization to kinetochores in prometaphase is also sensitive to inhibition of Aurora kinases. Aurora B plays a major role in the correction of k-MT attachment errors by facilitating the destabilization of k-MT attachments through regulation of a variety of substrates, including the microtubule depolymerase MCAK (Lampson and Cheeseman, 2011). A current model for how Aurora B mediates these effects is through the creation of a spatially defined phosphorylation gradient emanating from the inner centromere and reaching the kinetochore in prometaphase (Liu et al., 2009; Lampson and Cheeseman, 2011). This is an attractive model for the regulation of Kif2b (as proposed previously; Bakhoum et al., 2009a; Manning et al., 2010), and yet our quantitative mass spectrometry provided no evidence that Aurora B directly phosphorylates Kif2b. This can be reconciled in a model in which Kif2b is regulated directly by Plk1 and indirectly by Aurora B (Figure 7). The protein astrin has been shown to remove Kif2b from kinetochores in metaphase through competitive binding of CLASP1 (Manning et al., 2010). During prometaphase, Aurora B kinase activity prevents astrin from localizing to kinetochores (Manning et al., 2010; Schmidt et al., 2010). This permits Kif2b to localize to kinetochores to destabilize k-MT attachments to execute error correction through Plk1-dependent recruitment and activation. However, in metaphase, the separation between sister kinetochores increases by spindle-applied forces on bioriented chromosomes, altering the extent of the Aurora B phosphorylation gradient. This allows astrin to effectively compete for CLASP1 binding, thus removing Kif2b from kinetochores and stabilizing k-MT attachments. This model is contrary to how Aurora B directly regulates MCAK localization and activity and provides a molecular explanation for how Kif2b and MCAK activities are regulated in a temporally distinct manner (Bakhoum et al., 2009b).
MATERIALS AND METHODS
Cell culture
Human U2OS cells were maintained at 37°C in a 5% CO2 atmosphere in DMEM containing 10% fetal bovine serum (FBS), 50 IU/ml penicillin, and 50 μg/ml streptomycin. Tet-inducible GFP-Kif2b U2OS cells were maintained in DMEM containing 10% Tet system-approved FBS (Clontech, Mountain View, CA), 50 IU/ml penicillin, and 50 μg/ml streptomycin and selected with 500 μg/ml geneticin and 300 μg/ml hygromycin. For SILAC experiments, GFP-Kif2b U2OS cells were grown in arginine- and lysine-free DMEM with 10% dialyzed FBS supplemented with [13C6,15N2]lysine (75 mg/l) and [13C6,15N4]arginine (75 mg/l) (Cambridge Isotope Laboratories, Andover, MA; heavy population) or identical concentrations of isotopically normal lysine and arginine (light population) for six cell doublings.
Plasmids and site-directed mutagenesis
Plasmid encoding GFP-tagged Kif2b was a gift of L. Wordeman (University of Washington, Seattle, WA). Tet-inducible GFP-Kif2b expression constructs were created in the pTRE-Tight vector (Clontech). GFP-Kif2b was digested with NheI and NotI restriction enzymes and inserted into the NheI/NotI site of pTRE-Tight. Mutant forms of Kif2b were obtained by PCR amplification using the Change-It Multiple Site-Directed Mutagenesis Kit (US Biological, Swampscott, MA). All mutations were confirmed by DNA sequencing (Dartmouth Molecular Biology Core Facility, Hanover, NH).
Plasmid encoding the Kif2bW115-N544 truncation was generated by PCR using forward (5′-GCGGGATCCTGGGTTGCGATGATC-3′) and reverse (5′-GCGCTCGAGTTAATTTAATTTTTTTACTCTGTTTGC-3′) primers containing BamHI/XhoI restriction sites. The truncation was inserted into the BamHI/XhoI site of the pET-98 vector (gift of Jared Cochran, Indiana University, Bloomington, IN). The pET-98 vector is constructed from pET-16b digested with BamHI and NcoI and ligated with an adaptor sequence containing a hexahistidine tag followed by a Tobacco Etch Virus protease site and a multiple cloning site.
