

The conserved protein kinase Mps1 is required for the spindle assembly checkpoint (SAC). It is also involved in correction of erroneous attachments of kinetochores to the mitotic spindle before anaphase onset. Characterization of Drosophila Mps1 reveals yet another function: SAC-independent inhibition of sister chromatid separation.
Abstract
Monopolar spindle 1 (Mps1) is essential for the spindle assembly checkpoint (SAC), which prevents anaphase onset in the presence of misaligned chromosomes. Moreover, Mps1 kinase contributes in a SAC-independent manner to the correction of erroneous initial attachments of chromosomes to the spindle. Our characterization of the Drosophila homologue reveals yet another SAC-independent role. As in yeast, modest overexpression of Drosophila Mps1 is sufficient to delay progression through mitosis during metaphase, even though chromosome congression and metaphase alignment do not appear to be affected. This delay in metaphase depends on the SAC component Mad2. Although Mps1 overexpression in mad2 mutants no longer causes a metaphase delay, it perturbs anaphase. Sister kinetochores barely move apart toward spindle poles. However, kinetochore movements can be restored experimentally by separase-independent resolution of sister chromatid cohesion. We propose therefore that Mps1 inhibits sister chromatid separation in a SAC-independent manner. Moreover, we report unexpected results concerning the requirement of Mps1 dimerization and kinase activity for its kinetochore localization in Drosophila. These findings further expand Mps1's significance for faithful mitotic chromosome segregation and emphasize the importance of its careful regulation.
INTRODUCTION
Monopolar spindle 1 (Mps1) is required for the spindle assembly checkpoint (SAC), which inhibits exit from mitosis in the presence of misaligned chromosomes (Musacchio and Salmon, 2007). Moreover, Mps1 also contributes in a SAC-independent manner to the correction of initial erroneous chromosome attachments to the spindle during prometaphase (Jones et al., 2005; Maure et al., 2007; Jelluma et al., 2008b; Hewitt et al., 2010; Santaguida et al., 2010; Sliedrecht et al., 2010).
The mechanistic details of how Mps1 functions in mitotic regulation are complex and still poorly understood. The intracellular localization of Mps1 is highly dynamic (Fisk and Winey, 2001; Stucke et al., 2002; Liu et al., 2003; Fischer et al., 2004; Howell et al., 2004; Jelluma et al., 2010; Zhang et al., 2011). During interphase Mps1 resides primarily in the cytosol, with some enrichment on centrosomes and on the nuclear envelope becoming apparent in late G2. During prometaphase, a fraction of Mps1 accumulates rapidly and strongly on unattached kinetochores, with a residence time of only a few seconds (Howell et al., 2004; Jelluma et al., 2010). Attachment to the spindle correlates with disappearance of Mps1 from the kinetochore.
The high local concentration of Mps1 on unattached kinetochores makes a crucial contribution to SAC activation. It is likely to stimulate Mps1 kinase activity via Mps1–Mps1 protein interactions (Hewitt et al., 2010) and autophosphorylation (Kang et al., 2007; Mattison et al., 2007; Jelluma et al., 2008a; Dou et al., 2011). Apart from Mps1 itself, some additional proteins involved in kinetochore attachment and SAC function (like borealin, BubR1, Cenp-E, Dam1, Mad1, Mad2, and Ndc80) have been shown to be Mps1 kinase substrates (Hardwick et al., 1996; Shimogawa et al., 2006; Espeut et al., 2008; Huang et al., 2008; Jelluma et al., 2008b; Kemmler et al., 2009; Zich et al., 2012). Mps1 kinase activity is also known to regulate accumulation of the SAC components Mad1 and Mad2 at unattached kinetochores (Abrieu et al., 2001; Martin-Lluesma et al., 2002; Liu et al., 2003; Tighe et al., 2008; Hewitt et al., 2010; Kwiatkowski et al., 2010; Maciejowski et al., 2010; Santaguida et al., 2010; Sliedrecht et al., 2010). Moreover, Mps1 activity at the kinetochore has been proposed to stimulate Mad1-dependent conformational change of Mad2 from an open to a closed form (Hewitt et al., 2010; Maldonado and Kapoor, 2011) that is crucial for inhibition of the Cdc20-anaphase–promoting complex/cyclosome (Cdc20-APC/C; Luo et al., 2002; Sironi et al., 2002; DeAntoni et al., 2005; Mapelli et al., 2007; Fava et al., 2011), the ubiquitin ligase that controls anaphase onset.
Besides accumulation at unattached kinetochores, subsequent removal of Mps1 after correct spindle attachment appears to be important. Expression of Mps1 variants fused to protein domains enforcing persistent localization to kinetochores even after correct bipolar attachment to the spindle was shown to prevent timely SAC silencing and anaphase onset in both human cells and fission yeast (Jelluma et al., 2010; Ito et al., 2012). Finally, Mps1 has also been proposed to act in the cytosol, where it promotes the formation of Cdc20-inhibitory complexes, including the SAC component BubR1 (Maciejowski et al., 2010).
The molecular mechanisms controlling Mps1 kinetochore recruitment remain unclear. For example, some studies argued that Mps1's kinase activity is required for recruitment (Xu et al., 2009; Colombo et al., 2010), whereas others found that kinase activity stimulates its release from the kinetochore (Hewitt et al., 2010; Jelluma et al., 2010). The N-terminal region of human Mps1 is required for kinetochore localization, and the kinetochore protein Ndc80/Hec1 is clearly required for Mps1 accumulation at kinetochores (Martin-Lluesma et al., 2002; Saurin et al., 2011). In budding yeast, Mps1 binds directly to Ndc80 (Kemmler et al., 2009), but the corresponding interaction has not been observed in animal cells. Aurora B might stimulate the Ndc80–Mps1 interaction, as this kinase is required for recruitment of normal levels of Mps1 to kinetochores and its activity phosphorylates Ndc80 (Cheeseman et al., 2006; DeLuca et al., 2006; Ciferri et al., 2008), presumably in particular at unattached kinetochores that do not experience physical tension (Liu et al., 2009; Welburn et al., 2010). Mps1 disappearance after bipolar attachment of chromosomes to the spindle might therefore reflect reduced Ndc80 phosphorylation by Aurora B. Mps1 disappearance after kinetochore attachment presumably depends on additional pathways. After attachment, Mps1 is transported along kinetochore microtubules away from kinetochores like other SAC components (Pandey et al., 2007). This shedding of SAC components from the kinetochore is known to be dynein dependent (Howell et al., 2001; Wojcik et al., 2001). Moreover, in budding yeast, APC/C-dependent proteolytic degradation of Mps1 during exit from mitosis prevents SAC reactivation (Palframan et al., 2006). Similarly, Mps1 degradation during exit from mitosis has also been implicated in mammalian cells (Cui et al., 2010).
Here we characterize Mps1 function in Drosophila melanogaster. As revealed by our initial analyses (Fischer et al., 2004), Drosophila Mps1 is essential for SAC function. In addition, it appears to have SAC-independent functions, since the phenotype caused by Mps1-null mutations is more severe than that of mad2-null and bubR1KEN mutants. These last-named mutants clearly revealed that the SAC is not required for development into fertile adults in Drosophila (Buffin et al., 2007; Rahmani et al., 2009). Far fewer Mps1-null mutants develop into adults. Adult Mps1 mutant females missegregate chromosomes during meiosis (Gilliland et al., 2005, 2007). In fact, the first Mps1 allele to be isolated is altered disjunction1 (ald1), which results in chromosome missegregation during meiosis I (O'Tousa, 1982). Here we focus on mitotic functions. Using transgenic strains allowing expression of wild-type and mutant Mps1 versions in different genetic backgrounds, we analyze functional domains and reveal a novel SAC-independent role in the control of sister chromatid separation.
RESULTS
Kinase activity of Drosophila Mps1 is dispensable for self-association but required for kinetochore localization
In vivo imaging of a fully functional enhanced green fluorescent protein (EGFP)–Mps1 protein during the syncytial mitoses of early embryogenesis indicated that Drosophila Mps1 is localized to the kinetochore but only during the early mitotic stages, when the SAC is known to be active (Figure 1A; Fischer et al., 2004). We observed that the C-terminal kinase domain (amino acids 325–630) but not the N-terminal regulatory region (amino acids 1–332) localizes at kinetochores after expression of EGFP fusion proteins from transgenes under control of the normal Mps1 cis-regulatory region (Figure 1B). In contrast, the opposite behavior has been reported for the corresponding regions of human Mps1 (Liu et al., 2003; Stucke et al., 2004). However, consistent with the observations in human cells, a kinase-dead version (Mps1kd, i.e., Mps1D478A) tagged with EGFP displayed normal kinetochore localization (Figure 1B).
FIGURE 1:

Localization and self-interaction of wild-type and mutant Mps1. (A) Mitotic figures from a mitotic wave in a syncytial-stage Drosophila embryo expressing EGFP-Mps1 and the centromere protein Cenp-C-mRFP after fixation and DNA labeling reveal peak levels of Mps1 at kinetochores during prometaphase (left), followed by disappearance from the kinetochore during progression into anaphase (right). EGFP-Mps1 is also detectable on centrosomes and weakly on the spindle. (B) Prometaphase figures from syncytial Mps1+ embryos expressing the following EGFP-tagged Mps1 variants: wild-type (wt), N-terminal regulatory domain (N), C-terminal kinase domain (C), and kinase-dead Mps1kd (kd). Arrowheads indicate kinetochore localization. (C) Larval extracts were used for immunoprecipitation with anti-EGFP after coexpression of an EGFP- and a myc-tagged Mps1 variant during a developmental stage with minimal endogenous Mps1 expression. Immunoblotting of extracts (I) and immunoprecipitates (IP) with anti-EGFP and anti-myc revealed coimmunoprecipitation of the tagged variants. Anti-EGFP does not coimmunoprecipitate myc-Mps1kd from extracts of larvae expressing EGFP only instead of EGFP-Mps1, indicating the specificity of the Mps1 self-interaction. Loading was 1 and 15 larvae equivalents in I and IP lanes, respectively. (D) Prometaphase figures from syncytial Mps1− embryos expressing the EGFP-tagged Mps1 variants described in B. Arrowheads indicate kinetochore localization. In the case of EGFP-Mps1kd, some residual kinetochore localization is only apparent after contrast enhancement (rightmost panels at higher magnification). Bars, 5 μm.
For the interpretation of kinetochore localization of mutant Mps1 versions, it is important to consider the role of endogenous wild-type Mps1. Human myc-Mps1 and GFP-Mps1 can be coimmunoprecipitated (Hewitt et al., 2010), and additional evidence clearly supports Mps1–Mps1 interactions (Hached et al., 2011; Lee et al., 2012). Kinetochore localization of mutant Mps1 versions expressed in the presence of endogenous wild-type Mps1 might therefore reflect recruitment by the latter instead of binding to a distinct kinetochore-docking site. To determine whether Drosophila Mps1 also interacts in-trans with itself, we coexpressed myc-Mps1 and GFP-Mps1 in Drosophila S2R+ cells, followed by an analysis of coimmunoprecipitation (Supplemental Figure S1). These experiments clearly confirmed that Drosophila Mps1 dimerizes like human Mps1. Moreover, the C-terminal kinase domain but not the N-terminal regulatory region was found to associate with full-length Mps1 (Supplemental Figure S1). To determine whether kinase activity is required for this self-interaction in-trans, we coexpressed Mps1kd versions tagged with myc and GFP. These Mps1kd versions were ectopically expressed during larval stages where only a minor fraction of cells proliferate mitotically and express endogenous Mps1+ (Supplemental Figure S2A). The Mps1kd versions expressed from heat-inducible transgenes were found to be coimmunoprecipitated with efficiency similar to that of wild-type Mps1 (Figure 1C). We conclude, therefore, that Mps1 protein kinase activity is not required for self-interaction in-trans.
