

Abstract
We have examined peptide-based catalysts for the site-selective thiocarbonylation of a protected form of vancomycin. Several catalysts were identified that either enhanced or altered the inherent selectivity profile exhibited by the substrate. Two catalysts, one identified through screening, and another through rational design, were demonstrated to be effective on 0.50-gram scale. Deoxygenations led ultimately to two new deoxy-vancomycin derivatives, and surprising conformational consequences of deoxygenation were revealed for one of the new compounds. These effects were mirrored in the biological activities of the new analogs, and support a structural role for certain hydroxyls in the native structure.
Keywords: vancomycin, regioselective, thiocarbonylation, radical deoxygenation, peptide-catalyzed
Introduction
The selective alteration of complex, biologically active natural products is a long-standing objective for chemists. The allure of catalytic modification stems from the potential for one- or two-step access to new, complex analogs that might provide improved pharmacological properties. This potential is particularly significant when the natural products are readily accessible by fermentation. The identification of catalysts, whether they are enzymes or synthetic catalysts, for selective diversification of complex scaffolds represents a significant chemical challenge. Our laboratory has been pursuing this goal with peptide-based catalysts. To date we have identified catalysts for the selective derivatization of polyols, including selective acylations of simple sugars1 as well as erythromycin and apoptolidin.2
Site-selective deoxygenation has become an increasing focus of our attention, given the appeal of interrogation of the structure-activity-relationships (SAR) that might be associated with individual hydroxyl groups within a polyol. Hydroxyl groups have been implicated in drug specificity, toxicity, potency, and bioavailability.3 Furthermore, hydroxyls may be targeted by drug-resistant organisms as sites of acylation or phosphorylation for “deactivation” of the drug.4 Recent studies from our group demonstrated the chemical capacity to remove oxygen atoms in this sense in the context of simple sugars,5 and more recently in erythromycin A.6 We have now turned our attention to the significantly more complex glyocopeptide antibiotics, including vancomycin (1; Figure 1). These studies have culminated in strikingly selective catalysts for modification of a minimally protected form of 1. Moreover, these studies have revealed structural control elements within 1 that were unexpected. In light of other observations in our laboratory, we believe that site-selective catalysts may provide a general tool for studying not only (a) selectivity issues in catalysis and (b) SAR associated with individual hydroxyl groups, but also (c) the possible biosynthetic significance of individual functional groups, the deletion of which can cause significant perturbation of a natural product’s native structure.
Figure 1.

The structure of vancomycin and its protected form, 2
Vancomycin represents an important, complex scaffold that plays a significant role in the treatment of infectious disease. Analogs of vancomycin have been accessed through total synthesis,7 enzymatic derivatization8 and biosynthetic engineering.9 Semisynthesis of vancomycin analogs has been examined by many groups, and indeed has led to important, biologically active analogs.10 To our knowledge, systematic studies of hydroxyl group deletions have not been reported for 1. We thus describe studies of this type, and report catalyst-dependent syntheses of two previously unknown deoxy-vancomycins,11 their biological activity profiles, and intriguingly, unexpected aspects of their structure.
Results and Discussion
Our studies began with the examination of catalyst-dependent thiocarbonylation of vancomycin as a prelude to deoxygenation through the venerable Barton-McCombie method.12 Previous studies from our laboratory had shown that peptide-dependent regioselectivity was possible in the selective manipulation of simple sugar derivatives.5a As noted in Figure 2, regioselective thiocarbonylation of 3 could be achieved such that 4 or 5 could be efficiently accessed in a catalyst and condition-dependent fashion. Conventional deoxygenation of these compounds led to the corresponding deoxy-sugars. Extrapolation of these findings to vancomycin was then addressed. In order to apply this method directly, we elected to initially pursue the selective modification of a readily available vancomycin derivative.13 The known alloc- and allyl-protected vancomycin derivative 2 (Figure 1, above) proved readily amenable to study. Vancomycin derivative 2 retains six hydroxyl groups, relative to the nine of 1, reducing the site-selectivity challenge somewhat. Compound 2 also exhibits excellent solubility properties in the organic solvents where the peptide-catalyzed thiocarbonylation chemistry developed to date works best.
Figure 2.

Peptide-catalyzed thiocarbonylation of diol 3.
Vancomycin itself is readily available by fermentation,14 and we prepared its protected analog 2 on a 30-gram scale. We were then pleased to find that the conditions we had developed for the selective thiocarbonylation of simple sugars proved quite portable to the study of selective modification of 2. Figure 3 shows the results of our initial study. These conditions provided a negligible background reaction in the absence of a catalyst (entry 1). However, the achiral control catalyst N-methylimidazole (NMI, 8) did result in modest rate acceleration, yielding moderate conversion to a major and a minor product, each of which we were able to isolate from the reaction mixture.
