

Abstract
Previous clinical and epidemiological studies of vitamin E have used primarily α-tocopherol for the prevention of cancer. However, γ-tocopherol has demonstrated greater anti-inflammatory and anti-tumor activity than α-tocopherol in several animal models of cancer. This study assessed the potential chemopreventive activities of a tocopherol mixture containing 58%γ-tocopherol (γ-TmT) in an established rodent model of mammary carcinogenesis. Female ACI rats were utilized due to their sensitivity to 17β-estradiol (E2) to induce mammary hyperplasia and neoplasia. The rats were implanted subcutaneously with sustained release E2 pellets and given dietary 0.3% or 0.5% γ-TmT for 2 or 10 weeks. Serum E2 levels were significantly reduced by the treatment with 0.5% γ-TmT. Serum levels of inflammatory markers, prostaglandin E2 and 8-isoprostane, were suppressed by γ-TmT treatment. Histology of mammary glands showed evidence of epithelial hyperplasia in E2 treated rats. Immunohistochemical analysis of the mammary glands revealed a decrease in proliferating cell nuclear antigen (PCNA), cyclooxygenase-2 (COX-2) and estrogen receptor α (ERα), while there was an increase in cleaved-caspase 3, peroxisome proliferator activated receptor γ (PPARγ), and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) in γ-TmT treated rats. In addition, treatment with γ-TmT resulted in a decrease in the expression of ERα mRNA, whereas mRNA levels of ERβ and PPARγ were increased. In conclusion, γ-TmT was shown to suppress inflammatory markers, inhibit E2-induced cell proliferation, and upregulate PPARγ and Nrf2 expression in mammary hyperplasia, suggesting that γ-TmT may be a promising agent for human breast cancer prevention.
Keywords: vitamin E, tocopherol, breast cancer, estrogen receptor, peroxisome proliferator activated receptor γ
INTRODUCTION
Breast cancer is one of the most common malignancies affecting women and is the second leading cause of cancer death in women [1]. Although breast cancer is a disease that is prevalent in society, the etiology and pathogenesis remain poorly understood. Prevention of breast cancer is essential, and the success of prevention strategies depends on understanding the molecular mechanism of breast cancer initiation and progression.
Dietary intake of vitamin E, a fat-soluble vitamin, has been suggested to reduce cancer risk due to its antioxidant properties [2]. There are eight different forms of vitamin E which include four tocopherols (with a saturated phytyl tail) and four tocotrienols (with an unsaturated isoprenoid side chain), designated as α, β, γ, and δ variants [3] (Fig. 1A). γ-Tocopherol is most prominent in the American diet and is found in vegetable oils such as soybean, corn, and cottonseed [4]. However, α-tocopherol is the major tocopherol found in human blood and tissues, and has superior activity over the other tocopherols in the classic fertility restoration assay, it is therefore known as the “classic” vitamin E [5]. Consequently, α-tocopherol has been the most widely studied form of vitamin E for the prevention and treatment of cancer, but epidemiological evidence supporting an association between α-tocopherol and cancer prevention is inconclusive [5–8].
Figure 1.

(A) Structures of four tocopherol forms and short chain metabolites, CEHC: α-, β-, γ-, and δ-tocopherol. (B) Serum samples were analyzed for E2 (pg/ml) (n = 6). The data are presented as the mean ± S.E.M. Statistical significance, *p<0.05, †p<0.01. p-Values are compared to the E2 control of their respective time point. (C) Mammary glands were stained with H & E (200× total magnification). Representative sections from the control pellet, E2 control, and E2 treated animals fed either 0.3% or 0.5% γ-TmT diet are shown.
Higher concentrations of α-tocopherol have the paradoxical effects of decreasing the level of serum γ-tocopherol [8,9]. This may not be beneficial since γ-tocopherol has demonstrated significantly greater anti-inflammatory and anti-tumor activity than α-tocopherol in several different animal models of colon, breast, and prostate cancer [4,10]. In addition, γtocopherol is more effective in inhibiting the activity of cyclooxygenase and trapping reactive nitrogen species (RNS) than α-tocopherol [11–13]. Although the biological effects of vitamin E, mainly α-tocopherol, have been investigated over many decades, current understanding of mechanisms of action for different tocopherol forms in inhibiting breast cancer is limited. We have recently demonstrated that administration of 0.3% and 0.5% tocopherol mixture diet containing 58% γ-tocopherol (γ-TmT) suppressed mammary tumor growth in N-methyl-N-nitrosourea (NMU)-induced Sprague-Dawley rats by 50% and 80%, respectively [14]. Mammary tumors from NMU-treated rats are estrogen receptor (ER)-positive, which is classified as a luminal subtype of breast cancer [15]. Luminal tumors express ER, ER-responsive genes, and other genes that encode characteristic proteins of luminal epithelial cells of origin, and therefore respond to selective estrogen receptor modulators, such as tamoxifen, and have a better prognosis than other breast cancer subtypes [16].
Because the mammary tumor model in Sprague-Dawley rats uses a synthetic carcinogen, NMU, we utilized the ACI rats, which are known to be sensitive to estrogen in inducing mammary tumors that are more comparable to human breast cancer [17]. Female ACI rats rarely develop spontaneous mammary tumors [18] and shows a high incidence (80–100%) of mammary ductal adenocarcinomas within 24–36 weeks when exposed to low doses of exogenous estradiol (E2) [19,20]. Using this model, we examined the effects of E2 and γ-TmT during the early development of mammary hyperplasia. In the present study, we found that treatment with dietary γ-TmT inhibits cell proliferation, down-regulates the levels of ERα and circulating serum E2, suppresses serum levels of inflammatory markers, while upregulates peroxisome proliferator activated receptor γ (PPARγ) and nuclear factor (erythroid-derived 2)-like 2 (Nrf2) levels in mammary hyperplasia.
MATERIALS AND METHODS
Animals and experimental procedures
Female ACI rats were purchased from Harlan Sprague Dawley Inc. (Indianapolis, IN) at 7–8 weeks of age. The procedure to implant the pellet was adapted from a previously described method [20]. Briefly, an incision was made in a shaved area in the middle of the back between the scapulas. The animals were implanted subcutaneously with either a control pellet containing 20 mg of cholesterol or a pellet containing 2.5 mg of E2 and 17.5 mg of cholesterol. The pellets were purchased from Hormone Pellet Press (Shawnee Mission, KS). The animals were sacrificed 2 or 10 weeks after the surgical implantation of the pellet. Blood was collected; serum was prepared and stored in −80°C. Mammary glands from each animal were harvested at necropsy and fixed in 10% formalin or flash frozen and stored in −80°C. The liver was weighed and stored in −80°C for further analysis.