On the basis of the observed self-interaction properties, it is possible that the localization of the C-terminal kinase domain of Mps1 and Mps1kd to the kinetochore in wild-type embryos (Figure 1B) might reflect recruitment by endogenous Mps1. Moreover, Mps1 kinase activity has been implicated in the control of Mps1 kinetochore localization, although with some puzzling disagreement concerning the direction of its effect (Xu et al., 2009; Colombo et al., 2010; Hewitt et al., 2010; Jelluma et al., 2010). To evaluate whether endogenous wild-type Mps1 is required for the observed kinetochore localization of the mutant Mps1 versions, we expressed them in mutant embryos obtained from females with Mps1aldB4 germline clones. The Mps1aldB4 mutation results in a premature stop after the first 47 amino acids (Page et al., 2007). Neither the N- nor the C-terminal Mps1 fragment localized to kinetochores in the Mps1aldB4-mutant background (Figure 1D), even though these fragments were clearly expressed (Supplemental Figure S2B). In case of Mps1kd, kinetochore signals were dramatically decreased (Figure 1D). Quantification of signal intensities revealed a reduction of Mps1kd at prometaphase kinetochores to 2.5% when compared with wild-type Mps1 expressed analogously in the Mps1aldB4-mutant background (698 arbitrary units [a.u.] ± 71, n = 25, for Mps1, vs. 17 a.u. ± 5, n = 26, for Mps1kd). We conclude, therefore, that Mps1 kinase activity is required for kinetochore localization in Drosophila, in contrast to recent observations in mammalian cells (Hewitt et al., 2010; Jelluma et al., 2010; see Discussion). In case of the C-terminal domain of Drosophila Mps1, we cannot resolve whether its kinetochore localization in wild-type but not mutant background depends on recruitment by endogenous Mps1 or on Mps1 kinase activity.
Apart from kinetochore localization, the EGFP fusions of Drosophila Mps1, Mps1kd, and Mps1C (but not Mps1N) were also detected at the centrosome throughout mitosis after expression in an Mps1+ background (Figure 1, A and B). In the Mps1aldB4-mutant background, only EGFP-Mps1 was detected at centrosomes (Figure 1C).
Mps1–Mad1 interactions
In an attempt to identify the elusive kinetochore component that provides the docking site for Mps1, proteins coimmunoprecipitated by EGFP-Mps1 were characterized by mass spectrometry. Apart from Mps1, we detected a number of highly abundant cellular proteins, reflecting nonspecific associations in all likelihood, and also Mad1/TXBP181-like (unpublished data). Immunoprecipitation (IP) Western experiments clearly confirmed the specificity of the Mps1–Mad1 interaction, which has also been observed in human cells (Lince-Faria et al., 2009). From extracts of transgenic embryos, Mad1-GFP pulled down not only Mad2, as expected (Chen et al., 1999; Sironi et al., 2002), but also Mps1 (Figure 2A). CoIP of Mps1 and Mad1-GFP was also observed after IP with anti-Mps1 instead of anti-GFP (unpublished data). Moreover, both the N- and the C-terminal domains of Mps1 were able to coimmunoprecipitate mCherry-Mad1, although less efficiently than full-length Mps1 and Mps1kd (Figure 2B).
FIGURE 2:

Mps1–Mad1 interaction and kinetochore localization dependences. (A) Extracts from embryos expressing either Mad1-GFP or GFP only were used for immunoprecipitation with anti-EGFP. Immunoblotting of extracts (I) and immunoprecipitates (IP) with anti-EGFP, anti-Mps1, and anti-Mad2 revealed that Mps1 and Mad2 are coimmunoprecipitated specifically with Mad1-EGFP. Loading was 30 and 925 embryo equivalents in I and IP lanes, respectively. The position of molecular weight markers is indicated on the left. A cross-reaction of anti-Mps1 is marked with an asterisk. (B) Extracts from embryos expressing mCherry-Mad1 and EGFP-tagged wild-type (wt), kinase-dead (kd), N-terminal (N), or C-terminal domain (C) of Mps1 were used for immunoprecipitation with anti-EGFP. Immunoblotting of extracts (I) and immunoprecipitates (IP) with anti-EGFP (EGFP) and anti-mCherry (mCherry-Mad1) indicated that all of the Mps1 variants associate with Mad1, although with reduced efficiency in case of the N- and C-terminal domains. However, even less mCherry-Mad1 was coimmunoprecipitated when EGFP only instead of an Mps1 fusion was expressed. Loading was 30 and 300 embryo equivalents in I and IP lanes, respectively. (C) Kinetochore localization of EGFP-Mps1 during prometaphase was analyzed in syncytial embryos from mothers that were wild type (wt), mad1−, or mad2−. (D) Kinetochore localization of Mad1-GFP during prometaphase was analyzed in syncytial embryos from mothers that were wild type (wt), Mps1−, or mad2− (only in the germline in case of Mps1). Bars, 10 μm (C, D) . Arrowheads indicate kinetochore signals. Kinetochore signals were quantified (bar diagrams) using arbitrary units with error bars representing SD. Brackets indicate statistically significant differences (t test) with **p < 0.01 and ***p < 0.001.
To determine whether Mad1 is required for Mps1 localization to kinetochores, we expressed EGFP-Mps1 in mad11/Df(mad1) mutants (Figure 2C). These mutants, which are viable and fertile, do not express Mad1 protein (Emre et al., 2011). Loss of Mad1 resulted in a limited but significant decrease of EGFP-Mps1 on kinetochores according to our quantification during prometaphase of syncytial blastoderm mitoses (Figure 2C). Conversely, Mad1-GFP signals on kinetochores were found to be even more strongly dependent on the presence of Mps1. Apart from a weak, diffuse signal throughout the chromosome region, dot-like kinetochore signals were no longer apparent (Figure 2D). Although most reports from vertebrate cells clearly revealed a strong dependence of Mad1 kinetochore localization on both the presence and activity of Mps1 (Abrieu et al., 2001; Martin-Lluesma et al., 2002; Liu et al., 2003; Vigneron et al., 2004; Wong and Fang, 2006; Zhao and Chen, 2006; Jelluma et al., 2008b; Tighe et al., 2008; Hewitt et al., 2010; Kwiatkowski et al., 2010; Maciejowski et al., 2010; Santaguida et al., 2010; Sliedrecht et al., 2010), quantitative data have not been published to our knowledge. Moreover, the partial dependence of Mps1 kinetochore localization on Mad1 in Drosophila embryos indicates that the Mps1–Mad1 interactions are not strictly hierarchical, and the partial reduction of both EGFP-Mps1 and EGFP-Mad1 on kinetochores in mad2 mutants further emphasizes the complexity of the Mps1–Mad1–Mad2 interdependences.
Level and phosphorylation of Mps1 during progression through mitosis
The observed dependence of Mps1 kinetochore localization on Mad1 is relatively minor and unlikely to explain Mps1 localization dynamics during mitosis. However, APC/C-mediated degradation of Mps1 during exit from mitosis has been implicated in Mps1 regulation in yeast and human cells (Palframan et al., 2006; Cui et al., 2010). To analyze the total levels of Drosophila Mps1 during progression through mitosis, we applied a highly efficient synchronization procedure, including microscopic isolation of embryos in precisely defined mitotic stages. Immunoblotting did not reveal a difference in Mps1 levels before and after the metaphase-to-anaphase transition, whereas cyclin B amounts decreased dramatically, as expected (Figure 3A). We conclude, therefore, that disappearance of Drosophila Mps1 from kinetochores during exit from mitosis does not reflect overall degradation, as also suggested by similar, although less accurately staged analyses from vertebrates (Stucke et al., 2002; Liu et al., 2003; Grimison et al., 2006; Sun et al., 2010).
FIGURE 3:
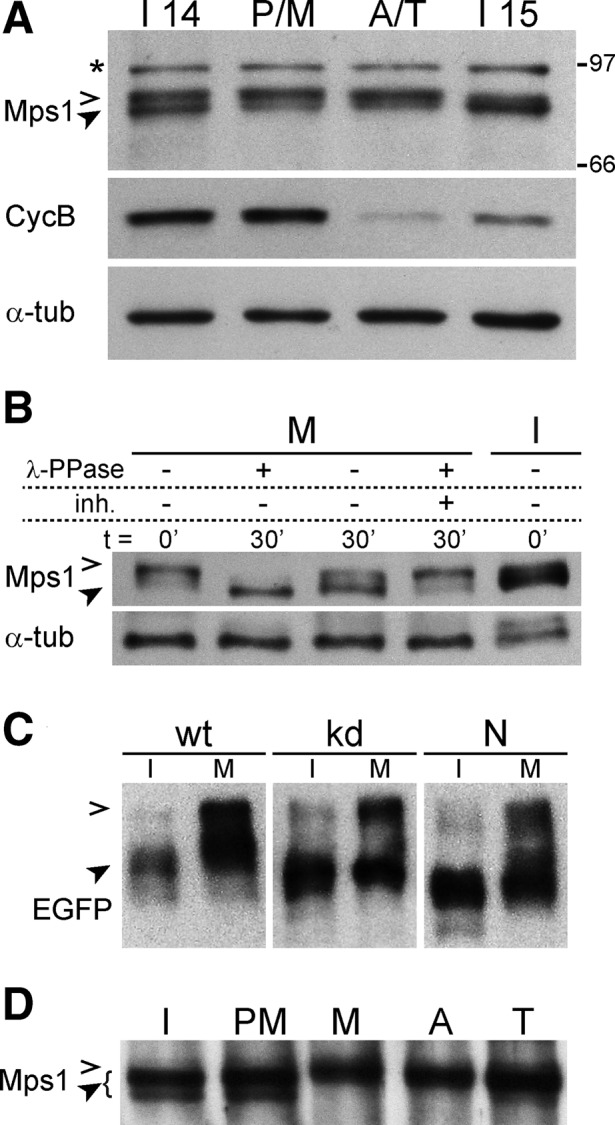
Mps1 stability and phosphorylation during mitosis. Progression through a synchronous mitosis 14 was induced in embryos. Extracts from microscopically selected embryos before mitosis (I14), in prophase and metaphase (P/M), in anaphase and telophase (A/T), and in the subsequent interphase (I15) were analyzed by immunoblotting with antibodies against Mps1, cyclin B, and α-tubulin (loading control) indicating that Mps1 levels do not decrease significantly during exit from mitosis. (B) Extracts from syncytial embryos in mitosis (M) or interphase (I) with (+) or without (–) pretreatment using λ-phosphatase (λ-PPase) and phosphatase inhibitors (inh.) for the indicated times were analyzed by immunoblotting with anti-Mps1 and anti–α-tubulin. (C) Extracts from Mps1-mutant embryos expressing EGFP-tagged wild-type (wt), kinase-dead (kd), or N-terminal domain (N) of Mps1 in interphase (I) or mitosis (M) were resolved on Phos-tag gels and analyzed by immunoblotting with anti-EGFP. (D) Extracts from microscopically selected syncytial embryos in interphase (I), prometaphase (PM), metaphase (M), anaphase (A), and telophase (T) were analyzed by immunoblotting with anti-Mps1. Open and filled arrowheads in A–D indicate phosphorylated low- and high-mobility forms of Mps1, respectively. A cross-reaction of anti-Mps1 is marked with an asterisk. Positions of molecular weight markers are indicated on the right.
Immunoblotting with anti-Mps1 revealed multiple isoforms in embryo extracts (Figure 3A), suggesting that Drosophila Mps1 is a phosphoprotein as previously observed in other organisms (Stucke et al., 2002; Grimison et al., 2006; Palframan et al., 2006; Zhao and Chen, 2006). Phosphatase treatment converted the isoforms with low electrophoretic mobility, which were predominant in M phase, into a faster-migrating species (Figure 3B), demonstrating that Drosophila Mps1 is hyperphosphorylated during mitosis. The corresponding phosphorylation sites are present in the N-terminal region, since only the N- but not the C-terminal region displayed decreased electrophoretic mobility during mitosis (unpublished data).
Autophosphorylation of human Mps1 is known to be required for full kinase activity and SAC function in vivo (Kang et al., 2007; Mattison et al., 2007; Jelluma et al., 2008b; Wang et al., 2009). To evaluate whether the observed hyperphosphorylation of Drosophila Mps1 reflects autophosphorylation, we expressed different EGFP-Mps1 variants in an Mps1-null mutant background and compared their electrophoretic mobility in interphase and mitosis after resolution of extracts on Phos-tag gels (Kinoshita et al., 2009). EGFP-Mps1kd and the N-terminal Mps1 region displayed a mobility shift in M phase similar to that of the wild-type version (EGFP-Mps1; Figure 3C). Therefore we conclude that protein kinases other than Mps1 are involved in mitotic hyperphosphorylation of Mps1.