Figure 3.

Background and achiral catalyst control reactions of 2 under the optimized conditions. (a) The reaction of 2 under the optimized thiocarbonylation conditions. (b) Tabulated and raw HPLC data revealing the product distributions observed when N-methylimidazole (NMI, 8) is employed as a catalyst.
The structures of the thionocarbonate products were identified by NMR spectroscopy (Figure 4). Using an edited HSQC experiment, we were able to observe clear changes in the chemical shifts of modified sites, relative to the spectrum recorded for 2 (Figure 4a). The faster-eluting peak was assigned as the G6-thionocarbonate (6), while the slower eluting peak was assigned as the Z6-thionocarbonate (7). In Figure 4b, we see a large change in the G6-methylene and smaller changes in the G5- and G4-methines. Likewise, in Figure 4c, we see a large change in the Z6-methine as well as smaller changes in the X6- and X5-methines.15
Figure 4.

1H-13C correlation with DEPT; methyls and methines shown in red, methylenes shown in blue. (a) Partial HSQC spectrum of 2. (b) Partial HSQC spectrum of 6 with 2 overlaid in grey. (c) Partial HSQC spectrum of 7 with 2 overlaid in grey. HSQC = heteronuclear single-quantum correlation. DEPT = distortionless enhancement by polarization transfer.
Having established a sense of the inherent reactivity, we evaluated peptide-based catalysts for perturbations in the product ratio. We initiated two approaches to catalyst discovery: one in which we would screen libraries of peptides, and one in which we would design peptides based on the natural binding site of vancomycin. Upon pursuing the first approach, one of our initial catalyst screens examined a library (25 members) of previously synthesized catalysts from historical projects focused on enantioselective or regioselective reactions of alcohols and amides.2c,16 Strikingly, from this small test library, we found two catalysts, peptide 9 and peptide 10, that could either reverse or enhance the product selectivity observed with NMI (Figure 5). Peptide 9 and the enantiomer of 10 have been reported previously for the enantioselective phosphorylation of tribenzyl-myo-inositol.17 Peptide 9 provided a clear reversal in selectivity in comparison to the selectivity exhibited by NMI, to give primarily the G6-thionocarbonate (entry 1). Catalyst 10, on the other hand, enhanced the Z6-selectivity afforded by NMI (entry 2). Moreover, both catalysts were significantly more active than NMI. Peptide 11, which differs from 10 by substituting the expensive DHyp(Ot-Bu) amino acid with DPro, showed similar Z6-selectivity (entry 3). While it is presently unclear exactly how these peptides interact with 2, one can imagine various hydrogen-bonding schemes that could lead to the site-selectivity observed.
Figure 5.

Tabulated and raw HPLC data revealing the product distributions observed when catalysts 9, 10 and 11 were employed as catalysts for the derivatization of 2.
On the other hand, the interaction between vancomycin and peptides that mimic its native binding site (the Lys-DAla-DAla terminus of the Gram-positive bacterial peptidoglycan) is well-known and has been observed in X-ray crystallographic (Figure 6a)18 and NMR studies.19 We hypothesized that this motif could enhance our ability to control the site of thiocarbonylation with judicious localization of the catalytic alkylimidazole moiety.20 We thus designed several catalysts based on incorporation of the catalytic histidine residue, Pmh (π-methylhistidine) in the D-form, as illustrated in Figure 6b. In particular, we assessed the catalyst scaffold based on Lys-DAla-DPmh. Our concept was that we might further enhance the targeting of the glucosyl residue by this strategy.
Figure 6.
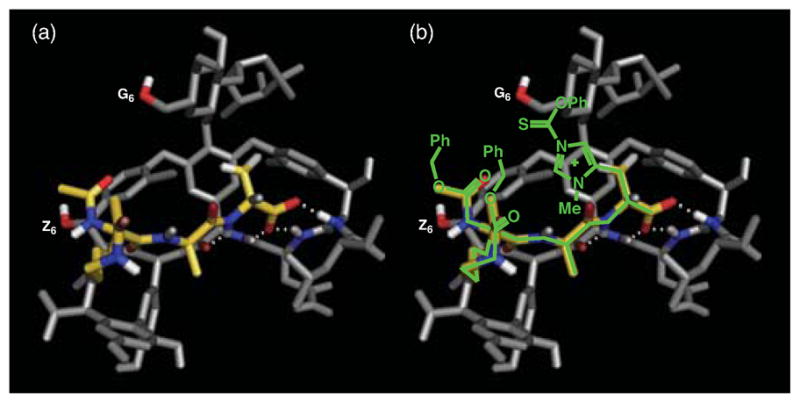
(a) The X-ray crystal structure of Ac-Lys(Ac)-DAla-DAla-O− (yellow) bound to vancomycin (grey). (b) The proposed model for delivery of phenyloxythiocarbonyl to the G6-hydroxyl by Cbz-Lys(Cbz)-DAla-DPmh-O− (green). X-ray structure adapted from ref 18, PDB # 1FVM.