Diet
γ-TmT was supplied by the Cognis Corporation (Cincinnati, OH) and contained 58% γ-tocopherol, 24% δ-tocopherol, 13% α-tocopherol, and 0.5% β-tocopherol. Semipurified modified AIN-93M diet was obtained from Research Diets Laboratory (New Brunswick, NJ) and used as the control diet. The test diet was prepared by Research Diets Laboratory by adding either 0.3% or 0.5% γ-TmT to the AIN-93M diet. The diets were stored at 4°C and the food cups were replenished with fresh pellets twice weekly.
Analysis of tocopherols in rat serum and mammary glands
The levels of tocopherols (α-, γ-, δ-) and their metabolites in the serum and mammary gland were analyzed by high performance liquid chromatography using methods as previously described [14,21,22]. Reference samples were prepared as previously reported [14,22].
Serum estradiol levels
The E2 levels in the serum were analyzed using an EIA kit from Enzo Life Sciences International, Inc (Plymouth Meeting, PA). Serum samples were purified and the assay was performed as described in the manufacture’s protocol.
Enzyme immunoassays for prostaglandin E2 and 8-isoprostane
EIA kits from Cayman Chemicals (Ann Arbor, MI) were used to analyze the levels of prostaglandin E2 (PGE2) and 8-isoprostane. Serum samples were prepared as previously described [21]. Briefly, the serum samples were mixed with ethyl acetate, vortexed and then centrifuged. The organic layer was collected and dried using a Speed Vacuum Evaporator (VWR International, Inc, West Chester, PA). Samples were then reconstituted in EIA buffer and the assay was performed as described in the manufacture’s protocol.
Histopathological analyses and immunostaining
Mammary glands from the 4th position on the left side were collected at necropsy, fixed in 10% formalin for 24 hours, and transferred to 70% ethanol. The mammary glands were embedded in paraffin (Electron Microscopy Sciences, Hatfield, PA) and then sectioned at 4 μm thickness. Tissue sections mounted on slides were stained with hematoxylin & eosin (H&E) for histopathological analysis. For immunohistochemistry, the sections were immunostained with antibodies to PPARγ (1:200 diluted, Santa Cruz Biotechnology, Santa Cruz, CA), ERα (1:50 diluted, ThermoScientific, Lafayette, CO), proliferating cell nuclear antigen (PCNA) (1:1000 diluted, BD Pharmingen, San Diego, CA), cleaved-caspase 3 (c-Casp3) (1:200 diluted, Cell Signaling, Beverly MA), Nrf2 (1:2000 diluted, Epitomics, Burlingame, CA) or cyclooxygenase-2 (COX-2) (1:500 diluted, Cayman Chemicals, Ann Arbor, MI). The slides were incubated with 3-diaminobenzamine substrate (Vector Labs, Burlingame, CA) and counterstained with Harris hematoxylin (Sigma, St. Louis, MO). Histopathological images were taken randomly with Zeiss AxioCam HRc camera fitted to a Zeiss Axioskop 2 Plus microscope (Carl Zeiss Microimaging, LLC, Thornwood, NY) at 200× total magnification. Immunohistochemical images were taken randomly with Nikon Eclipse E800 (Melville, N.Y) fitted to Nikon digital sight Ri1 at 600× total magnification. Quantification was performed by randomly selecting three animals per group and counting three sections from each animal for over 4000 cells per animal. Positive staining for PCNA, ERα, and PPARγ is indicated by brown staining in the nucleus. Positive staining for Nrf2 is found both in the cytoplasm and nuclei of the cells. Positive staining for COX-2 is found in the cytoplasm. The staining density for nuclear or cytoplasmic positive cells was determined by using Aperio® Scan Scope (Vista, CA). Positive c-Casp3 staining is indicated by light brown to dark brown precipitate in the cytoplasm or perinuclei area of the cells. Quantification of c-Casp3 was determined by NIS-Elements software (Melville, N.Y) where over 3000 cells were counted per animal (three animals/treatment group).
Western blot analysis
Liver tissue was homogenized; protein was extracted and electrophoresed as previously described [14]. The primary antibodies against Nrf2, kelch-like-ECH-associated protein 1 (Keap1), NAD(P)H dehydrogenase, quinone 1 (NQO1), (Santa Cruz Biotechnology, Santa Cruz, CA), UDP-glucuronosyltransferase (UGT) (Cell Signaling, Beverly MA), heme oxygenase (HO-1) (Epitomics, Burlingame, CA), β-actin (Sigma, St. Louis, MO), and secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) were used. Quantification of Western blots was analyzed by Image J program (NIH, USA).
Liver microsome analysis
Liver microsomes were made as described previously [23]. The final pellet was resuspended in 0.25 M sucrose by homogenization in a glass homogenizer with a Teflon pestle. Microsomal proteins, 10–20 μg per lane, were resolved on SDS-gels Criterion XT -10% BisTris and transferred to nitrocellulose BA83 pore 0.2 μm from Schleicher & Schuell Biosciences, Inc. (Keene, NH). Blots were probed with selected anti-cytochrome P450 monoclonal antibodies produced in mouse or polyclonal antibodies produced in rabbit as described previously [23]. Recognition of certain cytochrome P450 enzymes with these antibodies was revealed by probing with the appropriate secondary antibody coupled to alkaline phosphatase and visualized using nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate.
mRNA expression analysis using quantitative PCR
Mammary glands from the 4th position on the right side and liver tissue were harvested at necropsy, placed in 1 ml of trizol and stored in −80°C until RNA extraction. RNA extraction, reverse transcription, and quantitative PCR were carried out as previously reported [24]. Labeled primers for glyceraldehydes-3-phosphate dehydrogenase, ERα, ERβ, PPARγ, Nrf2, Keap1, Cyp 1A1, Cyp 1B1, UGT1A1, NQO1, glutathione s-transferase (GST) mu 1, LOC5011 (rat orthologue of GST A1), catechol-O-methyltransferase (COMT), glutamate cysteine ligase, modifier subunit (GClm), superoxide dismutase (SOD1), glutathione peroxidase (GPX1), HO-1, thioredoxin (TXN1), and catalase were obtained from Applied Biosystems (Carlsbad, CA).
Statistical analysis
Data was analyzed using Graph Pad Prism 4.0 (GraphPad Software Inc.). Statistical significance was evaluated by using one-way analysis of variance (ANOVA) followed by Dunnett’s post-test. The data presented represents the mean ± S.E.M. P values <0.05 were considered significant.
RESULTS
Estrogen increases body and liver weight
As shown in Table 1, there was no difference in the body weight among treatment groups at the 2-week time point. However, at the 10-week time point, the average body weight of the control pellet group was significantly lower than the E2 control group; no changes of body weight were shown with γ-TmT treatment. When administered E2, it is well documented in ACI and other rodent models that the liver weight increases [25]. Estrogen can induce proliferation and regulate MAPK activities, which result in cell survival and regeneration [25]. Thus, increased liver weight can be attributed to estrogen-induced growth. As seen in our study, E2 treated rats showed an increase in liver weight for both 2- and 10-week time points. At the 10-week time point, the liver weight of the 0.5% γ-TmT fed group was significantly less than the E2 control group.