Mitogen-activated protein kinase (MAPK) phosphorylates Mps1 in Xenopus, allowing kinetochore localization of Mps1 and other checkpoint proteins (Zhao and Chen, 2006). To analyze whether mitotic Mps1 hyperphosphorylation in Drosophila is temporally correlated with its localization on the kinetochore, we performed immunoblotting with extracts prepared from microscopically staged syncytial embryos, where thousands of nuclei progress synchronously through mitosis. In this manner, hyperphosphorylation was found to perdure to late mitotic stages (Figure 3D) when Mps1 is no longer detected on kinetochores and the SAC is inactive. We conclude that the mitotic phosphorylation that causes the Mps1 electrophoretic mobility shift is unlikely to be sufficient for Mps1 kinetochore localization and SAC activation.
Precise control of Mps1 localization and level is crucial for normal mitosis
Although the molecular basis for the disappearance of Mps1 from kinetochores during metaphase is not understood in detail, elegant experiments in human cells recently demonstrated that this process is crucial for SAC silencing and progression from metaphase into anaphase (Jelluma et al., 2010). In these experiments, the normal disappearance of Mps1 from kinetochores was prevented by expression of an Mps1 variant fused to Mis12, a kinetochore component that is present throughout mitosis. Recently similar experiments with an Mps1 variant fused to Ndc80 kinetochore protein were performed in fission yeast (Ito et al., 2012). In independently initiated work, we applied the same experimental strategy, although with different constitutive kinetochore targeting domains. We fused the C-terminal domain of Drosophila Cenp-C, which is sufficient for kinetochore localization (Heeger et al., 2005), and EGFP to Mps1. Apart from this EGFP-CenpCC-Mps1 fusion (EC-Mps1), we also expressed a kinase-dead variant (EC-Mps1kd) during interphase 14 of Drosophila embryogenesis (using the UAS/GAL4 system). When EC-Mps1kd was expressed, we did not observe abnormalities during progression through the subsequent mitosis 14 (Figure 4, A and B). In particular, we emphasize that anaphase and telophase figures were normal, that is, without chromatin bridges. Of importance, GFP but not anti-BubR1 signals were clearly detected at kinetochores during these late mitotic stages, indicating that at least a fraction of EC-Mps1kd indeed persists at the kinetochore throughout mitosis, as expected (Figure 4B). This localized fraction is evidently not sufficiently abundant to outcompete endogenous Cenp-C to an extent incompatible with normal mitosis. However, 10-fold-higher expression levels, as determined by quantitative immunoblotting, of only the EC part without Mps1 resulted in mitotic defects, as expected from a dominant-negative effect on Cenp-C (unpublished data; Heeger et al., 2005).
FIGURE 4:

Mitotic defects caused by alterations in localization and level of Mps1. The UAS/GAL4 system was used for expression of Mps1 variants in embryos before mitosis 14. (A, B) Embryos were fixed at the stage of mitosis 14. The epidermal regions with mitotic domain 10 (Foe, 1989) are displayed after labeling with anti-α-tubulin (α-tub, A), anti-BubR1 (BubR1, B), and a DNA stain (DNA, A and B). As in control embryos without a UAS transgene (w/o UAS), progression through mitosis 14 was normal after expression of kinase-dead EGFP-Cenp-CC-Mps1kd (EC-Mps1kd), which localizes to kinetochores beyond metaphase, as indicated by EGFP signals on kinetochores in anaphase (arrowhead in B). However, chromosome segregation defects during completion of mitosis were apparent after expression of EGFP-Cenp-CC-Mps1 (EC-Mps1), as revealed by DNA bridges with EGFP signals in anaphase and telophase figures (arrow in B). Whereas a strong delay in metaphase was observed after overexpression of EGFP-Mps1 (E-Mps1), progression through mitosis 14 was normal after overexpression of EGFP-Mps1kd (E-Mps1kd). Bars, 10 μm. (C, D) Expression levels of the Mps1 variants at the stage analyzed in A and B were determined by quantitative immunoblotting with antibodies against Mps1, EGFP, and lamin as loading control. Serial dilutions of embryo extracts were loaded with numbers representing embryo equivalents. (C) Compared to Mps1 in wild-type embryos (wt), EGFP-Mps1 (E-Mps1) levels were approximately fivefold higher. (D) Compared to EGFP-Mps1, EGFP-Cenp-CC-Mps1 (EC-Mps1) levels were ∼10-fold lower. A cross-reaction of anti-Mps1 is marked with an asterisk. The positions of wild-type Mps1 and EGFP fusions are indicated by open and filled arrowheads, respectively.
In contrast to EC-Mps1kd, expression of EC-Mps1 caused severe mitotic defects (Figure 4, A and B). Unexpectedly, however, EC-Mps1 did not cause an extended mitotic delay or arrest in metaphase, as predicted if persistent SAC activation was induced. Prophase and metaphase figures were normal in number and appearance. In contrast, cells in anaphase, telophase, and early in the next interphase displayed chromatin bridges (Figure 4, A and B). Centromeric EC-Mps1 signals were observed near these bridges (Figure 4B), indicating a failure of sister kinetochore segregation to spindle poles.
The chromosome segregation defects were obtained after EC-Mps1 expression in embryos with endogenous Mps1. An overall increase in Mps1 levels rather than persistent kinetochore localization might therefore explain the observed defects. To analyze whether excess Mps1 causes mitotic defects, we expressed E-Mps1 (lacking the Cenp-CC localization domain) on top of endogenous Mps1. Strikingly, this resulted in a pronounced mitotic delay during metaphase, as evidenced by a strong enrichment of metaphase figures in fixed embryos (Figure 4A). On the one hand, this result was unexpected. Metaphase delays were not observed in our earlier experiments with Drosophila strains having extra Mps1+ gene copies (Fischer et al., 2004). Similarly, Mps1/Ttk overexpression in mammalian cells does not result in mitotic delays (Stucke et al., 2002; Kang et al., 2007). On the other hand, MPS1 overexpression in budding yeast is clearly sufficient for SAC activation and mitotic arrest (Hardwick et al., 1996). The overexpression applied here resulted in E-Mps1 levels that were about fivefold higher than those of endogenous Mps1 (Figure 4C). Threefold instead of fivefold excess obtained with a weaker transgene insertion was insufficient to cause the same strong metaphase delays (Supplemental Figure S3).
High levels of E-Mps1kd expression did not result in metaphase enrichment or other mitotic defects (Figure 4A and Supplemental Figure S3), indicating that the metaphase delay caused by high levels of E-Mps1 depends on Mps1 kinase activity. Our quantitative immunoblotting experiments also showed that the expression level of EC-Mps1 was below the E-Mps1 level that caused a metaphase delay (Figure 4D), suggesting that EC-Mps1 is a relatively unstable variant.
Kinetochore levels observed after EC-Mps1 and E-Mps1 overexpression were quantified microscopically (Supplemental Figure S4). E-Mps1 kinetochore levels were found to be approximately twofold increased in prometaphase. The overwhelming majority of metaphase cells had such high levels of overexpressed E-Mps1 at kinetochores. In contrast, without overexpression E-Mps1 signals disappear rapidly from kinetochores during metaphase. In anaphase cells, E-Mps1 was no longer detectable on kinetochores even after overexpression. Overexpression of EC-Mps1 resulted in a more limited increase at kinetochores in prometaphase (∼1.8-fold), but these kinetochore signals were largely maintained during exit from mitosis (70% of prometaphase signal intensity during anaphase/telophase).
In summary, EC-Mps1, which persists at kinetochores throughout mitosis, caused chromosome segregation defects (but not a substantial metaphase delay) at an expression level comparable to that of endogenous wild-type Mps1, whereas E-Mps1 resulted in a different mitotic abnormality, that is, a strong metaphase delay, but only after approximately fivefold overexpression. Therefore we conclude that normal exit from mitosis in Drosophila depends critically on both normal localization and normal levels of Mps1.
Apart from Mps1, Bub1, BubR1, and Mad2 play prominent roles in the SAC. Similar to Mps1, these proteins also accumulate on kinetochores during prometaphase and decrease again after chromosome attachment to the spindle before anaphase onset also in Drosophila (Basu et al., 1999; Logarinho et al., 2004; Buffin et al., 2005). To evaluate whether localization and levels of these proteins are equally critical as in the case of Mps1, we performed analogous experiments. However, expression of E-Bub1 and E-BubR1, as well as an E-BubR1 version fused to the constitutive Cenp-CC localization domain, did not perturb progression through mitosis in Drosophila embryos (Supplemental Figure S5). In addition, E-mad2 overexpression did not cause mitotic abnormalities (Supplemental Figure S5).
The mitotic defects observed after enforcing persistent Mps1 maintenance at the kinetochore by EC-Mps1 expression in Drosophila embryos are different from those caused by Mis12-Mps1 in human cells, possibly because of localization differences. The positions of the Cenp-CC domain and of Mis12 within the kinetochore have been determined accurately along the spindle axis by superresolution (Schittenhelm et al., 2007), and Mps1 has been localized at the ultrastructural level (Dou et al., 2003). Accordingly, Mps1 is normally located within the outer corona, whereas Mis12 is further inward and Cenp-CC even more so. In an attempt to enforce Mps1 persistence at a kinetochore location that is as close as possible to normal, we performed experiments with an E-Mps1-Nuf2 fusion, which is predicted to localize Mps1 at the base of the outer corona (DeLuca et al., 2005; Schittenhelm et al., 2007). E-Mps1-Nuf2 expression resulted in metaphase delays. However, this observation cannot be viewed as support for the notion that Mps1 disappearance from the kinetochore is required for SAC silencing because most of the overexpressed E-Mps1-Nuf2 protein did not appear to be localized at the kinetochore, and overall this fusion protein was not more effective in causing metaphase delays than E-Mps1 (Supplemental Figure S6). Similarly, an experiment to address the effect of Mps1 dimerization after ectopic targeting to the cell membrane (Tor-E-Mps1 and Tor4021-E-Mps1) remained inconclusive because of technical complications (Supplemental Figure S6).
Excess Mps1 inhibits sister chromatid separation independent of the SAC
To characterize the metaphase delay resulting from excess Mps1 in further detail, we performed time-lapse in vivo imaging with embryos expressing fluorescent proteins for visualization of chromosomes, kinetochores, and spindles (combinations of histone H2Av–monomeric red fluorescent protein [mRFP] and Cenp-A/Cid-EGFP or Tomato-Cenp-C and Jupiter-GFP; Figure 5). These experiments confirmed that excess Mps1 delayed mitotic progression in metaphase. Although chromosome condensation and congression into the metaphase plate appeared to proceed with the same speed as in control embryos, metaphase was ∼10 times longer after Mps1 overexpression. Thereafter, completion of mitosis occurred with similar kinetics as in controls (Figure 5, A and D).
FIGURE 5:

SAC effects on mitosis in presence of excess Mps1. (A) The dynamics of progression through mitosis 14 was analyzed by time-lapse in vivo imaging of embryos without (wt, white bars) or with UAS/GAL4-mediated Mps1 overexpression (Mps1 OE, black bars). Because embryos also expressed histone H2Av-mRFP and the centromere protein Cenp-A/Cid-EGFP, the duration from prophase onset until metaphase-to-anaphase transition (start–M/A), as well as from metaphase-to-anaphase transition until telophase end (M/A– end), could be determined. Bars indicate average duration (±SD) obtained from >50 mitotic cells from at least six different embryos. (B, C) The width of metaphase plates (B) and separation of sister kinetochores (C) was determined after in vivo imaging of mitosis 14 in embryos expressing the centromere protein Tomato-Cenp-C and the spindle-associated protein Jupiter-GFP (see also D and E). Moreover, embryos were either mad2+ (+) or mad2− (–) and did (+) or did not (–) overexpress Mps1 (Mps1 OE). Box plots display values determined 2 min after nuclear envelope breakdown. The t tests failed to reveal significant differences between the different genotypes. (D, E) Entry into mitosis 14 until metaphase (top) and exit from mitosis 14 (bottom) was analyzed by in vivo imaging of embryos with the same genotypes as in B and C. Comparison of mad2+ embryos (D) either without (wt) or with Mps1 overexpression (Mps1 OE) indicated that excess Mps1 resulted in a substantial metaphase delay, eventually followed by almost normal sister chromatid segregation during anaphase. In contrast, comparison of mad2− embryos (E) either without (wt) or with Mps1 overexpression (Mps1 OE) revealed that the metaphase delay induced by excess Mps1 depends on SAC function. Moreover, sister chromatid segregation was almost completely inhibited during exit from mitosis in mad2− embryos with excess Mps1. Maximum projections of selected stacks at the indicated times are shown from representative cells with spindle poles indicated by open and kinetochores by closed arrowheads. Bars correspond to 5 μm. (F, G) The speed of poleward sister kinetochore segregation during anaphase (F) and their final separation at the end of mitosis (G) in embryos with the same genotypes as in B–E were quantified. Bars represent average (±SD) from 12 mitotic cells from at least three embryos. Brackets indicate statistically significant differences (t test) with ***p < 0.001.