Plasmid transfection
For stable inducible cell lines, U2OS cells were transfected with pTet-ON-Advanced vector (Clontech) using FuGENE 6 (Roche, Indianapolis, IN) according to the manufacturer's instructions and placed under 500 μg/ml geneticin selection for ∼21 d. Clones were isolated and screened for stable expression of pTet-ON by transient transfection of pTRE-Tight-GFP-Kif2b, followed by induction with 1 μg/ml doxycycline. Stably expressing pTet-ON clonal cell lines were transfected with pTRE-Tight-GFP-Kif2b constructs and placed under 500 μg/ml geneticin and 300 μg/ml hygromycin selection for ∼21 d. Clones were isolated and screened for stable, inducible expression of GFP-Kif2b. Two independent clones with the brightest expression were analyzed for each Kif2b construct.
For transient transfections, U2OS cells were transfected with eGFP-Kif2b vectors using FuGENE 6 and analyzed after 24 h.
Antibodies
The following antibodies were used: Kif2b-specific antibody (Manning et al., 2007), tubulin-specific DM1α (Sigma-Aldrich, St. Louis, MO), CREST antibody (gift of Kevin Sullivan, Scripps Institute, La Jolla, CA), actin-specific monoclonal antibody (gift of H. Higgs, Dartmouth Medical School, Hanover, NH), and astrin-specific antibody (Mack and Compton, 2001). Antibodies were used at dilutions of 1:1000.
Immunoblotting
Proteins were resolved by SDS–PAGE and transferred to polyvinylidene fluoride membrane (Millipore, Billerica, MA). Primary antibodies were incubated for 3 h at room temperature in 1% milk Tris-buffered saline (TBS). Primary antibody was detected using horseradish peroxidase–conjugated secondary antibodies (Bio-Rad, Hercules, Ca) diluted in TBS for 1 h at room temperature. The signal was detected using chemiluminescence.
Indirect immunofluorescence microscopy
Cell fixation and antibody staining were performed as described previously (Manning et al., 2007). Cells were fixed with ice-cold methanol. Images were acquired with a Hamamatsu Orca ER cooled charge-coupled device camera (Hamamatsu, Hamamatsu, Japan) mounted on a Nikon TE-2000E microscope (Nikon, Melville, NY) with a 60×, 1.4 numerical aperture objective. Image series in the z-axis were obtained using 0.2-μm optical sections. Image deconvolution and contrast enhancement was performed using Elements software (Nikon). Final images represent selected overlaid planes.
Small-molecule inhibitors and treatment of SILAC GFP-Kif2b U2OS cells
The following drugs were used: monastrol (100 μM; Tocris Bioscience, Ellisville, MO), nocodazole (0.1 ng/ml; Sigma-Aldrich), hesperadin (50 nM; Tarun Kapoor, The Rockefeller University, New York, NY), BI-2536 (200 nM; synthesized in house), and MG132 (5 μM; Sigma-Aldrich). Drug washouts were performed using three washes with PBS, followed by fresh growth medium.
To synchronize in prometaphase, heavy- and light-labeled GFP-Kif2b U2OS cells were treated with 0.1 ng/ml nocodazole for 16 h. To synchronize in metaphase, cells were treated with nocodazole and then washed out into 5 μM MG-132 for 1 h. Heavy-labeled cells were incubated with either 50 nM hesperadin or 200 nM BI-2536 at the indicated concentration along with nocodazole plus MG-132 or MG-132 alone for 30 min. After inhibitor treatment, cells were collected by mitotic shake-off and counted. Equal numbers of heavy and light cells were mixed and washed twice in PBS before lysis.
Immunoprecipitation
Immunoprecipitation was performed as previously described (Compton and Luo, 1995). Briefly, ∼107 GFP-Kif2b U2OS cells were lysed in 450 μl of extraction buffer (2% SDS, 50 mM Tris-HCl, pH 6.8, 1 mM EDTA, 2 mM ethylene glycol tetraacetic acid [EGTA], 1 mM dithiothreitol [DTT], 10 mM sodium fluoride, 10 mM sodium pyrophosphate) and heated to 100°C. Samples were clarified by centrifugation at 13,000 × g for 15 min. The supernatant was then transferred to a new tube and diluted eightfold with SDS-scavenging buffer (20 mM Tris-HCl, pH 7.4, 140 mM NaCl, 10 mM sodium pyrophosphate, 3.4% Triton X-100). Extracts were split in half and incubated with either 60 μg of rabbit preimmune immunoglobulin G or anti-Kif2b antibody at 4°C for 12 h with gentle agitation. Then 50 μl of protein A–conjugated agarose bead slurry (Roche) was added and the extracts were incubated for 2 h at 4°C with gentle agitation. Beads were collected by centrifugation and washed five times with 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7, 75 mM KCl, 1 mM MnCl2, 2 mM EGTA, 4 mM MgCl2, and 3 mM DTT. Protein was eluted by boiling in SDS–PAGE sample buffer and then reduced with 5 mM DTT at 55°C for 30 min and alkylated with 15 mM iodoacetamide at room temperature in the dark for 15 min. Samples were resolved by SDS–PAGE and visualized with Novex Colloidal Blue stain (Invitrogen, Carlsbad, CA) or immunoblotting.