Table 1.
Body and liver weights of female ACI rats at 2 and 10 weeks
| Treatment Group | Treatment Period (weeks) | No. of rats per group | Body Weight (g) | Liver Weight (g) | Liver/Body Wt Ratio |
|---|---|---|---|---|---|
| Control Pellet | 2 | 6 | 155.6 ± 3.5 | 5.3 ± 0.3† | 3.4 ± 0.1† |
| E2 Control | 2 | 7 | 151.8 ± 3.6 | 7.0 ± 0.2 | 4.6 ± 0.1 |
| E2 + 0.3% γ-TmT | 2 | 6 | 155.0 ± 2.5 | 7.0 ± 0.2 | 4.5 ± 0.2 |
| E2 + 0.5% γ-TmT | 2 | 6 | 149.4 ± 5.3 | 6.7 ± 0.3 | 4.5 ± 0.1 |
| Control Pellet | 10 | 6 | 177.7 ± 2.4† | 5.5 ± 0.1‡ | 3.1 ± 0.1‡ |
| E2 Control | 10 | 7 | 190.7 ± 1.8 | 9.0 ± 0.3 | 4.7 ± 0.1 |
| E2 + 0.3% γ-TmT | 10 | 6 | 191.0 ± 3.0 | 8.6 ± 0.3 | 4.5 ± 0.1 |
| E2 + 0.5% γ-TmT | 10 | 6 | 184.9 ± 3.8 | 8.2 ± 0.2* | 4.4 ± 0.1* |
Rats were treated with a control pellet, E2 pellet, E2 pellet and 0.3% γ-TmT diet, or E2 pellet and 0.5% γ-TmT diet for 2 or 10 weeks. Values presented are mean ± S.E.M. Statistical significance,
p<0.05,
p<0.01,
p<0.001.
p-Values are compared to the E2 control of their respective time point.
Administration of γ-TmT increases levels of γ- and δ-tocopherol in the serum and mammary glands
We examined the levels of α-, γ-, δ-tocopherol and their respective metabolites, carboxydimethyldecyl hydroxychromans (CDMDHCs), carboxymethylbutyl hydroxychromans (CMBHCs), and carboxyethyl hydroxychromans (CEHCs) in the serum and mammary gland (Table 2). The longer chain metabolites, α-, γ-, δ-CDMDHCs and α-, γ-, δ-CMBHCs, showed negligible levels in the serum and mammary gland (data not shown). When administered 0.3% and 0.5% γ-TmT diet for either 2 or 10 weeks of treatment, α-CEHC was increased, while the level of α-tocopherol remained relatively constant in both the serum and mammary gland. After the treatment with 0.3% and 0.5% γ-TmT for 2 weeks, serum levels of γ and δ-tocopherols and their short chain metabolite, CEHC, were significantly increased (Table 2): γ-tocopherol (6.4- and 7.4-fold, respectively), δ-tocopherol (5.0- and 6.0-fold, respectively),γ-CEHC (59- and 85-fold, respectively), and δ-CEHC (39- and 54-fold, respectively). After 10 weeks of 0.3% and 0.5% γ-TmT treatment, serum levels of tocopherols and CEHCs were higher than those of 2 weeks: γ-tocopherol (6.4- and 14-fold, respectively), δ-tocopherol (7.0- and 18-fold, respectively), γ-CEHC (91- and 108-fold, respectively), and δ-CEHC (40- and 54-fold, respectively). The levels of tocopherols in the mammary gland showed a similar trend of changes to the serum data. After 2 weeks of 0.3% and 0.5% γ-TmT treatment, levels of tocopherols and CEHCs in the mammary gland were increased: γ-tocopherol (4.2- and 6.4-fold, respectively), δ-tocopherol (24- and 37-fold, respectively), γ-CEHC (37- and 49-fold, respectively) and δ-CEHC (20- and 26-fold, respectively). After 10 weeks of 0.3% and 0.5% γ-TmT treatment, levels of tocopherols and CEHCs in the mammary gland increased: γ-tocopherol (8.0- and 11-fold, respectively) and δ-tocopherol (14- and 21-fold, respectively) and γ-CEHC (28- and 32-fold, respectively) and δ-CEHC (45- and 60-fold, respectively).
Table 2.
Analysis of tocopherol levels in the serum and mammary gland of ACI rats
| Serum
| ||||||
|---|---|---|---|---|---|---|
| α-T (μM) | γ-T (μM) | δ-T (μM) | α-CEHC (μM) | γ-CEHC (μM) | δ-CEHC (μM) | |
| 2 weeks | ||||||
| E2 Control | 33.8 ± 1.9 | 0.5 ± 0.1 | 0.1 ± 0.0 | 7.5 ± 1.0 | 0.3 ± 0.1 | 1.0 ± 0.2 |
| E2 + 0.3% γ-TmT | 44.6 ± 4.8* | 3.2 ± 0.2‡ | 0.5 ± 0.1‡ | 26.8 ± 2.6‡ | 17.8 ± 2.1‡ | 39.8 ± 6.2‡ |
| E2 + 0.5% γ-TmT | 34.2 ± 2.1 | 3.7 ± 0.5‡ | 0.6 ± 0.1‡ | 22.0 ± 1.1‡ | 25.6 ± 2.1‡ | 54.4 ± 4.2‡ |
| 10 weeks | ||||||
| E2 Control | 33.5 ± 1.5 | 0.5 ± 0.1 | 0.1 ± 0.0 | 4.7 ± 0.0 | 0.2 ± 0.0 | 0.6 ± 0.0 |
| E2 + 0.3% γ-TmT | 36.2 ± 0.6 | 3.2 ± 0.1‡ | 0.7 ± 0.1‡ | 14.0 ± 0.0‡ | 18.3 ± 0.0‡ | 32.3 ± 0.0‡ |
| E2 + 0.5% γ-TmT | 39.1 ± 1.7 | 7.0 ± 0.6‡ | 1.8 ± 0.2‡ | 16.1 ± 1.2‡ | 21.6 ± 2.8‡ | 44.1 ± 2.1‡ |
|
| ||||||
|
Mammary Gland
| ||||||
| α-T (μM) | γ-T (μM) | δ-T (μM) | α-CEHC (μM) | γ-CEHC (μM) | δ-CEHC (μM) | |
|
| ||||||
| 2 weeks | ||||||
| E2 Control | 25.5 ± 0.8 | 2.1 ± 1.2 | 0.1 ± 0.0 | 0.9 ± 0.1 | 0.0 ± 0.0 | 0.3 ± 0.1 |
| E2 + 0.3% γ-TmT | 37.1 ± 6.4 | 8.9 ± 2.4† | 2.4 ± 0.6† | 4.0 ± 0.9‡ | 3.7 ± 0.9‡ | 6.1 ± 1.6‡ |
| E2 + 0.5% γ-TmT | 47.0 ± 12.5 | 13.4 ± 2.8‡ | 3.7 ± 0.9‡ | 3.5 ± 0.3‡ | 4.9 ± 0.4‡ | 7.8 ± 0.5‡ |
| 10 weeks | ||||||
| E2 Control | 26.9 ± 0.8 | 0.8 ± 0.1 | 0.2 ± 0.1 | 0.4 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| E2 + 0.3% γ-TmT | 25.4 ± 0.8 | 6.4 ± 0.5‡ | 2.8 ± 0.2‡ | 1.4 ± 0.1‡ | 2.8 ± 0.2‡ | 4.5 ± 0.2‡ |
| E2 + 0.5% γ-TmT | 25.7 ± 1.6 | 9.1 ± 0.7‡ | 4.3 ± 0.3‡ | 1.8 ± 0.1‡ | 3.2 ± 0.2‡ | 6.0 ± 0.2‡ |
Serum and mammary gland samples were collected at necropsy and analyzed for α-, γ-, δ-tocopherol (α-, γ-, δ-T) and their short chain metabolites, carboxyethyl hydroxychromans (α-,γ-, δ-CEHC). β-Tocopherol is not shown because β-tocopherol and its metabolites were below the limit for detection. The data is expressed as the mean ± S.E.M (n = 6). Statistical significance,
p<0.05,
p<0.01,
p<0.001.