Yeast and human Mps1 are known to destabilize erroneous kinetochore attachments to spindle microtubules (Jones et al., 2005; Maure et al., 2007; Jelluma et al., 2008b; Tighe et al., 2008; Hewitt et al., 2010; Maciejowski et al., 2010; Santaguida et al., 2010; Sliedrecht et al., 2010). In Drosophila, accordingly, the delay of the metaphase-to-anaphase transition might be caused by excess Mps1 via a primary effect on kinetochore attachment and consequent secondary SAC activation. To evaluate this possibility, we analyzed metaphase plate formation. A general impairment of kinetochore attachment as a result of excess Mps1 is expected to result in a widening of the metaphase plate. Therefore we compared the width of metaphase plates in Mps1-overexpressing and control embryos 2 min after nuclear envelope breakdown (NEBD), that is, at a time point that is ∼30 s before anaphase onset in control embryos. Metaphase plates are expected to be widened if some or all chromosomes fail to congress into the metaphase plate (Oliveira et al., 2005). However, we failed to observe a significant difference (Figure 5B). Moreover, we also measured the separation between sister kinetochores, which is known to respond to physical tension resulting from biorientation. In Drosophila embryos, the separation between sister kinetochores in chromosomes with normal bipolar attachment is ∼1.6-fold higher than with unattached chromosomes (Heeger et al., 2005; Pandey et al., 2007). However, we were unable to detect a significant difference in the average sister kinetochore distance when comparing Mps1-overexpressing and control embryos 2 min after NEBD (Figure 5C).
On the basis of our analysis of metaphase plate width and sister kinetochore separation, it did not appear that excess Mps1 caused the observed mitotic delay via destabilization of kinetochore attachments. Therefore we considered the possibility that excess Mps1 might activate the SAC directly without affecting primarily spindle dynamics and/or kinetochore attachment. Accordingly, blocking SAC function downstream of Mps1 is expected to restore a largely normal progression through mitosis in Mps1-overexpressing embryos because the SAC is known to be dispensable for mitosis in Drosophila (Buffin et al., 2007). Therefore we compared progression through mitosis in mad2-null mutants (mad2P) (Buffin et al., 2007) in the presence and absence of Mps1 overexpression (Figure 5E). In mad2 mutants, excess Mps1 no longer resulted in a metaphase delay. We conclude, therefore, that the pause in metaphase caused by Mps1 overexpression in the presence of mad2+ function depends on SAC activity.
Although mitosis was no longer delayed after Mps1 overexpression in mad2-mutant embryos, the completion of mitosis was surprisingly abnormal. Chromosome movement during anaphase was almost completely abolished (Figure 5, E–G). Scoring of the metaphase-to-anaphase transition was therefore based on spindle appearance, which displayed a sudden although limited extension. In contrast, without excess Mps1, anaphases were mostly normal in mad2-mutant embryos (Figure 5, F and G).
The severe failure of chromosome segregation during anaphase resulting from Mps1 overexpression in mad2-mutant embryos prompted us to measure metaphase plate width and sister kinetochore separation also in these embryos. These measurements were performed 2 min after NEBD as previously in the mad2+ background. We did not detect statistically significant differences, indicating that also in the mad2-mutant background Mps1 overexpression did not interfere with chromosome alignment into the metaphase plate. The failure of chromosome segregation during anaphase in mad2-mutant embryos with excess Mps1 therefore is very unlikely to reflect a lack of stable bipolar sister kinetochore attachment.
Like Mps1, BubR1 is also present at maximal levels at unattached kinetochores in prometaphase and strongly diminished in metaphase (Taylor et al., 2001). Moreover, it was proposed that BubR1 levels at kinetochores are inversely correlated with physical tension (Skoufias et al., 2001; Logarinho et al., 2004). Therefore we analyzed the behavior of BubR1-GFP after Mps1 overexpression in mad2+ and mad2− embryos (Figure 6). The rapid initial accumulation of BubR1-GFP on kinetochores after entry into mitosis reached peak levels in prometaphase and was not affected by Mps1 overexpression (Figure 6, A, C, and D). Moreover, Mps1 overexpression did also not affect the initial phase of the subsequent disappearance of BubR1-GFP from kinetochores during metaphase in both mad2+ and mad2− embryos (Figure 6, B–D). However, the late phase of BubR1-GFP disappearance from kinetochores was slowed down by Mps1 overexpression (Figure 6, B–D), in particular from one or two exceptional kinetochores within a given metaphase cell (Figure 6B). At the time of the metaphase-to-anaphase transition BubR1-GFP signals were no longer detectable on kinetochores. These observations suggest that Mps1 overexpression might result in transient detachment of one or two occasional kinetochores, causing SAC activation and metaphase delay in mad2+ embryos. However, the observed BubR1-GFP behavior argues strongly against the interpretation that the severe anaphase defects caused by Mps1 overexpression in mad2− embryos reflect a global inability to form stable kinetochore attachments. The large majority of chromosomes appear to undergo normal attachment and biorientation also after Mps1 overexpression.
FIGURE 6:
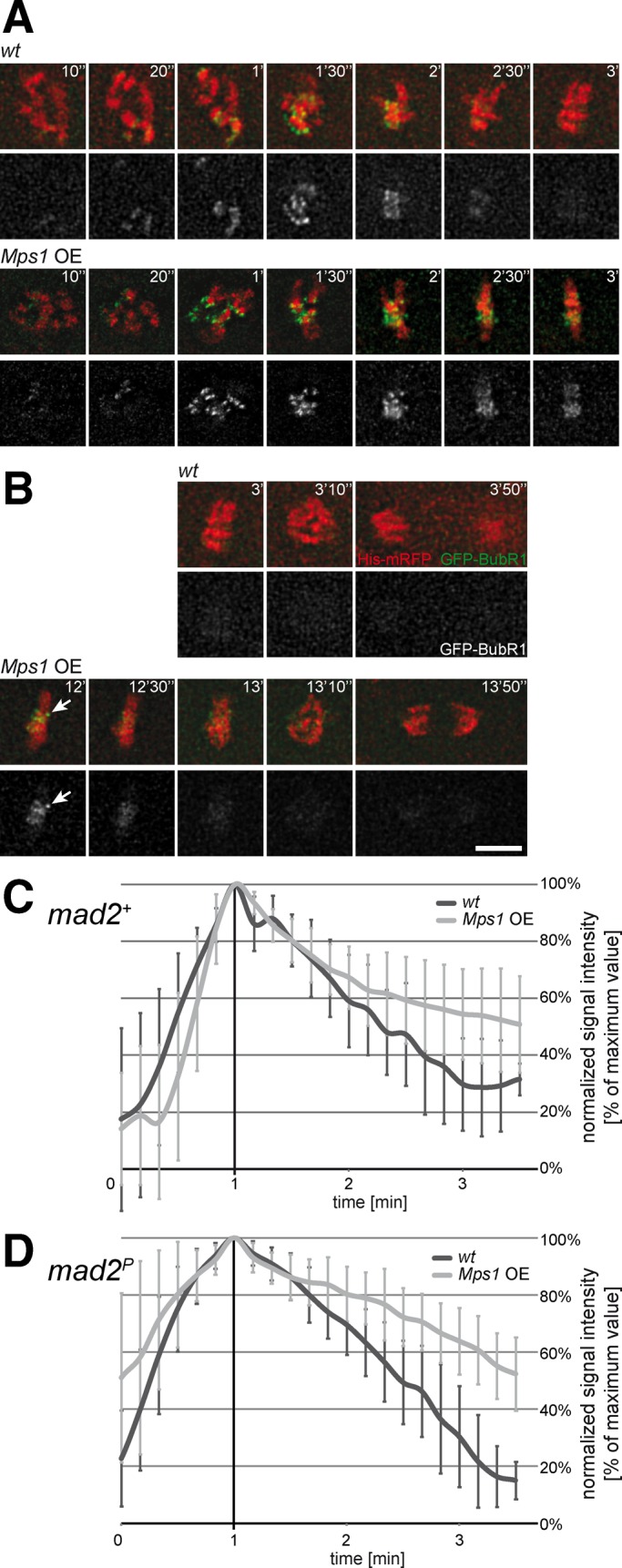
BubR1 kinetochore localization during mitosis in presence of excess Mps1. (A–C) Embryos with GFP-BubR1 and histone H2Av-mRFP either without (wt) or with UAS/GAL4-mediated Mps1 overexpression (Mps1 OE) were analyzed by time-lapse in vivo imaging. Maximum projections of selected stacks at the indicated times are shown from representative cells during (A) entry into mitosis and (B) exit from mitosis. Arrow indicates a single kinetochore retaining GFP-BubR1 during the metaphase delay induced by excess Mps1. Bar, 5 μm. (C) GFP-BubR1 signals on kinetochores were quantified over time, and curves were aligned at their peak and averaged (±SD). Although accumulation and subsequent initial disappearance of BubR1 from kinetochores was not affected by excess Mps1, this overexpression led to a slowdown of the late phase of BubR1 disappearance, presumably due to the one or two GFP-BubR1–retaining kinetochores characteristically observed during the metaphase delay caused by excess Mps1. (D) GFP-BubR1 signals on kinetochores were also analyzed in mad2-mutant embryos either without (wt) or with UAS/GAL4-mediated Mps1 overexpression (Mps1 OE). As in mad2+ embryos (A–C), excess Mps1 did not affect accumulation and subsequent initial disappearance of BubR1 from kinetochores.
To determine whether Mps1 overexpression also abolished chromosome segregation during anaphase when SAC function was impaired by mutations other than mad2P, we performed analogous experiments in bubR1KEN mutants. These mutants are also characterized by a complete loss of SAC function (Rahmani et al., 2009). Mps1 overexpression in these mutants was found to cause the same anaphase defects as in mad2 mutants (unpublished data).
If not insufficient kinetochore attachment, what else might cause the severe chromosome segregation defects during anaphase in the presence of excess Mps1 in SAC-deficient embryos? It is intriguing that Mps1 overexpression affected anaphase far less drastically in mad2+ than in mad2− embryos. In mad2+ embryos, Mps1-overexpressing cells eventually entered anaphase and completed mitosis. Sister chromatids were far more effectively transported poleward than in Mps1-overexpressing mad2-mutant embryos. Nevertheless, the speed and extent of chromosome separation were not entirely normal in comparison to control embryos without Mps1 overexpression (Figure 5, F and G). Anaphase quality, as well as overall survival of embryos, was dependent not only on mad2 function, but also to some extent on the particular combination of fluorescent marker proteins (Supplemental Figure S7). The different marker proteins (histone H2Av-mRFP, Cenp-A/Cid-EGFP, Tomato-Cenp-C, and Jupiter-GFP) might well have weak SAC-activating effects to variable degrees. All our observations were therefore consistent with the interpretation that SAC-dependent pausing in metaphase counteracts the negative effects of excess Mps1 on chromosome segregation during anaphase. Accordingly, we hypothesized that cohesion fatigue during metaphase extensions might oppose a SAC-independent Mps1 effect on sister chromatid resolution. Cohesion fatigue has been described recently in human cells, where spindle forces can, with time, rupture normal sister chromatid cohesion during extended metaphase arrest (Daum et al., 2011; Stevens et al., 2011). The efficiency of sister chromatid resolution might not be sufficient to allow poleward chromatid segregation after cohesion stabilization by excess Mps1, when anaphase is initiated early, as in mad2-mutant embryos (Figure 7E). However, after delayed anaphase onset, as in mad2+ embryos, sister chromatid resolution might be efficient enough to allow some segregation because cohesion fatigue might have canceled out the stabilizing effect of excess Mps1 on sister chromatid cohesion (Figure 7D).