Immunoprecipitation of endogenous Kif2b was as described, except that ∼108 U2OS cells synchronized with nocodazole were lysed in 4.5 ml of extraction buffer and diluted eightfold in SDS-scavenging buffer. Extracts were incubated with 120 μg of anti-Kif2b antibody and then 100 μl of protein A–conjugated agarose bead slurry.
Expression and purification of Kif2bW115-N544
Hexahistidine-tagged recombinant Kif2bW115-N544 protein was expressed in BL21-CodonPlus(DE3)-RIL Escherichia coli (Agilent, Santa Clara, CA). A single colony was grown in 2 ml of ZYP-0.8G (ZY media plus 2 mM MgSO4, 40% glucose, 25 mM NH4SO4, 50 mM KH2PO4, 25 mM Na2HPO4) plus 50 μg/ml ampicillin and 25 μg/ml chloramphenicol for at 37°C for 5 h at 300 rpm. The 2-ml culture was added to 800 ml of LB (lysogeny broth) plus 2 mM MgSO4, 50 μg/ml ampicillin, and 25 μg/ml chloramphenicol in a 3-l baffled flask and grown at 37°C at 300 rpm to reach and OD600 of 0.4–0.5. Protein expression was induced by adding 0.4 mM isopropyl-β-d-thiogalactopyranoside. The culture was continually grown at 20°C at 150 rpm for 16 h. The bacteria was harvested at 5000 rpm for 20 min at 4°C, washed with wash buffer (10 mM sodium phosphate buffer, pH 7.2, 20 mM NaCl, 1 mM EGTA, 2 mM MgCl2, 2 mM DTT, 2 mM phenylmethylsulfonyl fluoride), and centrifuged at 5000 rpm for 30 min at 4°C, and the bacterial pellet was stored at −80°C. The pellet was resuspended in 100 ml of ice-cold lysis buffer (wash buffer plus 0.1 mM ATP, 20 μg/ml lysozyme, 0.2% Triton X-100), and the cell mixture was stirred on ice for 30 min. The cell mixture was sonicated, and then centrifuged at 10,000 rpm at 4°C for 30 min. The enriched inclusion body pellet was resuspended in 40 ml of solubilization buffer (100 mM NaH2PO4, 10 mM Tris-Cl, 8 M urea, pH 8.0), stirred at room temperature for 1 h, and then sonicated. The solubilized mixture was centrifuged at 10,000 rpm at 20°C for 30 min. The supernatant was collected and incubated with 1 ml of nickel-nitriloacetic (Ni-NTA) acid agarose beads (Qiagen, Valencia, CA) and mixed at 4°C for 1 h. Beads were collected by centrifugation at 5000 rpm for 5 min and washed with 40 ml of wash buffer (solubilization buffer, pH 6.3) before loading onto a 5-ml column and further washing with 40 ml of wash buffer. Protein was eluted with 5 ml of elution buffer (solubilization buffer, pH 4.5). Pooled fractions were dialyzed against Plk1 kinase buffer (see later discussion), and protein concentration was determined by Bradford assay (Thermo Scientific, Waltham, MA).