p-Values are compared to the E2 control of their respective time points.
Serum levels of E2 are decreased by the administration of γ-TmT in rats
Circulating E2 levels were determined at 2- and 10-week time points in rats treated with the control pellet or with E2 pellets and their respective diets (Fig. 1B). We chose 2 weeks as a time point because previous experiments showed a peak of circulating E2 levels in the serum in E2 treated ACI rats (data not shown). In addition, Turan et al. showed that after 10 weeks of low dose E2 treatment, circulating levels of E2 remained constant [20]. In the present study, 2 weeks after implanting the pellets the average serum E2 levels of the control pellet group were 21.1 ± 2.1 pg/ml, whereas average serum E2 levels in the E2 control group were increased to 60.4 ± 2.8 pg/ml (p<0.01). When compared to the E2 control group, treatment with 0.5% γ-TmT for 2 weeks significantly decreased E2 serum levels to 45.3 ± 4.3 pg/ml (p<0.05). After 10 weeks of pellet implantation, the serum E2 level in the control pellet group (28.0 ± 3.6 pg/ml), E2 control group (25.8 ± 5.1 pg/ml), and 0.3% γ-TmT fed group (34.4 ± 5.1 pg/ml) were similar, suggesting that the serum E2 level spike shown at 2 weeks in the E2 control group reduced to the basal level by 10 weeks. Furthermore, treatment with 0.5% γ-TmT for 10 weeks showed a significant decrease in E2 levels to 11.6 ± 2.3 pg/ml (p<0.05).
Hyperplasia is evident in the mammary gland in the E2 treated groups
H&E staining was performed on mammary gland sections (Fig. 1C). The control pellet group showed normal mammary glands at both the 2- and 10-week time points. Molecular markers for the control pellet group were analyzed (Supplemental Figure 1). Based on histological evaluation, mild lobular hyperplasia was evident in the mammary gland in the E2 control group. Treatment with 0.3% and 0.5% γ-TmT at both 2- and 10-week time points showed no apparent effect on E2-induced mammary hyperplasia. Further evaluation was carried out to determine if γ-TmT had an effect on cell proliferation in mammary hyperplasia. Immunohistochemical evaluation of the hyperplastic mammary gland, as described below, revealed that cell proliferation was significantly decreased, while a significant increase of apoptotic cells was observed when administered γ-TmT.
Treatment with γ-TmT reduces proliferating cell nuclear antigen (PCNA) but increases cleaved-caspase 3 (c-Casp3) in the mammary gland
PCNA expression in the mammary gland was reduced after 2 and 10 weeks of treatment with γ-TmT (Fig. 2A). After 10 weeks of treatment, 0.3% or 0.5% γ-TmT-fed groups resulted in a 54% or 56% decrease in proliferation, respectively (p<0.05). As shown in Fig. 2B, after 2 weeks of treatment with 0.3% or 0.5% γ-TmT, c-Casp3 expression in the mammary gland showed 108% (p<0.01) or 54% (p<0.05) increase above the E2 control group, respectively. After 10 weeks of treatment, 0.3% or 0.5% γ-TmT diet-fed group showed an increase in c-Casp3 expression by 47% (p<0.05) and 66% (p<0.01), respectively, indicating an induction of apoptosis in the mammary gland by treatment with γ-TmT.
Figure 2.

ACI rats treated with estrogen pellets were fed the control, 0.3%, or 0.5% γ-TmT diet. A representative immunostaining of (A) PCNA and (B) c-Casp3 in the mammary gland is shown (600×). Three mammary glands from each group were randomly selected, and three representative areas from each mammary gland were analyzed for PCNA and c-Casp3 expression. Positive staining of PCNA is found in the nuclei of the cells. Positive staining of c-Casp3 is shown as a light brown to dark brown precipitate in the cytoplasm or perinuclei of the cells. The arrows indicate positive staining for c-Casp3. The data are presented as the mean ± S.E.M. Statistical significance, *p<0.05, †p<0.01.
γ-TmT treatment decreases estrogen receptor α (ERα) but increases peroxisome proliferator activated receptor γ (PPARγ) protein levels
As shown in Fig. 3A, after treatment of 0.3% and 0.5% γ-TmT for 2 weeks, there were 10% and 29% decrease in ERα positive cells in the mammary gland, respectively. After 10 weeks with treatment of 0.3% and 0.5% γ-TmT, there were 49% (p<0.05) and 50% (p<0.05) decrease in ERα expression, respectively. After 2 weeks of treatment, there was a 66% (p<0.05) increase above the E2 control group in expression of PPARγ in the 0.5% γ-TmT treated group (Fig. 3B). When the rats were treated with 0.5% γ-TmT for 10 weeks, the expression of PPARγ was increased by 67% (p<0.01) in the mammary gland.
Figure 3.

ACI rats treated with estrogen pellets were fed the control, 0.3%, or 0.5% γ-TmT diet. A representative immunostaining of (A) ERα and (B) PPARγ in the mammary gland is shown (600×). Three mammary glands from each group were randomly selected, and three representative areas from each mammary gland were analyzed for ERα and PPARγ expression. Positive staining of ERα and PPARγ are found in the nuclei of the cells. The data are presented as the mean ± S.E.M. Statistical significance, *p<0.05, †p<0.01.