FIGURE 7:

SAC-independent inhibition of sister chromatid separation by excess Mps1. (A, B) The UAS/GAL4 system was used for Mps1 overexpression in mad2-mutant embryos before mitosis 14. These embryos also expressed histone H2Av-mRFP and Cid-EGFP, allowing in vivo imaging of progression through mitosis 14. Moreover, instead of the wild-type cohesin subunit Rad21, these embryos expressed a Rad21 version with inserted internal TEV protease cleavage sites. In the presence of a UAS-TEV transgene, therefore, separase-independent cleavage of Rad21 occurred, resulting in elimination of sister chromatid cohesion (Pauli et al., 2008). (A) Maximum projections of selected stacks at the indicated times from representative cells of embryos without (–TEV) or with UAS-TEV (+TEV) reveal that TEV expression restores an essentially normal poleward sister chromatid segregation during anaphase in the presence of excess Mps1. Bar, 5 μm. (B) Quantification of the velocity of poleward sister chromatid segregation during anaphase in mad2P His2Av-mRFP Cid-EGFP embryos with maternally derived Gal4 and variable aspects of the genotype (UAS-Mps1, UAS-TEV, rad21TEV) indicated below the bars. Seventeen mitotic cells from at least four embryos were analyzed for each genotype. Brackets indicate statistically significant differences (t test) with ***p < 0.001. (C–F) Schematic summary of progression from metaphase (yellow) into anaphase (orange) in embryos with wild-type (C) or excess levels of Mps1 (D–F). In SAC-competent mad2+ embryos (D), excess Mps1 results in metaphase extension, followed by an almost normal sister chromatid separation during anaphase, presumably because cohesion fatigue during the SAC-dependent metaphase arrest largely counteracts an inhibition of sister chromatid separation by excess Mps1. Without the extended metaphase delay, as in the SAC-deficient mad2 mutant embryos (E), an essentially complete block of sister chromatid segregation occurs during anaphase, presumably because the SAC-independent inhibition of sister chromatid separation by excess Mps1 is no longer counteracted by cohesion fatigue. However, as predicted by our interpretation, TEV-mediated elimination of cohesion restores an essentially normal sister chromatid segregation in mad2-mutant embryos with excess Mps1 (F).
Our hypothesis that excess Mps1 might lead to stabilization of sister chromatid cohesion in a SAC-independent manner predicts that a more effective removal of sister chromatid cohesion should restore efficient poleward chromatid segregation during anaphase in mad2-mutant embryos with excess Mps1 (Figure 7F). To achieve a more effective resolution of sister chromatid cohesion, we performed experiments in embryos that had a cohesin variant with tobacco etch virus (TEV) protease cleavage sites within the Rad21 subunit. This cohesin variant can therefore be opened by TEV protease expression (Pauli et al., 2008). Indeed, TEV protease expression restored chromosome segregation to spindle poles very effectively in mad2-mutant embryos with excess Mps1 (Figure 7, A and B). In contrast, in the absence of TEV expression, the mad2-mutant embryos with excess Mps1 and TEV-cleavable cohesin displayed the same severe anaphase defects as observed with wild-type cohesin in mad2-mutant embryos with excess Mps1 (Figure 7, A and B). These results demonstrate that the failure of chromosome segregation during anaphase resulting from excess Mps1 is caused by inefficient resolution of sister chromatid cohesion and not by insufficient kinetochore attachment. After TEV-mediated cohesion removal, kinetochore attachments are clearly good enough for efficient poleward segregation of sister chromatids in mad2-mutant embryos with excess Mps1.
DISCUSSION
Our characterization of Drosophila Mps1 revealed a number of novel aspects. Protein kinase activity is not required for Mps1–Mps1 interactions according to our findings with Mps1kd. However, kinase activity seems to be required for Mps1 kinetochore localization in Drosophila. Moreover, accurate control of its level and localization is essential for successful mitosis. Both mistargeting and modest overexpression prevent successful mitosis. It is striking that, in addition to inappropriate maintenance of SAC activity, excess Mps1 stabilizes sister chromatid cohesion also in a SAC-independent manner.
In the case of human Mps1, Hewitt et al. (2010) demonstrated coimmunoprecipitation of GFP-Mps1 with myc-Mps1. Moreover, coimmunoprecipitation was also observed when both variants carried the D664A mutation, which eliminates kinase activity. Given that these kinase-dead variants were expressed in cells with endogenous wild-type Mps1, it is not entirely excluded that endogenous Mps1 activity might have provided an indispensable contribution for the observed coimmunoprecipitation of the kinase-dead combination. In our experiments, the kinase-dead combination was expressed in larvae, where the great majority of cells are postmitotic and no longer expresses Mps1. Nevertheless, it can still be argued that also in these experiments coimmunoprecipitation would not have occurred in the absence of the minute amounts of residual wild-type Mps1. It is of interest, however, that the kinase-dead combination coimmunoprecipitated in human cells no longer displayed the electrophoretic mobility shifts observed when at least one of the two coimmunoprecipitated versions had a wild-type kinase domain (Hewitt et al., 2010). The results from human cells and Drosophila therefore argue very strongly that the Mps1–Mps1 interaction does not depend on Mps1 kinase activity. In this regard human and Drosophila Mps1 appear to be identical.
In other respects, human and Drosophila Mps1 display some surprisingly different behaviors. The N-terminal regulatory region of human (Liu et al., 2003; Stucke et al., 2004; Xu et al., 2009) but not of Drosophila Mps1 is sufficient for kinetochore localization. According to Xu et al. (2009), this localization domain, as well as kinase-dead Mps1, is unable to localize to the kinetochore after endogenous Mps1 depletion in human cells. However, several publications reported an opposite result, that is, a more efficient kinetochore localization of human Mps1kd after depletion of endogenous Mps1 (Tighe et al., 2008; Hewitt et al., 2010; Jelluma et al., 2010). Moreover, Xenopus Mps1kd still localizes to the kinetochore after immunodepletion of endogenous Mps1 in egg extracts (Abrieu et al., 2001). In addition, several different pharmacological inhibitors of human Mps1 kinase activity have been shown to induce higher levels of Mps1 accumulation at kinetochores (Hewitt et al., 2010; Jelluma et al., 2010; Maciejowski et al., 2010; Santaguida et al., 2010). Thus the majority of studies in vertebrates report kinetochore localization in the absence of Mps1 kinase activity, in contrast to our observations with Drosophila Mps1kd.
Although we analyzed a kinase-dead mutation that is identical to the one used in vertebrates, we cannot exclude that this same mutation has different structural consequences in the context of the Drosophila protein. Observations with a human Mps1 variant carrying a peroxisome-targeting signal indicated that kinase activation might be linked to substantial conformational changes (Jelluma et al., 2010). The Mps1 variant with the peroxisome-targeting signal was only localized to the peroxisome when it also contained the kinase-dead mutation or when kinase inhibitor was applied. The conformation of active Mps1 kinase therefore appears to be incompatible with a productive interaction between an engineered C-terminal peroxisome-targeting signal and the peroxisome translocation machinery (Jelluma et al., 2010). It seems likely, therefore, that complex conformational changes with decisive effects on localization and activity are involved in Mps1 regulation. In that context, we point out that Drosophila Mps1kd is fully capable of interacting with itself in-trans, as well as with Mad1, suggesting that its conformation is unlikely to be completely abnormal. Apart from hypothetical differences in conformational behavior, the discordant results concerning dependence of kinetochore localization on Mps1 kinase activity might perhaps also reflect different assay conditions. Nocodazole and MG132 were applied in all the experiments in which increased kinetochore localization of Mps1kd or pharmacologically inhibited Mps1 was observed but not in those in which kinetochore localization was absent (Figure 1D; Xu et al., 2009).
Another difference between human and Drosophila Mps1 concerns the consequences of overexpression. As in yeast (Hardwick et al., 1996), modest overexpression of Drosophila Mps1 (approximately fivefold) is sufficient for SAC hyperactivation and consequent extension of metaphase by more than 10-fold, whereas comparable overexpression in human cells does not have an effect on mitosis (Stucke et al., 2002; Kang et al., 2007; Jelluma et al., 2010). Excess Mps1 in Drosophila appears to hyperactivate the SAC directly rather than indirectly via destabilization of kinetochore attachments (Figures 5 and 6). We point out that persistent kinetochore targeting of human Mps1 via fusion to Mis12 also does not destabilize kinetochore attachments (Jelluma et al., 2010), even though it has been demonstrated that human Mps1 can promote transformation of syntelic into amphitelic attachments (Jelluma et al., 2008b; Hewitt et al., 2010; Santaguida et al., 2010; Sliedrecht et al., 2010), similar to yeast Mps1 (Maure et al., 2007).
Experiments in human cells and fission yeast with Mps1 variants (Mis12–Mps1 and Ndc80–Mph1, respectively) that persist at the kinetochore throughout mitosis (Jelluma et al., 2010; Ito et al., 2012) provided strong arguments that the release of Mps1 from kinetochores that occurs during metaphase of unperturbed mitoses is essential for mitotic checkpoint silencing and the transition from metaphase to anaphase. Our experiments in Drosophila with similar Mps1 variants (Cenp-CC-Mps1 and Nuf2-Mps1) that also persist at the kinetochore failed to confirm this notion. Our negative evidence might reflect technical difficulties. The fact that the SAC is readily hyperactivated by wild-type Mps1 overexpression in Drosophila (but not in human cells) clearly complicated our experiments. The amount and activity of Mps1 that was persistently localized at the kinetochore might have been insufficient. Given that EGFP-Cenp-CC-Mps1 resulted in a failure of sister chromatid segregation during anaphase, our results agree with the findings concerning human Mis12–Mps1 to the extent that both demonstrate the importance of normal Mps1 subcellular localization dynamics.
Failure of sister chromatid segregation during anaphase was not only caused by EGFP-Cenp-CC-Mps1 but also by overexpression of wild-type Mps1 in the absence of Mad2. Because sister chromatid segregation was fully restored when cohesion was experimentally removed by TEV expression, we conclude that excess Mps1 inhibits sister chromatid separation. The final separation of sister chromatids immediately before the onset of anaphase is brought about by separase-mediated cleavage of the Rad21 subunit of the cohesion complex (Uhlmann et al., 2000). However, a large fraction of cohesin is released from chromosomes already during prophase and prometaphase in response to Polo and Aurora B kinase activity (Waizenegger et al., 2000). It remains to be analyzed whether Mps1 interferes with these identified pathways of cohesion removal. Our preliminary analyses failed to detect an unusual behavior of Rad21, separase, and Polo. With regard to the inhibitory effect of Mps1 on sister chromatid separation, it also remains to be shown that it is of physiological relevance and not entirely dependent on overexpression. We consider it very likely that a SAC-independent inhibition of sister chromatid separation by endogenous Mps1 is of physiological relevance, as it could provide additional protection against premature sister chromatid separation and consequential aneuploidy.
In Drosophila, SAC activation can proceed with breathtaking speed. The time from the start of mitosis until anaphase onset is <3 min during the syncytial cycles of early embryogenesis. Nevertheless, hypoxia, which can be induced very rapidly and causes a relatively subtle effect on microtubule dynamics, triggers a reliable SAC arrest even when generated after mitosis has already started (Fischer et al., 2004; Pandey et al., 2007). SAC-mediated mitotic arrest induction, therefore, requires <3 min in these conditions. In general, rapid and reliable responses to small signals (like a single unattached kinetochore or the slight reduction of microtubule dynamics) involve amplification steps that in turn create a major challenge for timely reversion (Ciliberto and Shah, 2009). We propose that the functional properties of Drosophila Mps1 might reflect an evolutionary tuning of SAC speed and efficiency. As a result, even modest overexpression of Mps1 is already sufficient for SAC activation in Drosophila. In addition, the apparent SAC-independent stabilization of sister chromatid cohesion by Drosophila Mps1 is likely to provide extra support in the fight against premature sister chromatid separation and consequent aneuploidy. An improved mechanistic understanding of the SAC might also resolve whether the comparatively rapid SAC adaptation in the presence of persistent spindle damage in Drosophila reflects design constraints. Moreover, whether Mps1 contributes also in human cells to the control of sister chromatid cohesion in a SAC-independent manner is yet another interesting question for the future.