Plk1 purification
Plk1 was purified as previously described (Kettenbach et al., 2011). Briefly, Plk1 was amplified from a sequenced cDNA clone and cloned into a modified version of the pFastBac vector (Invitrogen) containing a 10-histidine tag. For bacmid generation, pFastBac constructs were transformed into DH10Bac E. coli (Invitrogen). Recombinant bacmid DNA was purified, and recombination was confirmed by PCR. Recombinant bacmid DNA was transfected into Sf9 cells using Cellfectin (Invitrogen) according to the manufacturer's instructions. Five days after transfection, P1 virus stock was isolated and further amplified. For protein expression, Sf9 cells were infected with amplified virus stocks, and cells were harvested 72 h after infection. Three hours before harvesting, cells were treated with 100 nM okadaic acids (LC Labs, Woburn, MA). Ten-histidine–tagged Plk1 was purified using Ni-NTA agarose (Qiagen) according to the manufacturer's instructions. Purified proteins were dialyzed overnight against 10 mM HEPES pH 7.7, 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT, and 10% glycerol and stored at −80°C.
In vitro kinase reactions
For Plk1 in vitro kinase reactions, 7.5 μg of Kif2b115-544 was incubated with 150 ng of Plk1 kinase in 50 μl of Plk1 kinase buffer (20 mM HEPES, pH 7.5, 20 mM MgCl2, 0.1 mM DTT, 2.5 mM β-glycerophosphate, and 250 μM ATP) at 30°C for 2 h. For control reactions, 7.5 μg of Kif2b115-544 was incubated alone in Plk1 kinase buffer. Samples were prepared for mass spectrometry as described.
LC-MS/MS
Kif2b gel bands were excised, destained, trypsin digested, and analyzed by nanoscale microcapillary LC-MS/MS on a LTQ-Orbitrap (Thermo Scientific) equipped with an Agilent 1100 capillary HPLC, FAMOS autosampler (LC Packings, San Francisco, CA) and nanospray source (Thermo Fisher Scientific). Raw data were searched using SEQUEST (Eng et al., 1994; Faherty and Gerber, 2010) with a precursor mass tolerance of ±1 Da and requiring fully tryptic peptides with up to two miscleavages, carbamidomethylcysteine as a fixed modification, and oxidized methionine and phosphorylated serine, threonine, and tyrosine as variable modifications. Final search filtering criteria required peptide identifications to have a precursor mass tolerance of <2.5 ppm from theoretical XCorr values of >1.8 and >2.6 for +2 and +3 charge state ions, respectively. SILAC quantification was performed using a highly in-house–modified version of the XPRESS algorithm (http://tools.proteomecenter.org; Han et al., 2001). All heavy-labeled sample/light-labeled sample (H/L) ratios were log2 transformed and adjusted to a calculated, experiment-specific distribution offset (determined by analysis of several abundant whole-cell lysate proteins separated by SDS–PAGE before immunoaffinity purification of Kif2b). For most of these phosphopeptides, sufficient site-determining ions were obtained in the MS2 fragmentation spectra to determine the localization of the phosphorylation site by SEQUEST. In addition, phosphorylation site localization was manually evaluated. For analysis of endogenous Kif2b, phosphopeptide enrichment was performed as previously described (Kettenbach and Gerber, 2011).
Quantification of GFP-Kif2b intensity at kinetochores
Images of U2OS cells stably expressing wild-type or mutant GFP-Kif2b and stained for DNA and CREST were obtained. Kinetochores were identified by CREST staining and circumscribed using the circle tool in Elements software. Total voxel intensity of GFP-Kif2b at kinetochores was measured. Circles of the same size were used for all measurements, and each measurement was performed five times and averaged. Total voxel intensities of GFP-Kif2b in cytosol was measured using the same-sized circle and measured at five different places in the cell. Total voxel intensity of each kinetochore was divided by the total voxel intensity of the cytosol from the same cell. Ratios were averaged from at least 75 kinetochores from at least 10 cells for each condition.
Quantification of microtubule depolymerase activity
Microtubule quantification was performed as previously described (Manning et al., 2007). Briefly, enhanced GFP (eGFP) and eGFP-Kif2b fusion constructs were transiently transfected into cultured cells. Cells were then treated with monastrol (16 h) or monastrol washed out into MG-132 (2 h) and then fixed and stained for tubulin and DNA. Images in the z-axis were obtained containing expressing and nonexpressing cells in the same field of view. The lasso tool in Elements software was used to circumscribe microtubules, and total voxel intensities were obtained. Each measurement was performed three times and averaged. After subtracting background, the total voxel intensity for each expressing cell was divided by the total voxel intensity of a nonexpressing cell in the same field of view. Ratios of expressing to nonexpressing microtubule intensities were averaged in 30 cells from three experiments for each condition.