We were initially concerned that the affinity of 2 for catalysts based on the Lys-DAla-DAla motif might be too high to allow for catalyst turnover. Thus, we examined these catalysts with a full (100 mol%) loading (Figure 7). Despite the increase in catalyst loading, the first two peptides we examined, with amide and ester functionality at the C-terminal position (e.g., 12 and 13, entries 1–2), showed low conversions. In addition, the slight selectivity that was observed favored the Z6-position, contrary to our hypothesis (Figure 6b). However, maintaining the C-terminal carboxylate that is critical to the biological binding of vancomycin to the Lys-DAla-DAla motif proved decisive. Thus, the organic soluble tetrabutylammonium salt 14 led to a dramatic increase in reactivity. Furthermore, selectivity for the G6-thionocarbonate proved to be very high (entry 3). Also of significant note, the reaction is very efficient with a 20 mol% catalyst loading (entry 4). Indeed, we found the reaction proceeded to higher conversion while maintaining excellent selectivity, allaying concerns about a possible lack of catalyst turnover.21 Importantly, control experiments revealed that tetrabutylammonium chloride is not a catalyst for the reaction. A combination of equimolar tetrabutylammonium chloride and NMI provided results that were essentially indistinguishable from those observed with NMI alone. Moreover, reactions catalyzed by peptide 14, performed in the presence of Cbz-Lys(Cbz)-DAla-DAla/NBu4 as a putative competitive inhibitor resulted in lower levels of conversion and selectivity.22
Figure 7.

Tabulated and raw HPLC data revealing the product distributions observed when Lys-DAla-DPmh catalysts 12–14 were employed as catalysts for the derivatization of 2.
With selective catalysts 11 and 14 in hand, we wished to assess their performance on a significant scale. These experiments would set the stage for deoxygenation and deprotection experiments. We were pleased to find that the reactions proceeded with equivalent or better efficiency when conducted on 0.50-g scale, as shown in Figure 8. While the results for catalyst 14 were almost identical to the smaller scale reaction, we observed a significant improvement in conversion and selectivity for catalyst 11.
Figure 8.
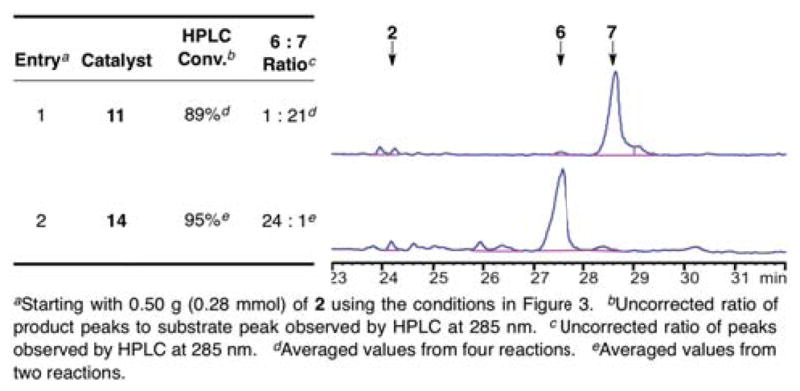
Tabulated and raw HPLC data revealing the product distributions observed when catalysts 11 and 14 were employed as catalysts for the derivatization of 2 on half-gram scale.
We then turned our attention to deoxygenation and deprotection. We expected that the G6- (6) and Z6-thionocarbonate (7) might require different conditions for radical deoxygenation since 6 is a primary thionocarbonate while 7 is benzylic. Under traditional Barton-McCombie conditions,12 we observed complete reaction of the primary G6-thionocarbonate at 80 °C, whereas the reduction of the benzylic Z6-thionocarbonate proceeded at 70 °C (Figure 9). Deoxygenation of 7 was also performed at room temperature using BEt3/air as the radical initiator.23,24 Allyl- and alloc-removal from the protected deoxy-vancomycins proceeded smoothly under conditions similar to those in the literature.13b,25 Notably, HSnBu3 could be substituted with PhSiH3 without loss of reaction efficiency. Thus, compounds 15 (G6-deoxy-vancomycin) and 16 (Z6-deoxy-vancomycin) were obtained, each in 5 efficient steps from 1.
Figure 9.

Deoxygenation and deprotection of thionocarbonates 6 and 7 to yield the corresponding deoxy-vancomycins 15 and 16.