γ-TmT treatment suppresses the expression of ERα mRNA while inducing the expression of ERβ and PPARγ mRNA in mammary glands
The mRNA levels of ERα, ERβ, and PPARγ were significantly changed by γ-TmT in mammary glands (Fig. 4). When given γ-TmT, the mRNA level of ERα was decreased: the 0.5% γ-TmT treated group for the 2- and 10-week time points were reduced by a fold change of 3.6 ± 0.1 (p<0.05) and 2.5 ± 0.1, respectively (Fig. 4A). The mRNA expression for ERβ showed fold increase of 2.3 ± 0.5 (p<0.05) for the 0.3% γ-TmT treated group at 10 weeks (Fig. 4B). After treatment with 0.3% and 0.5% γ-TmT for 2 weeks, PPARγ mRNA level was increased by a fold change of 3.9 ± 0.6 (p<0.05) and 2.5 ± 0.3, respectively (Fig. 4C). After γ-TmT treatment for 10 weeks, there was further fold increase of 5.0 ± 1.1 (p<0.05) and 3.2 ± 0.6 when fed 0.3% and 0.5% γ-TmT, respectively.
Figure 4.
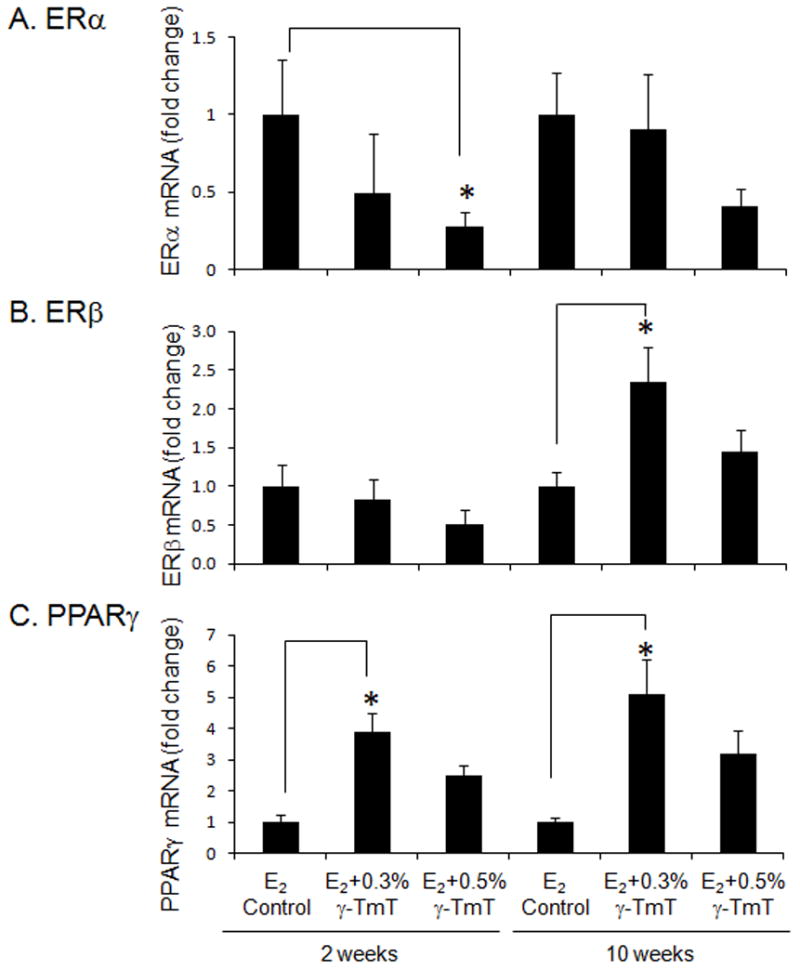
ACI rats treated with estrogen pellets were fed the control, 0.3%, or 0.5% γ-TmT diet. At least four mammary glands from each treatment group were analyzed for mRNA levels of (A) ERα, (B) ERβ, and (C) PPARγ by quantitative PCR (n = 4–6). The data are presented as the mean ± S.E.M. Statistical significance, *p<0.05.
Treatment with γ-TmT increases the expression of Nrf2 in mammary glands and upregulates protein levels of Nrf2, NQO1, UGT, and HO-1 in the liver
As shown in Fig. 5A, after 2 weeks of treatment with 0.3% or 0.5% γ-TmT, Nrf2 levels in the mammary gland showed 12% (p<0.05) or 20% (p<0.01) increase above the E2 control group, respectively. After 10 weeks of treatment, 0.3% or 0.5% γ-TmT diet-fed group showed an increase in Nrf2 levels by 11% (p<0.05) and 17% (p<0.05), respectively. In the liver, the protein level of Nrf2 is increased when administered 0.3% and 0.5% γ-TmT in the diet for 2 weeks, while Keap1 levels remain constant (Fig. 5B). At 10 weeks of γ-TmT treatment, NQO1, UGT, and HO-1 protein levels were increased in the liver.
Figure 5.
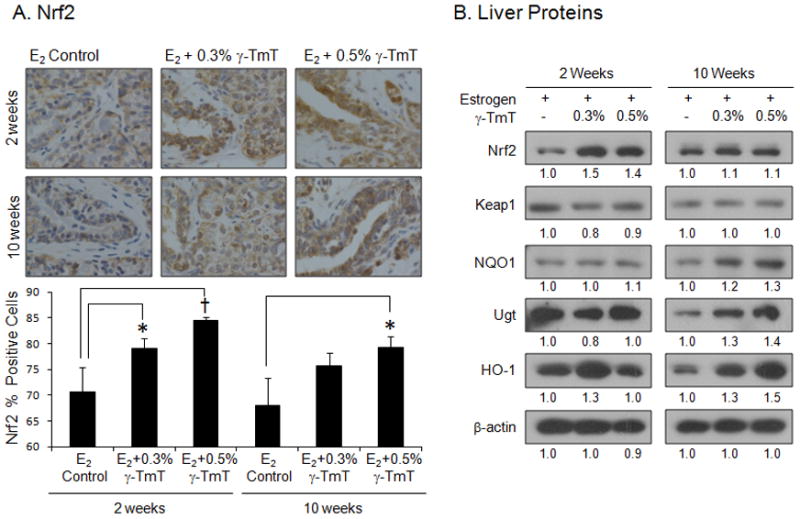
ACI rats treated with estrogen pellets were fed the control, 0.3%, or 0.5% γ-TmT diet. (A) A representative immunostaining of Nrf2 in the mammary gland is shown (600×). Three mammary glands from each group were randomly selected, and three representative areas from each mammary gland were analyzed for levels of Nrf2. Positive staining for Nrf2 is found both in the cytoplasm and nuclei of the cells. The data are presented as the mean ± S.E.M. Statistical significance, *p<0.05, †p<0.01. (B) Regulation of liver protein expression by the treatment with γ-TmT. Liver samples were homogenized and pooled together (n=3). Western blots are shown. Quantification of Western blot was performed by Image J 1.45s (NIH), and the numbers are provided at the bottom of each Western blot.