Supplementary Material
Acknowledgments
We thank Samuel Bakhoum and Amity Manning for the design and construction of some expression constructs and Chris Stroupe, Dean Madden, and Jared Cochran for advice and reagents. This work was supported by National Institutes of Health Grants GM51542 (D.A.C.) and RR01878 (S.A.G.).
Abbreviations used:
- CIN
chromosomal instability
- k-MT
kinetochore-microtubule
- Plk1
polo-like kinase 1
- SILAC
stable isotope labeling of amino acids in cell culture
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-1013) on April 25, 2012.
REFERENCES
- Ahonen LJ, Kallio MJ, Daum JR, Bolton M, Manke IA, Yaffe MB, Stukenberg PT, Gorbsky GJ. Polo-like kinase 1 creates the tension-sensing 3F3/2 phosphoepitope and modulates the association of spindle-checkpoint proteins at kinetochores. Curr Biol. 2005;15:1078–1089. doi: 10.1016/j.cub.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Andrews PD, Ovechkina Y, Morrice N, Wagenbach M, Duncan K, Wordeman L, Swedlow JR. Aurora B regulates MCAK at the mitotic centromere. Dev Cell. 2004;6:253–268. doi: 10.1016/s1534-5807(04)00025-5. [DOI] [PubMed] [Google Scholar]
- Bader JR, Kasuboski JM, Winding M, Vaughan PS, Hinchcliffe EH, Vaughan KT. Polo-like kinase1 is required for recruitment of dynein to kinetochores during mitosis. J Biol Chem. 2011;286:20769–20777. doi: 10.1074/jbc.M111.226605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Kettenbach AN, Loros JJ, Gerber SA, Dunlap JC. Quantitative proteomics reveals a dynamic interactome and phase-specific phosphorylation in the Neurospora circadian clock. Mol Cell. 2009;34:354–363. doi: 10.1016/j.molcel.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Genovese G, Compton DA. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol. 2009a;19:1937–1942. doi: 10.1016/j.cub.2009.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Thompson SL, Manning AL, Compton DA. Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat Cell Biol. 2009b;11:27–35. doi: 10.1038/ncb1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol. 2001;153:517–528. doi: 10.1083/jcb.153.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton DA, Luo C. Mutation of the predicted p34cdc2 phosphorylation sites in NuMA impair the assembly of the mitotic spindle and block mitosis. J Cell Sci. 1995;108:621–633. doi: 10.1242/jcs.108.2.621. [DOI] [PubMed] [Google Scholar]
- Elowe S, Hümmer S, Uldschmid A, Li X, Nigg EA. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 2007;21:2205–2219. doi: 10.1101/gad.436007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ems-McClung SC, Walczak CE. Kinesin-13s in mitosis: key players in the spatial and temporal organization of spindle microtubules. Semin Cell Dev Biol. 2010;21:276–282. doi: 10.1016/j.semcdb.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Faherty BK, Gerber SA. MacroSEQUEST: efficient candidate-centric searching and high-resolution correlation analysis for large-scale proteomics data sets. Anal Chem. 2010;82:6821–6829. doi: 10.1021/ac100783x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley EA, Maldonado M, Kapoor TM. Formation of stable attachments between kinetochores and microtubules depends on the B56-PP2A phosphatase. Nat Cell Biol. 2011;13:1265–1271. doi: 10.1038/ncb2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19:946–951. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauf S, Cole RW, LaTerra S, Zimmer C, Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL, Peters J-M. The small molecule hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C-Y, Coppinger JA, Seki A, Yates J 3rd, Fang G. Plk1 and Aurora A regulate the depolymerase activity and the cellular localization of Kif2a. J Cell Sci. 2009;122:1334–1341. doi: 10.1242/jcs.044321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJPL, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- Kettenbach AN, Gerber SA. Rapid and reproducible single-stage phosphopeptide enrichment of complex peptide mixtures: application to general and phosphotyrosine-specific phosphoproteomics experiments. Anal Chem. 2011;83:7635–7644. doi: 10.1021/ac201894j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of aurora and polo-like kinase activities in mitotic cells. Sci Signal. 2011;4:rs5. doi: 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline-Smith SL, Khodjakov AL, Hergert PJ, Walczak CE. Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol Biol Cell. 2004;15:1146–1159. doi: 10.1091/mbc.E03-08-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton AL, Lan W, Stukenberg PT. Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr Biol. 2006;16:1705–1710. doi: 10.1016/j.cub.2006.07.057. [DOI] [PubMed] [Google Scholar]