Preparative HPLC purification of deoxy-vancomycins 15 and 16 provided access to high-purity samples for structural characterization and biological evaluation. G6-Deoxy-vancomycin 15, perhaps unsurprisingly, revealed close analogy to vancomycin structurally. As shown in Figure 10a, samples could be obtained in significant quantity and in high purity. NMR analysis, exemplified by the HSQC spectrum in Figure 10b, revealed a highly homogeneous structure, for which analogy to the spectra of 1 itself was excellent.15
Figure 10.

(a) HPLC trace of 15 after purification. (b) Partial HSQC spectrum of 15.
Z6-Deoxy-vancomycin 16 presented surprising differences from 1 and 15. HPLC purification and LCMS analysis of 16 consistently revealed the presence of two peaks (Figure 11a), of equivalent mass. Moreover, isolation of the fast-eluting peak, and reinjection for HPLC analysis revealed the same doubling of the peaks. Isolation and reinjection of the slow-eluting peak produced the identical HPLC trace. These observations led us to conclude that the Z6-deoxy-vancomycin (16) exists at room temperature as a conformationally mobile species of at least two interconverting states, in stark contrast to either 1 or 15. The conformational heterogeneity was supported by NMR studies, which revealed doubling of cross-peaks in the aromatic and α-CH regions of the HSQC spectrum (e.g., Figure 11b).
Figure 11.

(a) HPLC trace of 16 after purification. (b) Partial HSQC spectrum of 16.
The exact nature of the conformational equilibrium exhibited by 16 is difficult to establish unambiguously. We have considered several possibilities. Our first proposal, stimulated by conformational studies and of synthetic vancomycin fragments in the literature,26 was that of possible atropisomerism about the chloroarene ring-6 of 16 (Figure 12a). However, when 16 was subjected to dechlorination conditions (Pd/C, H4N2, NH4OH, MeOH/H2O),27 the HPLC trace exhibited by didechloro-Z6-deoxy-vancomycin 17 retains its apparent conformational heterogeneity (Figure 12b).
Figure 12.
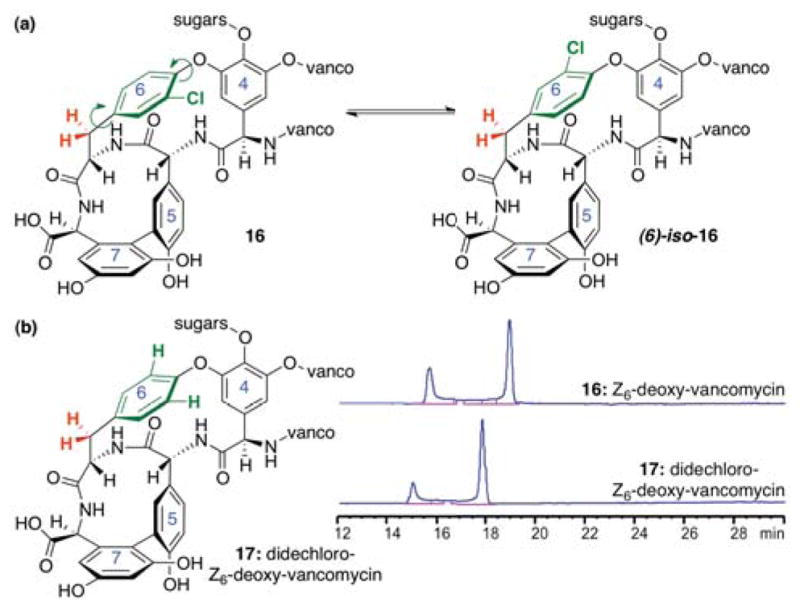
(a) Proposed atropisomers about the chloroarene ring-6 of 16. (b) HPLC analysis of didechloro-Z6-deoxy-vancomycin (17).
There are at least three other possibilities that one can consider for the observed conformational equilibrium: (a) a cis-amide/trans-amide isomerization about the C5-W6 amide bond (Figure 13a);28 (b) medium ring isomerization associated with the allylic strain conformers induced by the C6-W7 amide bond (Figure 13b);29 and (c) atropisomerization about the biaryl moiety of the 5- and 7-arenes (not shown).30 Of course, these issues could also operate simultaneously and synergistically. What is remarkable about this observation of conformational heterogeneity exhibited by 16 is that our catalytic studies have unveiled conformational programming induced by the Z6-hydroxyl in native vancomycin. Thus, its presence is not associated with a simple SAR, but carries with it significant conformational consequences. Our current assessment of the role of the hydroxyl is that it may form a hydrogen bond with the C6-amide carbonyl, stabilizing the peptidoglycan-binding conformation, as illustrated in Figure 13c (the OC6-OZ6 distance is 3.2 Å in the X-ray structure of vancomycin complexed with Ac-Lys(Ac)-DAla-DAla).18 We also note in passing that this structural programming of native natural product conformations has surfaced in our previous studies of catalytic polyol modification, with analogous structural reorganizations of erythromycin upon site-selective acylation of individual hydroxyl groups.2c,31
Figure 13.