MATERIALS AND METHODS
Drosophila genetics
All Drosophila strains are described in Supplemental Table S1. Genotypes of animals that were used for the collection of data displayed in the figures are specified in Supplemental Table S2. The null alleles of Mps1 (Page et al., 2007), mad1 (Emre et al., 2011), mad2 (Buffin et al., 2007), and vtd/rad21 (Pauli et al., 2008) that were used for our analyses have been described before, as have the bubR1KEN mutants, which are SAC deficient (Rahmani et al., 2009). A chromosome with P{neoFRT}82B and Mps1aldB4, which allowed for induction of germline clones (Chou and Perrimon, 1996; Fischer et al., 2004), was kindly provided by William D. Gilliland (DePaul University, Chicago, IL).
For expression of UAS transgenes in embryos during interphase 14 and analysis of phenotypic consequences during the subsequent mitosis 14, we crossed males carrying such transgenes with mat-GAL4 females. These females produce eggs containing maternally derived Gal4, which results in activation of strong expression from paternally contributed UAS transgenes at onset of zygotic expression during interphase 14.
Hs-GAL4 was used for heat-induced expression of UAS transgenes in larvae. For induction of a synchronous progression through embryonic mitosis 14, we used w*; stg7B, P{w+, Hs-stg}/TM3 as described (Sauer et al., 1995).
For marking chromosomes, centromeres, and spindles for some of the cytological analyses, we used the transgenes Jupiter-GFP (Morin et al., 2001), His2Av-mRFP and Cid-EGFP (Schuh et al., 2007), mRFP-Cenp-C (Schittenhelm et al., 2007), and Tom-Cenp-C (this study).
For expression of SAC proteins fused to fluorescent tags, we used GFP-bubR1 (Buffin et al., 2005), mad1-GFP, mCherry-mad1 (Emre et al., 2011), and gEGFP-Mps1 (Fischer et al., 2004). The cis-regulatory regions of the corresponding genes control expression of these transgenes.
Additional transgenic lines (gEGFP-Mps1kd, gEGFP-Mps1N, gEGFP-Mps1C, UAS-Mps1, UAS-EGFP-Mps1, UAS-EGFP-Mps1kd, UAS-10myc-Mps1, UAS-10myc-Mps1kd, UAS-EGFP-Cenp-CC-Mps1, UAS-EGFP-Cenp-CC-Mps1kd, UAS-EGFP-Mps1N, UAS-EGFP-Mps1C, UAS-EGFP-Mps1Ckd, UAS-EGFP-Mps1-Nuf2, UAS-EGFP-Mps1kd-Nuf2, UAS-Torso(EC/TM)-EGFP-Mps1, UAS-Torso(EC/TM)4021-EGFP-Mps1, UAS-EGFP-bub1, UAS-EGFP-bubR1, UAS-EGFP-bubR1kd, UAS-EGFP-Cenp-CC-bubR1, UAS-EGFP-mad2) were obtained after P element–mediated germline transformation with the constructs described in the Supplemental Material. pUAST constructs were generated for Gal4-mediated expression (Brand and Perrimon, 1993). pCaSpeR4 (Thummel and Pirrotta, 1992) was used for transgene constructs under control of the corresponding normal cis-regulatory region. Mps1kd codes for the D478A mutation. This mutation abolishes kinase activity (Lince-Faria et al., 2009). Mps1N and Mps1C code for amino acids 1–332 (N-terminal regulatory region) and 325–630 (C-terminal kinase domain), respectively. The C-terminal domain of Cenp-C (Cenp-CC), which was used to enforce constitutive kinetochore localization of SAC proteins, comprises the amino acids 1009–1411 of the Cenp-C protein. This domain is sufficient for kinetochore localization (Heeger et al., 2005).
For the experiments involving experimental elimination of sister chromatid cohesion before mitosis 14 by TEV expression in mad2 mutants with excess Mps1, we collected embryos (+TEV, +Rad21TEV, +Mps1 OE) from a cross of 2xrad21TEV/mat-GAL4, His2Av-mRFP, Cid-EGFP; rad21ex3, mad2P females with UAS-TEV, UAS-Mps1 II.4; rad21ex3, mad2P/TM6 males. For the collection of control embryos without TEV expression (–TEV, +Rad21TEV, +Mps1 OE), the same females were crossed with UAS-Mps1 II.4; rad21ex3, mad2P/TM6 males. Additional control embryos (+TEV, –Rad21TEV, +Mps1 OE) were collected from a cross of mat-GAL4, His2Av-mRFP, Cid-EGFP/+; rad21ex3, mad2P females with UAS-TEV, UAS-Mps1 II.4; rad21ex3; mad2P/TM6 males. Finally, a cross of mat-GAL4, His2Av-mRFP, Cid-EGFP/CyO; mad2P females with mad2P males was used for the collection of yet other control embryos (–TEV, –Rad21TEV, –Mps1 OE). After in vivo imaging, embryos were genotyped individually by PCR to distinguish embryos homozygous for rad21ex3 and mad2P from embryos carrying the balancer chromosome TM6. Before genotyping, embryos were removed manually from the glass slides, and genomic DNA was prepared using 133 μg/ml proteinase K (Fermentas, Glen Burnie, MD) for 30 min at 50°C in 6 μl of GC reaction buffer for Phusion polymerase (Fermentas) supplied with 0.17% Nonidet P40 Substitute and 0.17% Tween-20. The resulting suspension was used as PCR template. The genetic system for experimental elimination of sister chromatid cohesion before mitosis 14 by TEV expression and its application in mad2+ embryos has been described (Pauli et al., 2008).
Meiotic recombination was used for the construction of chromosomes with multiple mutations or transgenes. Particular chromosomes were combined by standard crossing schemes to arrive at the stocks used in the experiments (see the Supplemental Material). Because all transgenes were established in a white mutant background, we used w1 flies as wild-type control stock unless otherwise specified.
Microscopy
Embryos were collected on apple juice agar plates and aged to the desired stage. In vivo imaging, fixation, and immunolabeling of Drosophila embryos were done essentially as described (Pandey et al., 2005). For in vivo imaging, embryos were dechorionated, immobilized on glass slides, and covered with halocarbon oil. Before fixation, embryos were first dechorionated for 2 min in 7% NaOCl. Embryos were fixed during 1 min with a 1:1 mixture of heptane and methanol. A modified fixation procedure was applied in case of anti-tubulin labeling (Figure 4), which involved preincubation of the dechorionated embryos in 0.73 μM Taxol (Sigma-Aldrich, St. Louis, MO) during 2 min in a 2:1 mixture of heptane and 100 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 10 mM EDTA, and 20 mM MgSO4 (PEM) before fixation. Subsequently embryos were fixed during 20 min in a 1:1 mixture of heptane and 4% formaldehyde in PEM. The vitelline membrane was removed by shaking for 1 min in a 1:1 mixture of heptane and 90% methanol/50 mM ethylene glycol tetraacetic acid. For immunofluorescence staining, we used rabbit anti-BubR1 (kindly provided by C. Sunkel, Universidade do Porto, Porto, Portugal) at 1:2000 and mouse monoclonal antibody DM1A anti–α-tubulin (Sigma-Aldrich) at 1:8000. DNA was stained with Hoechst 33258 at 1 μg/ml in phosphate-buffered saline. Embryos were mounted in 70% glycerol, 50 mM Tris-HCl, pH 8.5, 5 mM p-phenylenediamine, and 50 mM n-propylgallate.
Images were acquired with a Zeiss CellObserver HS system (Carl Zeiss, Jena, Germany). For in vivo imaging, we used a 470-nm (GFP) and a 555-nm (mRFP/Tomato) light-emitting diode (LED) as fast-switchable light sources. The 555-nm LED was used in combination with a 550/32 bandpass filter (BrightLine HC; Semrock, Rochester, NY). Emitted signals were detected with a dual bandpass filter (Zeiss 56HE without excitation filter). A 63×/1.4 oil immersion objective was used. Number and spacing of focal planes per time point, as well as time intervals and total duration of the in vivo imaging, were adjusted according to experimental needs.
Single images and movies were processed and evaluated using AxioVision (Zeiss) for region of interest selection, Huygens Remote Manager, version 1.2.3 (Ponti et al., 2007), for deconvolution, Imaris (Bitplane Scientific Software, Zurich, Switzerland) and ImageJ (National Institutes of Health, Bethesda, MD) for measurement of metaphase plate width, sister kinetochore separation, kinetochore velocity, dynamics of progression through mitosis, and kinetochore signal intensities, and Photoshop (Adobe, San Jose, CA) for still-frame processing. Fluorescence intensities of kinetochore signals were measured by integrating pixel intensities within a square encompassing all of the kinetochore signals of a given cell, followed by subtraction of local background values as described previously (Hoffman et al., 2001; Schittenhelm et al., 2010). For width of a metaphase plate, we measured the shortest distance between two parallel lines that were perpendicular to the mitotic spindle and had all of the kinetochore signals in between. The intersister kinetochore distance during early mitosis and the kinetochore segregation distance during exit from mitosis were measured using the distance measurement tool in the Imaris software. The kinetochore segregation velocity was measured by subtracting the interkinetochore distance determined 50 s after anaphase onset from the interkinetochore distance determined at anaphase onset, followed by division of the resulting distance by 50 s.
Extract preparations and immunoprecipitation
Extracts from Drosophila embryos for immunoprecipitation were prepared as described (Jäger et al., 2001). Extracts from larvae were prepared analogously using one larva as the equivalent of 50 embryos. For induction of Hs-GAL4–mediated UAS transgene expression before extract preparation, bottles with larvae were incubated for 2 h in a 37°C water bath, followed by 1 h of recovery at 25°C. Protein extracts from transiently transfected S2R+ cells were prepared as described (Furrer et al., 2010). For transient transfection, 300,000 cells were seeded in a T25 flask and cotransfected 24 h later with a combination of pCaSpeR4-Actin5c-GAL4 and appropriate pUAST plasmids using FuGENE HD transfection reagent (Roche, Indianapolis, IN). Extracts were prepared 2 d after transfection. All buffers used for extract preparation were supplemented with protease inhibitor cocktail (P8340; Sigma-Aldrich). For phosphatase treatment, 50 μl of protein extract was incubated for 30 min at 30°C with 1600 U of λ-phosphatase (NEB). We used 50 mM NaF and 10 mM Na3VO4 (activated as described; Gordon, 1991) as phosphatase inhibitors. Immunoprecipitation was performed using GFP-Trap coupled to agarose beads (ChromoTek, Martinsried, Germany) or mouse monoclonal antibody 9E10 anti–c-myc (Evan et al., 1985) in combination with protein A–Sepharose beads.
Immunoblotting
Total extracts from Drosophila embryos for direct analysis by immunoblotting were prepared by homogenizing the embryos in SDS–PAGE sample buffer. For the preparation of extracts from embryos at defined mitotic stages, we collected eggs from w*; stg7B, P{w+, Hs-stg}/TM3 flies for 1 h, followed by aging at 25°C for 3 h. Hs-stg expression was induced by 15 min of incubation at 37°C. Aliquots of embryos were fixed with methanol either immediately or after recovery at 25°C for 10 and 20 min, respectively. Alternatively, syncytial embryos were collected for 1 h and aged for an additional hour at 25°C, followed by fixation in methanol. After DNA staining with Hoechst 33258, embryos were suspended in a 1:1 mixture of glycerol and EB buffer (Edgar et al., 1994). Embryos in distinct mitotic stages were identified using an inverted fluorescence microscope and pooled before solubilization in SDS–PAGE sample buffer. Discontinuous PAGE was performed according to standard protocols. To increase electrophoretic mobility shifts caused by phosphorylations, Phostag (Wako Chemicals USA, Richmond, VA) was added.