To determine whether the levels of hepatic cytochrome P450 isozymes were altered by implanting estrogen pellets or concurrent feeding of γ-TmT, we also examined microsomal preparations by SDS-PAGE and Western blots using antibodies specific for 7 individual P450 isozymes. At the 2-week time point, the levels of P450 2B1/2 and 2C7 per mg protein decreased 10–15% in the group treated with 0.5% γ-TmT compared to E2 control group but this change was not statistically significant (data not shown). The levels of P450 2E1, 2A1 and 2C12, a female specific isozyme, were unchanged by estrogen pellet implants or dietary γ-TmT. At the 10-week time point, the levels of P450 2B1/2, 2A1, 2C12 and 2C7 were not altered by the treatments; however, levels of P450 2E1 decreased 10–20% by 0.3% and 0.5% γ-TmT treatment, but these changes were not significant (data not shown). Neither cytochrome P450 3A1 nor 3A2 were detected at either the 2- or 10-week time points regardless of treatment. This was not unexpected since P450 3A2 is male specific and P450 3A1 levels are very low and not easily detected unless the rats have been exposed previously to a P450 3A inducer such as dexamethasone, other steroids, or barbiturates like phenobarbital [23]. However, this suggests that the E2 released from the implanted pellets nor the γ-TmT in the diet at 0.3% or 0.5% were sufficient to elevate P450 3A1 levels which is the rat homolog of human P450 3A4.
Administration of γ-TmT induces the expression of phase II detoxifying enzymes mRNA in the mammary gland and the liver
Nrf2 activation leads to induction of enzymes involved in anti-oxidative functions and phase II detoxification, such as GST, NQO1, HO-1, and UGT to reduce cellular stress, which may contribute to prevention of carcinogenesis. Quantitative RT-PCR was performed on mammary gland (Supplemental Data Table 1) and liver tissues (Supplemental Data Table 2) for Nrf2 and related markers. In the mammary gland after 10 weeks of treatment, the mRNA level of Cyp 1A1 was significantly reduced when fed 0.3% γ-TmT. In the mammary gland, UGT1A1, GSTm1, and LOC5O11 were induced by γ-TmT treatment. Antioxidant enzymes did not show a significant change when given γ-TmT diet (Supplemental Data Table 1). In the liver, phase II enzymes, UGT1A1, NQO1, GSTm1, and GClm, were increased with γ-TmT treatment. Among the antioxidant enzymes, only HO-1 was significantly increased in the liver when administered γ-TmT diet (Supplemental Data Table 2).
Inflammatory markers are reduced by the treatment with γ-TmT
Immunohistochemical staining for cyclooxygenase-2 (COX-2) was performed on mammary hyperplastic tissues (Fig. 6A). After treatment of 0.3% and 0.5% γ-TmT for 2 weeks, there were 39% (p<0.01) and 45% (p<0.01) decrease in COX-2 positive cells in the mammary gland, respectively. After 10 weeks with treatment of 0.3% and 0.5% γ-TmT, there were 24% (p<0.05) and 30% (p<0.05) decrease in levels of COX-2, respectively. PGE2 is a downstream product of the enzyme COX-2. Serum levels of PGE2 were decreased by γ-TmT treatment, with which significant reduction of serum PGE2 was shown by 10 week treatment with 0.5% γ-TmT by 71% (p<0.05) (Fig. 6B). The serum level of another inflammatory marker, 8-isoprostane, was also examined. At 10 weeks, the E2 control group had serum 8-isoprostane levels of 637 ± 107 pg/ml, and treatment with 0.5% γ-TmT decreased by 64% to 230 ± 31 pg/ml (p<0.05) (Fig. 6B).
Figure 6.
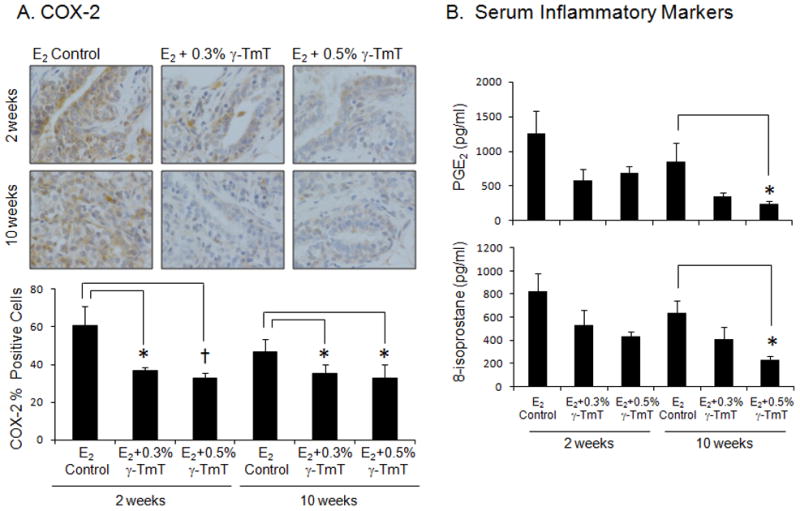
ACI rats treated with estrogen pellets were fed the control, 0.3%, or 0.5% γ-TmT diet. (A) A representative immunostaining of COX-2 in the mammary gland is shown (600×). Three mammary glands from each group were randomly selected, and three representative areas from each mammary gland were analyzed for levels of COX-2. Positive staining for COX-2 is found in the cytoplasm. (B) Serum was collected at necropsy and analyzed for PGE2 and 8-isoprostane (n=6). The data are presented as the mean ± S.E.M. Statistical significance, *p<0.05, †p<0.01.
DISCUSSION
The ACI rat strain is known to be sensitive to estrogen treatment and serves as an appropriate model to study estrogen-induced mammary carcinogenesis [18]. Unlike synthetic carcinogen models such as NMU, ACI rats exposed to elevated levels of estrogen provide a more natural representation of human breast carcinomas. In the present study, mammary gland hyperplasia was evident as early as 2 weeks after the E2 pellets were implanted into the animals. Thus, we utilized the ACI model to have a better representation of early biomarkers in mammary gland hyperplasia for ER-positive breast cancers. The ACI rat mammary model has a known limitation. Long-term exposure to high doses of E2 (27 mg) induces pituitary tumors which can affect mammary tumor prevention and treatment. Our study utilizes a lower dose of estrogen to induce mammary hyperplasia. The lower dose of estrogen could reduce the enlargement of the pituitary gland and consequential pituitary gland morbidity of the animal. Recently, it has been demonstrated that an ACI.COP-Ept2 rat model, still sensitive to E2 to induce mammary tumors, shows reduced pituitary morbidities [26]. This ACI.COP-Ept2 strain may be utilized for future long-term mammary cancer studies.