(a) cis/trans-Isomerization of the C5-W6 amide bond. (b) syn/anti-Conformational change within the (5-6-7)-macrocycle about the C6-W7 amide. (c) A proposed hydrogen bond that may rigidify the syn/anti-isomer shown.
Of note, the conformational mobility of the Z6-deoxy-vancomycin 16 appears to have biological consequences. The minimum inhibitory concentrations (MICs) exhibited by 1 and the new compounds were measured as an indicator of antibiotic activity (Table 1). Interestingly, G6-deoxy-vancomycin (15) showed identical activity against all tested bacterial strains. Z6-Deoxy-vancomycin (16), however, proved to be consistently less active; this result likely reflects the conformational heterogeneity, and the differential activity (high and low) associated with each interconverting structure.32
Table 1.
MIC values for deoxy-vancomycins 15 and 16 against selected bacterial strains.
| Entry | Compound | MSSAa,b | MRSAc | VSEd | VRE (VanB)e | VRE (VanA)f |
|---|---|---|---|---|---|---|
| 1 | 1 | 0.5 | 1 | 2 | 16 | >64 |
| 2 | 15 | 0.5 | 1 | 2 | 16 | >64 |
| 3 | 16 | 2 | 4 | 8 | >64 | >64 |
MIC values reported in μg/mL.
MSSA = methicillin-susceptible S. aureus, ATCC 29213.
MRSA = methicillin-resistant S. aureus, ATCC 43300.
VSE = vancomycin -susceptible enterococci, ATCC 29212.
VRE = vancomycin-resistant enterococci, ATCC 51299.
MMX 486. See Supporting Information for additional details.
Conclusions
In summary, we have discovered site-selective catalysts that allow hydroxyl group-selective thiocarbonylation of a minimally protected version of 1. These catalysts ultimately allowed preparation of two deoxy-vancomycins that differ from 1 only in that a singular oxygen atom is missing from either 15 or 16. We have shown that two methods of catalyst discovery, library screening and binding site-based design, are both viable strategies to selective modification of natural product derivatives. The prospects for generalizing this catalytic approach seem encouraging, given the substantially divergent site-selectivity exhibited by catalysts 11 and 14. The observation of near total conformational homology between 1 and 15 is mirrored in their essentially indistinguishable biological profiles. On the other hand, the removal of the O-atom from the Z6-position of 1 revealed a pronounced conformational role for the Z6-hydroxyl group. The biological effects of reducing the inherent rigidity of 1 manifest in the attenuated activity of 16 in the MIC assays. Perhaps most notable, however, is the capacity of site-selective catalysis to unveil conformational programming in complex structures, perhaps encoded by biosynthesis,33 which we will now look for in systematic ways.
Supplementary Material
Acknowledgments
Funding Sources
We are grateful to the National Institutes of General Medical Sciences of the National Institute of Health (GM-068649) for support. KML was supported by a post-doctoral fellowship from the German Academic Exchange Service (DAAD).
We also thank Dr. Bret E. Huff and Eli Lilly and Company for providing samples of 1.
Footnotes
The authors declare no competing financial interest.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Procedures and characterization data for all of the experiments described in the manuscript are available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Lewis CA, Sculimbrene BR, Xu Y, Miller SJ. Org Lett. 2005;7:3021–3023. doi: 10.1021/ol051061a. [DOI] [PubMed] [Google Scholar]; (b) Griswold KS, Miller SJ. Tetrahedron. 2003;59:8869–8875. [Google Scholar]
- 2.(a) Lewis CA, Longcore KE, Miller SJ, Wender PA. J Nat Prod. 2009;72:1864–1869. doi: 10.1021/np9004932. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lewis CA, Merkel J, Miller SJ. Bioorg Med Chem Lett. 2008;18:6007–6011. doi: 10.1016/j.bmcl.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lewis CA, Miller SJ. Angew Chem Int Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 3.(a) Gray KC, Palacios DS, Dailey I, Endo MM, Uno BE, Wil-cock BC, Burke MD. Proc Nat Acad Sci U S A. 2012;109:2234–2239. doi: 10.1073/pnas.1117280109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hanessian S, Giguère A, Grzyb J, Maianti JP, Saavedra OM, Aggen JB, Linsell MS, Goldblum AA, Hildebrandt DJ, Kane TR, Dozzo P, Gliedt MJ, Matias RD, Feeney LA, Arm-strong ES. ACS Med Chem Lett. 2011;2:924–928. doi: 10.1021/ml200202y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Szpilman AM, Cereghetti DM, Manthorpe JM, Wurtz NR, Carreira EM. Chem—Eur J. 2009;15:7117–7128. doi: 10.1002/chem.200900231. [DOI] [PubMed] [Google Scholar]; (d) Fujisawa KI, Hoshiya T, Kawaguchi H. J Antibiot. 1974;27:677–681. doi: 10.7164/antibiotics.27.677. [DOI] [PubMed] [Google Scholar]
- 4.For reviews on the topic, see: Wright GD. Adv Drug Delivery Rev. 2005;57:1451–1470. doi: 10.1016/j.addr.2005.04.002.Walsh C. Nature. 2000;406:775–781. doi: 10.1038/35021219.