Immunoblots were probed with affinity-purified rabbit antibodies against Mps1 (Rb1) (Pandey et al., 2007) diluted 1:5000, against Mad2 (IS793; a kind gift from D. Sharp, Albert Einstein College of Medicine, New York, NY) diluted 1:1500, against GFP (IS28; Schittenhelm et al., 2007) diluted 1:3000, against GFP (Torrey Pines Biolabs, Secaucus, NJ) diluted 1:5000, and against mRFP (IS743) diluted 1:3000. These antibodies against mRFP also react with mCherry and are therefore designated as anti-mCherry in the Results section. Moreover, we also used mouse monoclonal antibody 9E10 against c-myc diluted 1:15, DM1A against α-tubulin (Sigma-Aldrich) diluted 1:50,000, F2 against cyclin B (Knoblich and Lehner, 1993) diluted 1:3, and ADL67.10 (Stuurman et al., 1996) against lamin diluted 1:200. ADL67.10 was obtained from the Developmental Studies Hybridoma Bank (Department of Biology, University of Iowa, Iowa City, IA). Signals were detected by the ECL system (Amersham, Piscataway, NJ). For quantification of proteins by immunoblotting, a dilution series of a reference extract was loaded onto the same gel as the test samples. Nonsaturated exposures, in which the signals obtained for the dilution series were linearly correlated with the loaded amounts, were used for the quantification of the signals in test samples with intensities within the range of those of the dilution series. ImageJ was used for the determination of background-corrected signal intensities.
Supplementary Material
Acknowledgments
We are grateful to D. Beuchle, B. Jaunich, S. Moser, C. Sollmann, D. Sharp, C. Sunkel, and W. Gilliland for technical support, antibodies, and fly strains. This study was supported by Swiss National Science Foundation Grant 310003A-120276 (C.F.L.) and the French Agence Nationale de la Recherche (ANR-08-BLAN-0006-01; R.E.K).
Abbreviations used:
- EGFP
enhanced green fluorescent protein
- Mps1
monopolar spindle 1
- SAC
spindle assembly checkpoint
- TEV
tobacco etch virus
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-02-0117) on May 2, 2012.
REFERENCES
- Abrieu A, Magnaghi-Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW, Labbe JC. Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 2001;106:83–93. doi: 10.1016/s0092-8674(01)00410-x. [DOI] [PubMed] [Google Scholar]
- Basu J, Bousbaa H, Logarinho E, Li Z, Williams BC, Lopes C, Sunkel CE, Goldberg ML. Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila. J Cell Biol. 1999;146:13–28. doi: 10.1083/jcb.146.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Buffin E, Emre D, Karess RE. Flies without a spindle checkpoint. Nat Cell Biol. 2007;9:565–572. doi: 10.1038/ncb1570. [DOI] [PubMed] [Google Scholar]
- Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE. Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Curr Biol. 2005;15:856–861. doi: 10.1016/j.cub.2005.03.052. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127:983–997. doi: 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- Chen RH, Brady DM, Smith D, Murray AW, Hardwick KG. The spindle checkpoint of budding yeast depends on a tight complex between the Mad1 and Mad2 proteins. Mol Biol Cell. 1999;10:2607–2618. doi: 10.1091/mbc.10.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TB, Perrimon N. The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics. 1996;144:1673–1679. doi: 10.1093/genetics/144.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, et al. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133:427–439. doi: 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberto A, Shah JV. A quantitative systems view of the spindle assembly checkpoint. EMBO J. 2009;28:2162–2173. doi: 10.1038/emboj.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, et al. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010;70:10255–10264. doi: 10.1158/0008-5472.CAN-10-2101. [DOI] [PubMed] [Google Scholar]
- Cui Y, Cheng X, Zhang C, Zhang Y, Li S, Wang C, Guadagno TM. Degradation of the human mitotic checkpoint kinase Mps1 is cell cycle-regulated by APC-cCdc20 and APC-cCdh1 ubiquitin ligases. J Biol Chem. 2010;285:32988–32998. doi: 10.1074/jbc.M110.140905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Potapova TA, Sivakumar S, Daniel JJ, Flynn JN, Rankin S, Gorbsky GJ. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr Biol. 2011;21:1018–1024. doi: 10.1016/j.cub.2011.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAntoni A, Sala V, Musacchio A. Explaining the oligomerization properties of the spindle assembly checkpoint protein Mad2. Philos Trans R Soc Lond B Biol Sci. 2005;360:637–647. doi: 10.1098/rstb.2004.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Dong Y, Hergert P, Strauss J, Hickey JM, Salmon ED, McEwen BF. Hec1 and nuf2 are core components of the kinetochore outer plate essential for organizing microtubule attachment sites. Mol Biol Cell. 2005;16:519–531. doi: 10.1091/mbc.E04-09-0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca JG, Gall WE, Ciferri C, Cimini D, Musacchio A, Salmon ED. Kinetochore microtubule dynamics and attachment stability are regulated by hec1. Cell. 2006;127:969–982. doi: 10.1016/j.cell.2006.09.047. [DOI] [PubMed] [Google Scholar]
- Dou Z, Sawagechi A, Zhang J, Luo H, Brako L, Yao XB. Dynamic distribution of TTK in HeLa cells: insights from an ultrastructural study. Cell Res. 2003;13:443–449. doi: 10.1038/sj.cr.7290186. [DOI] [PubMed] [Google Scholar]
- Dou Z, von Schubert C, Korner R, Santamaria A, Elowe S, Nigg EA. Quantitative mass spectrometry analysis reveals similar substrate consensus motif for human Mps1 kinase and Plk1. PLoS One. 2011;6:e18793. doi: 10.1371/journal.pone.0018793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA, Sprenger F, Duronio RJ, Leopold P, O'Farrell PH. Distinct molecular mechanisms regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes Dev. 1994;8:440–452. doi: 10.1101/gad.8.4.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre D, Terracol R, Poncet A, Rahmani Z, Karess RE. A mitotic role for Mad1 beyond the spindle checkpoint. J Cell Sci. 2011;124:1664–1671. doi: 10.1242/jcs.081216. [DOI] [PubMed] [Google Scholar]
- Espeut J, Gaussen A, Bieling P, Morin V, Prieto S, Fesquet D, Surrey T, Abrieu A. Phosphorylation relieves autoinhibition of the kinetochore motor Cenp-E. Mol Cell. 2008;29:637–643. doi: 10.1016/j.molcel.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Kaulich M, Nigg EA, Santamaria A. Probing the in vivo function of Mad1:C-Mad2 in the spindle assembly checkpoint. EMBO J. 2011;30:3322–3336. doi: 10.1038/emboj.2011.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MG, Heeger S, Hacker U, Lehner CF. The mitotic arrest in response to hypoxia and of polar bodies during early embryogenesis requires Drosophila Mps1. Curr Biol. 2004;14:2019–2024. doi: 10.1016/j.cub.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Fisk HA, Winey M. The mouse Mps1p-like kinase regulates centrosome duplication. Cell. 2001;106:95–104. doi: 10.1016/s0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- Foe VE. Mitotic domains reveal early commitment of cells in Drosophila embryos. Development. 1989;107:1–22. [PubMed] [Google Scholar]
- Furrer M, Balbi M, Albarca-Aguilera M, Gallant M, Herr W, Gallant P. Drosophila Myc interacts with host cell factor (dHCF) to activate transcription and control growth. J Biol Chem. 2010;285:39623–39636. doi: 10.1074/jbc.M110.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland WD, Hughes SE, Cotitta JL, Takeo S, Xiang Y, Hawley RS. The multiple roles of Mps1 in Drosophila female meiosis. PLoS Genet. 2007;3:e113. doi: 10.1371/journal.pgen.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland WD, Wayson SM, Hawley RS. The meiotic defects of mutants in the Drosophila mps1 gene reveal a critical role of Mps1 in the segregation of achiasmate homologs. Curr Biol. 2005;15:672–677. doi: 10.1016/j.cub.2005.02.062. [DOI] [PubMed] [Google Scholar]
- Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- Grimison B, Liu J, Lewellyn AL, Maller JL. Metaphase arrest by cyclin E-Cdk2 requires the spindle-checkpoint kinase Mps1. Curr Biol. 2006;16:1968–1973. doi: 10.1016/j.cub.2006.08.055. [DOI] [PubMed] [Google Scholar]
- Hached K, Xie SZ, Buffin E, Cladiere D, Rachez C, Sacras M, Sorger PK, Wassmann K. Mps1 at kinetochores is essential for female mouse meiosis I. Development. 2011;138:2261–2271. doi: 10.1242/dev.061317. [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Weiss E, Luca FC, Winey M, Murray AW. Activation of the budding yeast spindle assembly checkpoint without mitotic spindle disruption. Science. 1996;273:953–956. doi: 10.1126/science.273.5277.953. [DOI] [PubMed] [Google Scholar]
- Heeger S, Leismann O, Schittenhelm R, Schraidt O, Heidmann S, Lehner CF. Genetic interactions of separase regulatory subunits reveal the diverged Drosophila Cenp-C homolog. Genes Dev. 2005;19:2041–2053. doi: 10.1101/gad.347805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt L, Tighe A, Santaguida S, White AM, Jones CD, Musacchio A, Green S, Taylor SS. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1-C-Mad2 core complex. J Cell Biol. 2010;190:25–34. doi: 10.1083/jcb.201002133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DB, Pearson CG, Yen TJ, Howell BJ, Salmon ED. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol Biol Cell. 2001;12:1995–2009. doi: 10.1091/mbc.12.7.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol. 2001;155:1159–1172. doi: 10.1083/jcb.200105093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, Moree B, Farrar EM, Stewart S, Fang G, Salmon ED. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr Biol. 2004;14:953–964. doi: 10.1016/j.cub.2004.05.053. [DOI] [PubMed] [Google Scholar]
- Huang H, Hittle J, Zappacosta F, Annan RS, Hershko A, Yen TJ. Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J Cell Biol. 2008;183:667–680. doi: 10.1083/jcb.200805163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Saito Y, Matsumoto T. Centromere-tethered Mps1 pombe homolog (Mph1) kinase is a sufficient marker for recruitment of the spindle checkpoint protein Bub1, but not Mad1. Proc Natl Acad Sci USA. 2012;109:209–214. doi: 10.1073/pnas.1114647109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger H, Herzig A, Lehner CF, Heidmann S. Drosophila separase is required for sister chromatid separation and binds to PIM and THR. Genes Dev. 2001;15:2572–2584. doi: 10.1101/gad.207301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelluma N, Brenkman AB, McLeod I, Yates JR, 3rd, Cleveland DW, Medema RH, Kops GJ. Chromosomal instability by inefficient Mps1 auto-activation due to a weakened mitotic checkpoint and lagging chromosomes. PLoS One. 2008a;3:e2415. doi: 10.1371/journal.pone.0002415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH, Kops GJ. Mps1 phosphorylates borealin to control Aurora B activity and chromosome alignment. Cell. 2008b;132:233–246. doi: 10.1016/j.cell.2007.11.046. [DOI] [PubMed] [Google Scholar]
- Jelluma N, Dansen TB, Sliedrecht T, Kwiatkowski NP, Kops GJ. Release of Mps1 from kinetochores is crucial for timely anaphase onset. J Cell Biol. 2010;191:281–290. doi: 10.1083/jcb.201003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MH, Huneycutt BJ, Pearson CG, Zhang C, Morgan G, Shokat K, Bloom K, Winey M. Chemical genetics reveals a role for Mps1 kinase in kinetochore attachment during mitosis. Curr Biol. 2005;15:160–165. doi: 10.1016/j.cub.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Kang J, Chen Y, Zhao Y, Yu H. Autophosphorylation-dependent activation of human Mps1 is required for the spindle checkpoint. Proc Natl Acad Sci USA. 2007;104:20232–20237. doi: 10.1073/pnas.0710519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmler S, Stach M, Knapp M, Ortiz J, Pfannstiel J, Ruppert T, Lechner J. Mimicking Ndc80 phosphorylation triggers spindle assembly checkpoint signalling. EMBO J. 2009;28:1099–1110. doi: 10.1038/emboj.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Koike T. Separation and detection of large phosphoproteins using Phos-tag SDS–PAGE. Nat Protoc. 2009;4:1513–1521. doi: 10.1038/nprot.2009.154. [DOI] [PubMed] [Google Scholar]