Previous studies employing primarily α-tocopherol for chemoprevention have provided inconsistent results [3,5]. In a recent study, treatment with δ-tocopherol was found to be more effective than γ-TmT and γ-tocopherol in reducing tumor growth in lung xenograft tumors, while α-tocopherol did not inhibit tumor growth [27]. The serum and tissue levels of α-tocopherol and metabolites did not change from the control diet and the γ-TmT treatment groups [27]. In addition, γ-TmT diet inhibited NMU-induced mammary tumors in Sprague-Dawley rats where α tocopherol serum levels were similar to the control diet, but γ- and δ-tocopherol levels increased in a dose-response manner [14]. In the present study, increased levels of γ- and δ-tocopherol and their short chain metabolites were evident in both the serum and mammary gland of the γ-TmT treatment groups, while α-tocopherol levels remained relatively constant in both the serum and mammary gland of all treatment groups. The α-tocopherol-transfer protein preferentially transfers α-tocopherol from the liver to the blood, while γ- and δ-tocopherol remain in the liver and are metabolized [28]. Cytochrome P450 4F2 catalyzes the initial step in the vitamin E-ω-hydroxylase pathway followed by β-oxidation, which removes 2 carbons from the side chain in each cycle ending in the short chain metabolite, CEHC [28]. When administered γ-TmT diet, γ-tocopherol, γ-CEHC, δ-tocopherol, and δ-CEHC showed a significant increase in the serum and mammary gland. We believe the cancer preventive activity of γ-TmT treatment may be mostly due to the increased levels of γ- and δ-tocopherol. The activity of individual metabolites remains to be determined.
Estrogens have been implicated in breast cancer. One theory suggests that oxidative stress from estrogen metabolism plays a key role in mammary cancer development [29]. ER is a nuclear receptor that stimulates cell growth and proliferation [29]. Vitamin E has been shown to inhibit ER-positive cell proliferation and work as antagonists of estrogen signaling [30]. Previously, we reported that γ- and δ-tocopherols, but not α-tocopherol, inhibited proliferation of ER-positive human breast cancer cells [14]. In this study, we found that γ-TmT decreased the expression of ERα in hyperplastic mammary tissues. In addition, γ-TmT in the diet reduced circulating levels of E2 in the serum, suggesting that γ-TmT could modify the response to estrogen. Similar to the finding in the NMU-induced breast cancer model in Sprague-Dawley rats [14], PPARγ was increased at both the protein and mRNA level in the mammary gland of ACI rats when treated with γ-TmT while ERα expression was decreased. Since PPARγ transactivation can be suppressed by ERα binding to the PPAR response element [31], the inhibition of ERα expression by tocopherols may result in the activation of PPARγ.
PPARγ is expressed in breast, prostate, and colon epithelium, and involved in lipid and glucose metabolism, cell proliferation and apoptosis, differentiation, and cell survival [32]. When the nuclear receptor PPARγ is ligand-activated, it forms heterodimers with the retinoid X receptor [32]. The chromanol ring of tocopherol is structurally similar to the one in troglitazone [10], a known PPARγ ligand. Because of this similarity, tocopherols may function as a PPARγ ligand. However, γ-tocopherol does not directly bind to PPARγ [33]. Recently, γ-tocopherol was shown to enhance the formation of 15-S-hydroxyeicosatetraenoic acid, an endogenous PPARγ ligand [33]. PPARγ plays a role in fatty acid uptake and transport and acts to control inflammation that can arise from increased adipocyte differentiation and proliferation [34]. These actions are altered in malignancy, and cancer cells derive their energy increasingly from anaerobic glycolysis [35]. PPARγ signaling in cancer has been shown to upregulate differentiation markers, CDK inhibitors (p21 and p27), and apoptosis markers while PPARγ has been shown to inhibit inflammatory markers (COX-2, cytokines, and inducible nitric oxide synthase), PI3K/Akt activity, and angiogenesis [34]. Specifically in breast cancer, stimulation of PPARγ increases the degradation of cell cycle genes (cyclin D1), interferes with estrogen receptor signaling, and NF-κB signaling cascades [33,36]. Thus, activation of PPARγ by tocopherols in breast tissue may have anti-estrogenic effects, inhibit cell cycle progression, and induce apoptosis to prevent breast cancer.
Nrf2 is a transcription factor which regulates cellular antioxidant and detoxification enzymes. Under basal conditions in the cytoplasm, Nrf2 is bound to Keap1. Keap1 inhibits Nrf2 signaling by promoting Nrf2 degradation through the proteasomal pathway [37]. Oxidative stress (reactive oxygen species/RNS) or chemopreventive agents can react with Keap1, and Keap1 undergoes covalent modification which allows the release of Nrf2 [37]. Nrf2 translocates into the nucleus, dimerizes with small Maf proteins, and binds to the antioxidant-responsive element (ARE) to stimulate gene expression of antioxidant enzymes (TXN, SOD, HO-1, catalase, and GPx) and phase II detoxification enzymes (UGTs, sulfotransferases, GSTs, and NQO1) [37,38]. Thus, Nrf2 plays a critical role in protecting cells from neoplastic transformation since detoxifying and antioxidant enzymes are stimulated to maintain oxidative stress homeostasis [38]. A loss of Nrf2 may lead to a decrease in cellular defense against oxidative stress which may result in tumorigenesis. Recently, it was shown that Nrf2 expression was suppressed in prostrate tumors of transgenic adenocarcinoma mouse prostrate (TRAMP) [39]. γ-TmT treatment upregulated the expression of Nrf2 and detoxifying enzymes and inhibited tumor development in TRAMP mice [38]. Yao et al. reported that inhibition of estrogen signaling activates the Nrf2 pathway in breast cancer [40]. In our study with estrogen-treated ACI rats, the protein expression level of Nrf2 was increased in the mammary gland and liver. Protein levels of phase II enzymes were increased in the liver by γ-TmT treatment. The mRNA expression in the mammary gland and liver showed that phase II detoxifying enzymes were also induced by γ-TmT treatment, suggesting that γ-TmT increases the transcription of Nrf2-ARE-target genes and exhibits protective defense against estrogen-induced oxidative stress.
In the present study, serum levels of PGE2 and 8-isoprostane as well as COX-2 levels in the mammary gland were reduced when treated with γ-TmT. In a lung tumor model using A/J mice, Lu et al. reported a decreased level of PGE2 and leukotriene B4 (LTB4) in the serum when given γ-TmT diet [41]. In a colon cancer model with azoxymethane/dextran sulfate sodium treated mice, treatment with γ-TmT reduced the levels of PGE2, LTB4, and 8-isoprostane in the serum [21]. Consistent with anti-inflammatory effects of tocopherols over several cancer models, γ-TmT treatment may reduce inflammation in an estrogen-induced model of mammary hyperplasia and tumorigenesis. In conclusion, we have shown that γ-TmT inhibits the cell proliferation of mammary hyperplasia, suppresses the expression of inflammatory markers, and upregulates PPARγ and Nrf2, while down-regulating ERα, suggesting that γ-TmT may be beneficial for human breast cancer prevention.