- 5.Sánchez-Roselló M, Puchlopek ALA, Morgan AJ, Miller SJ. J Org Chem. 2008;73:1774–1782. doi: 10.1021/jo702334z.For a pioneering application of thiocarboylation of carbohydrates for deoxygenation, see: Haque ME, Kikuchi T, Kanemitsu K, Tsuda Y. Chem Pharm Bull. 1987;35:1016–1029.
- 6.Jordan PA, Miller SJ. Angew Chem Int Ed. 2012;51:2907–2911. doi: 10.1002/anie.201109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Xie J, Pierce JG, James RC, Okano A, Boger DL. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crane CM, Boger DL. J Med Chem. 2009;52:1471–1476. doi: 10.1021/jm801549b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Crowley BM, Boger DL. J Am Chem Soc. 2006;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nicolaou KC, Mitchell HJ, Jain NF, Winssinger N, Hughes R, Bando T. Angew Chem Int Ed. 1999;38:240–244. [Google Scholar]
- 8.Many research groups have examined the enzymatic generation of vancomycin analogs. For examples, see: Oh TJ, Kim DH, Kang SY, Yamaguchi T, Sohng JK. J Antibiot. 2011;64:103–109. doi: 10.1038/ja.2010.131.Thayer DA, Wong CH. Chem—Asian J. 2006;1:445–452. doi: 10.1002/asia.200600084.Oberthür M, Leimkuhler C, Kruger RG, Wei Lu, Walsh CT, Kahne D. J Am Chem Soc. 2005;127:10747–10752. doi: 10.1021/ja052945s.Kruger RG, Lu W, Oberthür M, Tao J, Kahne D, Walsh CT. Chem Biol. 2005;12:131–140. doi: 10.1016/j.chembiol.2004.12.005.Malnar I, Sih CJ. J Mol Catal B: Enzym. 2000;10:545–549.
- 9.Mutasynthetic studies have been performed in the biosynthesis of balhimycin, a vancomycin-type glycopeptide: Weist S, Kittel C, Bischoff D, Bister B, Pfeifer V, Nicholson GJ, Wohlleben W, Süssmuth RD. J Am Chem Soc. 2004;126:5942–5943. doi: 10.1021/ja0499389.Weist S, Bister B, Puk O, Bischoff D, Pelzer S, Nicholson GJ, Wohlleben W, Jung G, Süssmuth RD. Angew Chem Int Ed. 2002;41:3383–3385. doi: 10.1002/1521-3773(20020916)41:18<3383::AID-ANIE3383>3.0.CO;2-R.
- 10.For examples, see: Crane CM, Pierce JG, Leung SSF, Tirado-Rives J, Jorgensen WL, Boger DL. J Med Chem. 2010;53:7229–7235. doi: 10.1021/jm100946e.Leadbetter MR, Adams SM, Bazzini B, Fatheree PR, Karr DE, Krause KM, Lam BMT, Linsell MS, Nodwell MB, Pace JL, Quast K, Shaw JP, Soriano E, Trapp SG, Villena JD, Wu TX, Christensen BG, Judice JK. J Antibiot. 2004;57:326–336. doi: 10.7164/antibiotics.57.326.Griffin JH, Linsell MS, Nodwel MB, Chen Q, Pace JL, Quast KL, Krause KM, Farrington L, Wu TX, Higgins DL, Jenkins TE, Christensen BG, Judice JK. J Am Chem Soc. 2003;125:6517–6531. doi: 10.1021/ja021273s.Ritter TK, Mong KKT, Liu H, Nakatani T, Wong CH. Angew Chem Int Ed. 2003;42:4657–4660. doi: 10.1002/anie.200351534.Nicolaou KC, Cho SY, Hughes R, Winssinger N, Smethurst C, Labischinski H, Enderman R. Chem—Eur J. 2001;7:3798–3823. doi: 10.1002/1521-3765(20010903)7:17<3798::aid-chem3798>3.0.co;2-6.Ge Min, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507.Cooper RDG, Snyder NJ, Zweifel MJ, Staszak MA, Wilkie SC, Nicas TI, Mullen DL, Butler TF, Rodriguez MJ, Huff BE, Thompson RC. J Antibiot. 1996;49:575–581. doi: 10.7164/antibiotics.49.575.Cristofaro MF, Beauregard DA, Yan H, Osborn NJ, Williams DH. J Antibiot. 1995;48:805–810. doi: 10.7164/antibiotics.48.805.Harris CM, Fesik SW, Thomas AM, Kannan R, Harris TM. J Org Chem. 1986;51:1509–1513.