- Knoblich JA, Lehner CF. Synergistic action of Drosophila cyclin A and cyclin B during the G2-M transition. EMBO J. 1993;12:65–74. doi: 10.1002/j.1460-2075.1993.tb05632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, et al. Small-molecule kinase inhibitors provide insight into Mps1 cell cycle function. Nat Chem Biol. 2010;6:359–368. doi: 10.1038/nchembio.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Thebault P, Freschi L, Beaufils S, Blundell TL, Landry CR, Bolanos-Garcia VM, Elowe S. Characterization of spindle checkpoint kinase Mps1 reveals domain with functional and structural similarities to tetratricopeptide repeat motifs of Bub1 and BubR1 checkpoint kinases. J Biol Chem. 2012;287:5988–6001. doi: 10.1074/jbc.M111.307355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lince-Faria M, Maffini S, Orr B, Ding Y, Claudia F, Sunkel CE, Tavares A, Johansen J, Johansen KM, Maiato H. Spatiotemporal control of mitosis by the conserved spindle matrix protein megator. J Cell Biol. 2009;184:647–657. doi: 10.1083/jcb.200811012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Vader G, Vromans MJ, Lampson MA, Lens SM. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science. 2009;323:1350–1353. doi: 10.1126/science.1167000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ST, Chan GK, Hittle JC, Fujii G, Lees E, Yen TJ. Human MPS1 kinase is required for mitotic arrest induced by the loss of CENP-E from kinetochores. Mol Biol Cell. 2003;14:1638–1651. doi: 10.1091/mbc.02-05-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logarinho E, Bousbaa H, Dias JM, Lopes C, Amorim I, Antunes-Martins A, Sunkel CE. Different spindle checkpoint proteins monitor microtubule attachment and tension at kinetochores in Drosophila cells. J Cell Sci. 2004;117:1757–1771. doi: 10.1242/jcs.01033. [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell. 2002;9:59–71. doi: 10.1016/s1097-2765(01)00435-x. [DOI] [PubMed] [Google Scholar]
- Maciejowski J, George KA, Terret ME, Zhang C, Shokat KM, Jallepalli PV. Mps1 directs the assembly of Cdc20 inhibitory complexes during interphase and mitosis to control M phase timing and spindle checkpoint signaling. J Cell Biol. 2010;190:89–100. doi: 10.1083/jcb.201001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M, Kapoor TM. Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat Cell Biol. 2011;13:475–482. doi: 10.1038/ncb2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mapelli M, Massimiliano L, Santaguida S, Musacchio A. The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell. 2007;131:730–743. doi: 10.1016/j.cell.2007.08.049. [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA. Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science. 2002;297:2267–2270. doi: 10.1126/science.1075596. [DOI] [PubMed] [Google Scholar]
- Mattison CP, Old WM, Steiner E, Huneycutt BJ, Resing KA, Ahn NG, Winey M. Mps1 activation loop autophosphorylation enhances kinase activity. J Biol Chem. 2007;282:30553–30561. doi: 10.1074/jbc.M707063200. [DOI] [PubMed] [Google Scholar]
- Maure JF, Kitamura E, Tanaka TU. Mps1 kinase promotes sister-kinetochore bi-orientation by a tension-dependent mechanism. Curr Biol. 2007;17:2175–2182. doi: 10.1016/j.cub.2007.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci USA. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- O'Tousa J. Meiotic chromosome behavior influenced by mutation-altered disjunction in Drosophila melanogaster females. Genetics. 1982;102:503–524. doi: 10.1093/genetics/102.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RA, Coelho PA, Sunkel CE. The condensin I subunit Barren/CAP-H is essential for the structural integrity of centromeric heterochromatin during mitosis. Mol Cell Biol. 2005;25:8971–8984. doi: 10.1128/MCB.25.20.8971-8984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page SL, Nielsen RJ, Teeter K, Lake CM, Ong S, Wright KR, Dean KL, Agne D, Gilliland WD, Hawley RS. A germline clone screen for meiotic mutants in Drosophila melanogaster. Fly (Austin) 2007;1:172–181. doi: 10.4161/fly.4720. [DOI] [PubMed] [Google Scholar]
- Palframan WJ, Meehl JB, Jaspersen SL, Winey M, Murray AW. Anaphase inactivation of the spindle checkpoint. Science. 2006;313:680–684. doi: 10.1126/science.1127205. [DOI] [PubMed] [Google Scholar]
- Pandey R, Heeger S, Lehner CF. Rapid effects of acute anoxia on spindle kinetochore interactions activate the mitotic spindle checkpoint. J Cell Sci. 2007;120:2807–2818. doi: 10.1242/jcs.007690. [DOI] [PubMed] [Google Scholar]
- Pandey R, Heidmann S, Lehner CF. Epithelial re-organization and dynamics of progression through mitosis in Drosophila separase complex mutants. J Cell Sci. 2005;118:733–742. doi: 10.1242/jcs.01663. [DOI] [PubMed] [Google Scholar]
- Pauli A, Althoff F, Oliveira RA, Heidmann S, Schuldiner O, Lehner CF, Dickson BJ, Nasmyth K. Cell-type-specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Dev Cell. 2008;14:239–251. doi: 10.1016/j.devcel.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponti A, Gulati A, Bäcker V, Schwarb P. Huygens remote manager: a web interface tool for high-volume batch deconvolution. Imag Microsc. 2007;9:57–58. [Google Scholar]
- Rahmani Z, Gagou ME, Lefebvre C, Emre D, Karess RE. Separating the spindle, checkpoint, and timer functions of BubR1. J Cell Biol. 2009;187:597–605. doi: 10.1083/jcb.200905026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer K, Knoblich JA, Richardson H, Lehner CF. Distinct modes of cyclin E/cdc2c kinase regulation and S phase control in mitotic and endoreduplication cycles of Drosophila embryogenesis. Genes Dev. 1995;9:1327–1339. doi: 10.1101/gad.9.11.1327. [DOI] [PubMed] [Google Scholar]
- Saurin AT, van der Waal MS, Medema RH, Lens SM, Kops GJ. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat Commun. 2011;2:316. doi: 10.1038/ncomms1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schittenhelm RB, Althoff F, Heidmann S, Lehner CF. Detrimental incorporation of excess Cenp-A/Cid and Cenp-C into Drosophila centromeres is prevented by limiting amounts of the bridging factor Cal1. J Cell Sci. 2010;123:3768–3779. doi: 10.1242/jcs.067934. [DOI] [PubMed] [Google Scholar]
- Schittenhelm RB, Heeger S, Althoff F, Walter A, Heidmann S, Mechtler K, Lehner CF. Spatial organization of a ubiquitous eukaryotic kinetochore protein network in Drosophila chromosomes. Chromosoma. 2007;116:385–402. doi: 10.1007/s00412-007-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh M, Lehner CF, Heidmann S. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. Curr Biol. 2007;17:237–243. doi: 10.1016/j.cub.2006.11.051. [DOI] [PubMed] [Google Scholar]
- Shimogawa MM, et al. Mps1 phosphorylation of Dam1 couples kinetochores to microtubule plus ends at metaphase. Curr Biol. 2006;16:1489–1501. doi: 10.1016/j.cub.2006.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, Musacchio A. Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a “safety belt” binding mechanism for the spindle checkpoint. EMBO J. 2002;21:2496–2506. doi: 10.1093/emboj/21.10.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoufias DA, Andreassen PR, Lacroix FB, Wilson L, Margolis RL. Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc Natl Acad Sci USA. 2001;98:4492–4497. doi: 10.1073/pnas.081076898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliedrecht T, Zhang C, Shokat KM, Kops GJ. Chemical genetic inhibition of Mps1 in stable human cell lines reveals novel aspects of Mps1 function in mitosis. PLoS One. 2010;5:e10251. doi: 10.1371/journal.pone.0010251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens D, Gassmann R, Oegema K, Desai A. Uncoordinated loss of chromatid cohesion is a common outcome of extended metaphase arrest. PLoS One. 2011;6:e22969. doi: 10.1371/journal.pone.0022969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucke VM, Baumann C, Nigg EA. Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps1. Chromosoma. 2004;113:1–15. doi: 10.1007/s00412-004-0288-2. [DOI] [PubMed] [Google Scholar]
- Stucke VM, Sillje HH, Arnaud L, Nigg EA. Human Mps1 kinase is required for the spindle assembly checkpoint but not for centrosome duplication. EMBO J. 2002;21:1723–1732. doi: 10.1093/emboj/21.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuurman N, Sasse B, Fisher PA. Intermediate filament protein polymerization: molecular analysis of Drosophila nuclear lamin head-to-tail binding. J Struct Biol. 1996;117:1–15. doi: 10.1006/jsbi.1996.0064. [DOI] [PubMed] [Google Scholar]
- Sun T, Yang X, Wang W, Zhang X, Xu Q, Zhu S, Kuchta R, Chen G, Liu X. Cellular abundance of Mps1 and the role of its carboxyl terminal tail in substrate recruitment. J Biol Chem. 2010;285:38730–38739. doi: 10.1074/jbc.M110.177642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci. 2001;114:4385–4395. doi: 10.1242/jcs.114.24.4385. [DOI] [PubMed] [Google Scholar]
- Thummel CS, Pirrotta V. Technical Notes: new pCaSpeR P-element vectors. Drosophila Inf Newsl. 1992;71:150. [Google Scholar]
- Tighe A, Staples O, Taylor S. Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol. 2008;181:893–901. doi: 10.1083/jcb.200712028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F, Wernic D, Poupart MA, Koonin EV, Nasmyth K. Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell. 2000;103:375–386. doi: 10.1016/s0092-8674(00)00130-6. [DOI] [PubMed] [Google Scholar]
- Vigneron S, Prieto S, Bernis C, Labbe JC, Castro A, Lorca T. Kinetochore localization of spindle checkpoint proteins: who controls whom? Mol Biol Cell. 2004;21:21. doi: 10.1091/mbc.E04-01-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103:399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. Structural and mechanistic insights into Mps1 kinase activation. J Cell Mol Med. 2009;13:1679–1694. doi: 10.1111/j.1582-4934.2008.00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn JP, Vleugel M, Liu D, Yates JR, 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38:383–392. doi: 10.1016/j.molcel.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik E, Basto R, Serr M, Scaerou F, Karess R, Hays T. Kinetochore dynein: its dynamics and role in the transport of the Rough deal checkpoint protein. Nat Cell Biol. 2001;3:1001–1007. doi: 10.1038/ncb1101-1001. [DOI] [PubMed] [Google Scholar]
- Wong OK, Fang G. Loading of the 3F3/2 antigen onto kinetochores is dependent on the ordered assembly of the spindle checkpoint proteins. Mol Biol Cell. 2006;17:4390–4399. doi: 10.1091/mbc.E06-04-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Zhu S, Wang W, Zhang X, Old W, Ahn N, Liu X. Regulation of kinetochore recruitment of two essential mitotic spindle checkpoint proteins by Mps1 phosphorylation. Mol Biol Cell. 2009;20:10–20. doi: 10.1091/mbc.E08-03-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yin Q, Ling Y, Zhang Y, Ma R, Ma Q, Cao C, Zhong H, Liu X, Xu Q. Two LXXLL motifs in the N terminus of Mps1 are required for Mps1 nuclear import during G(2)/M transition and sustained spindle checkpoint responses. Cell Cycle. 2011;10:2742–2750. doi: 10.4161/cc.10.16.15927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Chen RH. Mps1 phosphorylation by MAP kinase is required for kinetochore localization of spindle-checkpoint proteins. Curr Biol. 2006;16:1764–1769. doi: 10.1016/j.cub.2006.07.058. [DOI] [PubMed] [Google Scholar]
- Zich J, Sochaj AM, Syred HM, Milne L, Cook AG, Ohkura H, Rappsilber J, Hardwick KG. Kinase activity of fission yeast mph1 is required for mad2 and mad3 to stably bind the anaphase promoting complex. Curr Biol. 2012;22:296–301. doi: 10.1016/j.cub.2011.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.