- Lampson MA, Cheeseman IM. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol. 2011;21:133–140. doi: 10.1016/j.tcb.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson MA, Kapoor TM. The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nat Cell Biol. 2005;7:93–98. doi: 10.1038/ncb1208. [DOI] [PubMed] [Google Scholar]
- Lan W, Zhang X, Kline-Smith SL, Rosasco-Nitcher SE, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Walczak CE, Stukenberg PT. Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr Biol. 2004;14:273–286. doi: 10.1016/j.cub.2004.01.055. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- Lénárt P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters J-M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Li H, Liu XS, Yang X, Wang Y, Wang Y, Turner JR, Liu X. Phosphorylation of CLIP-170 by Plk1 and CK2 promotes timely formation of kinetochore-microtubule attachments. EMBO J. 2010;29:2953–2965. doi: 10.1038/emboj.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vader G, Vromans M, Lampson MA, Lens SMA. Sensing chromosome bi-orientation by spatial separation of Aurora B kinase from kinetochore substrates. Science. 2009;323:1350–1353. doi: 10.1126/science.1167000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack G, Compton DA. Analysis of mitotic microtubule-associated proteins using mass spectrometry identifies astrin, a spindle-associated protein. Proc Natl Acad Sci USA. 2001;98:14434–14439. doi: 10.1073/pnas.261371298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney T, Hunter AW, Wagenbach M, Wordeman L. Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J Cell Biol. 1998;142:787–801. doi: 10.1083/jcb.142.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Bakhoum SF, Maffini S, Correia-Melo C, Maiato H, Compton DA. CLASP1, astrin and Kif2b form a molecular switch that regulates kinetochore-microtubule dynamics to promote mitotic progression and fidelity. EMBO J. 2010;29:3531–3543. doi: 10.1038/emboj.2010.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Ganem NJ, Bakhoum SF, Wagenbach M, Wordeman L, Compton DA. The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol Biol Cell. 2007;18:2970–2979. doi: 10.1091/mbc.E07-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklas RB, Ward SC. Elements of error correction in mitosis: microtubule capture, release, and tension. J Cell Biol. 1994;126:1241–1253. doi: 10.1083/jcb.126.5.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi R, Burbank K, Liu Q, Mitchison TJ. Nonredundant functions of kinesin-13s during meiotic spindle assembly. Curr Biol. 2007;17:953–959. doi: 10.1016/j.cub.2007.04.057. [DOI] [PubMed] [Google Scholar]
- Ong SE. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Petronczki M, Lénárt P, Peters J-M. Polo on the rise—from mitotic entry to cytokinesis with Plk1. Dev Cell. 2008;14:646–659. doi: 10.1016/j.devcel.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Kung C, Shokat KM, Biggins S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol. 2006;8:78–83. doi: 10.1038/ncb1341. [DOI] [PubMed] [Google Scholar]
- Salmon ED, Cimini D, Cameron LA, Deluca JG. Merotelic kinetochores in mammalian tissue cells. Philos Trans R Soc Lond B Biol Sci. 2005;360:553–568. doi: 10.1098/rstb.2004.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Kiyomitsu T, Hori T, Backer CB, Fukagawa T, Cheeseman IM. Aurora B kinase controls the targeting of the Astrin-SKAP complex to bioriented kinetochores. J Cell Biol. 2010;191:269–280. doi: 10.1083/jcb.201006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumara I, Giménez-Abián JF, Gerlich D, Hirota T, Kraft C, de la Torre C, Ellenberg J, Peters J-M. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14:1712–1722. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–672. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci USA. 2011;108:17974–17978. doi: 10.1073/pnas.1109720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, et al. PLK1 phosphorylates mitotic centromere-associated kinesin and promotes its depolymerase activity. J Biol Chem. 2011;286:3033–3046. doi: 10.1074/jbc.M110.165340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.