- 11.To the best of our knowledge, Z2-deoxy-vancomycin is the only reported deoxy derivative: Xu B, Haison X, Yu H, Mao W, Ye W, Zhejiang CN. U.S. Patent Application 2010/0222252. 2010:A1.
- 12.Barton DHR, McCombie SW. J Chem Soc Perkin Trans I. 1975:1574–1585. [Google Scholar]
- 13.(a) Griffith BR, Krepel C, Blanchard S, Ahmed A, Edmiston CE, Thorson JS. J Am Chem Soc. 2007;129:8150–8155. doi: 10.1021/ja068602r. [DOI] [PubMed] [Google Scholar]; (b) Thompson C, Ge M, Kahne D. J Am Chem Soc. 1999;121:1237–1244. [Google Scholar]
- 14.McCormick MH, McGuire JM. Eli Lilly and Company, Indian-apolis; IN, USA: U.S. Patent 3067099. 1962For more recent studies on this topic, see: Jung HM, Kim SY, Moon HJ, Oh DK, Lee JK. Appl Microbiol Biotechnol. 2007;77:789–795. doi: 10.1007/s00253-007-1221-4.Padma PN, Rao AB, Yadav JS, Reddy G. Appl Biochem Biotechnol. 2002;102–103:395–405. doi: 10.1385/abab:102-103:1-6:395.
- 15.See the Supporting Information for full spectra and additional details.
- 16.(a) Fowler BS, Mikochik PJ, Miller SJ. J Am Chem Soc. 2010;132:6651–6653. doi: 10.1021/ja9107897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fiori KW, Puchlopek ALA, Miller SJ. Nat Chem. 2009;1:630–634. doi: 10.1038/nchem.410. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2008;130:16358–16365. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Miller SJ. Acc Chem Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- 17.Intriguingly, catalysts 9 and ent-10 deliver, with high enantioselectivity, inositol-1-phosphate and inositol-3-phosphate, respectively. Correspondingly, 9 and 10 would both lead to phosphorylation of the same position of tribenzyl-myo-inositol; interestingly, however, these catalysts selectively thiocarbonylate different sites of vancomycin. Sculimbrene BR, Morgan AJ, Miller SJ. J Am Chem Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m.
- 18.Nitanai Y, Kikuchi T, Kakoi K, Hanamaki S, Fujisawa I, Aoki K. J Mol Biol. 2009;385:1422–1432. doi: 10.1016/j.jmb.2008.10.026. (PDB: 1FVM) [DOI] [PubMed] [Google Scholar]
- 19.(a) Williams DH, Maguire AJ, Tsuzuki W, Westwell MS. Science. 1998;280:711–714. doi: 10.1126/science.280.5364.711. [DOI] [PubMed] [Google Scholar]; (b) Li D, Sreenivasan U, Juranic N, Macura S, Puga FJ, II, Frohnert PM, Axelsen PH. J Mol Recognit. 1997;10:73–87. doi: 10.1002/(SICI)1099-1352(199703/04)10:2<73::AID-JMR347>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]; (c) Hawkes GE, Molinari H, Singh S. J Magn Reson. 1987;74:188–192. [Google Scholar]; (d) Molinari H, Pastore A, Lian LY, Hawkes GE, Sales K. Biochemistry. 1990;29:2271–2277. doi: 10.1021/bi00461a010. [DOI] [PubMed] [Google Scholar]; (e) Kannan R, Harris CM, Harris TM, Waltho JP, Skelton NJ, Williams DH. J Am Chem Soc. 1988;110:2946–2953. [Google Scholar]; (f) Williams DH, Williamson MP, Butcher DW, Hammond SJ. J Am Chem Soc. 1983;105:1332–1339. [Google Scholar]
- 20.We recently applied a related strategy for targeting a peptide to the binding site of vancomycin with the goal of inducing selective bromination. See: Pathak TP, Miller SJ. J Am Chem Soc. 2012;134:6120–6123. doi: 10.1021/ja301566t.
- 21.We note that when catalyst 14 is applied to the attempted thiocarbonylation of native 1 (i.e., without the protecting groups found in 2) under phase-transfer conditions (pH=7 phosphate buffer/CH2Cl2:THF), a sluggish reaction results, presumably due to competitive hydrolysis of the reagent under these conditions.
- 22.Reactions catalyzed by peptide 14, as well as those catalyzed by NMI, in the presence of Cbz-Lys(Cbz)-DAla-DAla/NBu4, are described in the Supporting Information.
- 23.Nozaki K, Oshima K, Utimoto K. Tetrahedron Lett. 1988;29:6125–6126.See the Supporting Information for details.
- 24.In general, alternative H-atom sources such as silanes or dimethyl-phosphite led to sluggish reactions or no reaction at temperatures up to 80°C Barton DHR, Jang DO, Jaszberenyi JC. J Org Chem. 1993;58:6838–6842.Barton DHR, Jang DO, Jaszberenyi JC. Tetrahedron. 1993;49:2793–2804.Barton DHR, Jang DO, Jaszberenyi JC. Tetrahedron Lett. 1990;31:4681–4684.
- 25.(a) Dessolin M, Guillerez MG, Thieriet N, Guibé F, Loffet A. Tetrahedron Lett. 1995;36:5741–5744. [Google Scholar]; (b) Guibé F, Dangles O, Balavoine G. Tetrahedron Lett. 1986;27:2365–2368. [Google Scholar]
- 26.For atropisomerization studies in synthetic vancomycin precursors: Boger DL, Castle SL, Miyazaki S, Wu JH, Beresis RT, Loiseleur O. J Org Chem. 1999;64:70–80. doi: 10.1021/jo980880o.Boger DL, Loiseleur O, Castle SL, Beresis RT, Wu JH. Bioorg Med Chem Lett. 1997;7:3199–3202. doi: 10.1016/s0960-894x(98)00110-3.For related isomerization observed in the rearranged vancomycin derivative CDP-I: Harris CM, Kopecka H, Harris TM. J Am Chem Soc. 1983;105:6915–6922.
- 27.This procedure was derived from the following two precedents: Hoke JB, Gramiccioni GA, Balko EN. Appl Catal B. 1992:285–296.Harris CM, Kannan R, Kopecka H, Harris TM. J Am Chem Soc. 1985;107:6652–6658.See the Supporting Information for details.
- 28.For amide isomerization in the vancomycin-type antibiotic, UK-69542: Skelton NJ, Williams DH, Rance MJ, Ruddock JC. J Am Chem Soc. 1991;113:3757–3765.For amide isomerization studies in synthetic vancomycin precursors: Evans DA, Dinsmore CJ, Watson PS, Wood MR, Richardson TI, Trotter BW, Katz JL. Angew Chem Int Ed. 1998;37:2704–2707. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2704::AID-ANIE2704>3.0.CO;2-1.Evans DA, Dinsmore CJ. Tetrahedron Lett. 1993;34:6029–6032.For the proposed amide isomerization in Z6-deoxy-teicoplanin derivatives, see: Malabarba A, Ciabatti Kettenring J, Ferrari P, Scotti R, Goldstein BP, Denaro M. J Antibiot. 1994;47:1493–1506. doi: 10.7164/antibiotics.47.1493.
- 29.A similar conformational change has been proposed about the C2-W3 amide at room temperature. This presumably becomes one conformation upon hydrogen bonding with the DAla-DAla motif: Waltho JP, Williams DH, Stone DJM, Skelton NJ. J Am Chem Soc. 1988;110:5638–5643.Williams DH, Waltho JP. Biochem Pharmacol. 1988;37:131–141. doi: 10.1016/0006-2952(88)90765-4.This conformational change was also proposed for Z6-deoxy-teicoplanin: see reference 28d.
- 30.For atropisomerization of the 5–7 biaryl in the related semi-synthetic glycopeptide, oritavancin: Zhou CC, Stoner EJ, Kristensen EW, Stewart KD, Rasmussen RR, Hollis LS, Wittenberger SJ, Matayoshi ED, Christesen AC, Brill GM. Tetrahedron. 2004;60:10611–10618.For atropisomerization studies in synthetic vancomycin precursors: see references 28b and 28c.
- 31.Everett JR, Hunt E, Tyler JW. J Chem Soc Perkin Trans. 1991;2:1481–1487. [Google Scholar]
- 32.(a) Whether or not the attenuated biological activity is due to the canonical binding of the vancomycin derivative to the DAla-DAla target has not been established at this time. Analysis of binding of Ac-Lys(Ac)-DAla-DAla to vancomycin and the synthetic analogs reveals high similarity between 1 and 15, and significant differences between 1 and 16, consistent with the differential biological activities. Please see Supporting Information for the details. (b) For a pioneering study of alternative mechanisms of action, see reference 10f.
- 33.Walsh CT, Fischbach MA. J Am Chem Soc. 2010;132:2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.