Supplementary Material
Acknowledgments
Financial support
This work was supported in part by NIH R03 CA141756 [to N.S.]; the Trustees Research Fellowship Program at Rutgers, The State University of New Jersey [to N.S.]; and from the NIEHS Center Grant P30 ES005022 (Helmut Zarbl, P.I.).
We thank the Laboratory of Animal Service at the Department of Chemical Biology for taking care of the animals, Drs. You-Rong Lou and Guang Xun Li for their suggestions on immunohistochemical evaluation, and Dr. Allan Conney for helpful advice on our manuscript.
Abbreviations
- γ-TmT
γ-enriched mixed tocopherol
- c-Casp3
cleaved-caspase 3
- CDMDHC
carboxydimethyldecyl hydroxychroman
- CEHC
carboxyethyl hydroxychroman
- CMBHC
carboxymethylbutyl hydroxychroman
- COMT
catechol-O-methyltransferase
- COX-2
cyclooxygenase-2
- E2
estradiol
- ER
estrogen receptor
- GClm
glutamate cysteine ligase, modifier subunit
- GPX
glutathione peroxidase
- GST
glutathione s-transferase
- HO-1
heme oxygenase-1
- Keap1
kelch-like-ECH-associated protein 1
- NMU
N-methyl-N-nitrosourea
- NQO1
NAD(P)H dehydrogenase, quinone 1
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PCNA
proliferating cell nuclear antigen
- PGE2
prostaglandin E2
- PPARγ
peroxisome proliferator activated receptor γ
- RNS
reactive nitrogen species
- SOD
superoxide dismutase
- TXN
thioredoxin
- UGT
UDP-glucuronosyltransferase
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61(4):212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Taylor PR, Qiao YL, Abnet CC, et al. Prospective study of serum vitamin E levels and esophageal and gastric cancers. J Natl Cancer Inst. 2003;95(18):1414–1416. doi: 10.1093/jnci/djg044. [DOI] [PubMed] [Google Scholar]
- 3.Constantinou C, Papas A, Constantinou AI. Vitamin E and cancer: An insight into the anticancer activities of vitamin E isomers and analogs. Int J Cancer. 2008;123(4):739–752. doi: 10.1002/ijc.23689. [DOI] [PubMed] [Google Scholar]
- 4.Jiang Q, Christen S, Shigenaga MK, Ames BN. gamma-tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr. 2001;74(6):714–722. doi: 10.1093/ajcn/74.6.714. [DOI] [PubMed] [Google Scholar]
- 5.Brigelius-Flohe R, Kelly FJ, Salonen JT, Neuzil J, Zingg JM, Azzi A. The European perspective on vitamin E: current knowledge and future research. Am J Clin Nutr. 2002;76(4):703–716. doi: 10.1093/ajcn/76.4.703. [DOI] [PubMed] [Google Scholar]
- 6.Lee IM, Cook NR, Gaziano JM, et al. Vitamin E in the primary prevention of cardiovascular disease and cancer: the Women’s Health Study: a randomized controlled trial. JAMA. 2005;294(1):56–65. doi: 10.1001/jama.294.1.56. [DOI] [PubMed] [Google Scholar]
- 7.Bairati I, Meyer F, Gelinas M, et al. A randomized trial of antioxidant vitamins to prevent second primary cancers in head and neck cancer patients. J Natl Cancer Inst. 2005;97(7):481–488. doi: 10.1093/jnci/dji095. [DOI] [PubMed] [Google Scholar]
- 8.Lippman SM, Klein EA, Goodman PJ, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2009;301(1):39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang HY, Appel LJ. Supplementation of diets with alpha-tocopherol reduces serum concentrations of gamma- and delta-tocopherol in humans. J Nutr. 2003;133(10):3137–3140. doi: 10.1093/jn/133.10.3137. [DOI] [PubMed] [Google Scholar]
- 10.Campbell SE, Stone WL, Whaley SG, Qui M, Krishnan K. Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer. 2003;3:25. doi: 10.1186/1471-2407-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Q, Ames BN. Gamma-tocopherol, but not alpha-tocopherol, decreases proinflammatory eicosanoids and inflammation damage in rats. FASEB J. 2003;17(8):816–822. doi: 10.1096/fj.02-0877com. [DOI] [PubMed] [Google Scholar]
- 12.Jiang Q, Elson-Schwab I, Courtemanche C, Ames BN. gamma-tocopherol and its major metabolite, in contrast to alpha-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc Natl Acad Sci U S A. 2000;97(21):11494–11499. doi: 10.1073/pnas.200357097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christen S, Woodall AA, Shigenaga MK, Southwell-Keely PT, Duncan MW, Ames BN. gamma-tocopherol traps mutagenic electrophiles such as NO(X) and complements alpha-tocopherol: physiological implications. Proc Natl Acad Sci U S A. 1997;94(7):3217–3222. doi: 10.1073/pnas.94.7.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HJ, Ju J, Paul S, et al. Mixed tocopherols prevent mammary tumorigenesis by inhibiting estrogen action and activating PPAR-gamma. Clin Cancer Res. 2009;15(12):4242–4249. doi: 10.1158/1078-0432.CCR-08-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan MM, Lu X, Merchant FM, Iglehart JD, Miron PL. Gene expression profiling of NMU-induced rat mammary tumors: cross species comparison with human breast cancer. Carcinogenesis. 2005;26(8):1343–1353. doi: 10.1093/carcin/bgi100. [DOI] [PubMed] [Google Scholar]
- 16.Blows FM, Driver KE, Schmidt MK, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010;7(5):e1000279. doi: 10.1371/journal.pmed.1000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvell DM, Strecker TE, Tochacek M, et al. Rat strain-specific actions of 17beta-estradiol in the mammary gland: correlation between estrogen-induced lobuloalveolar hyperplasia and susceptibility to estrogen-induced mammary cancers. Proc Natl Acad Sci U S A. 2000;97(6):2779–2784. doi: 10.1073/pnas.050569097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shull JD, Spady TJ, Snyder MC, Johansson SL, Pennington KL. Ovary-intact, but not ovariectomized female ACI rats treated with 17beta-estradiol rapidly develop mammary carcinoma. Carcinogenesis. 1997;18(8):1595–1601. doi: 10.1093/carcin/18.8.1595. [DOI] [PubMed] [Google Scholar]
- 19.Li SA, Weroha SJ, Tawfik O, Li JJ. Prevention of solely estrogen-induced mammary tumors in female aci rats by tamoxifen: evidence for estrogen receptor mediation. J Endocrinol. 2002;175(2):297–305. doi: 10.1677/joe.0.1750297. [DOI] [PubMed] [Google Scholar]
- 20.Turan VK, Sanchez RI, Li JJ, et al. The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17beta-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16alpha-hydroxyestradiol, and 4-hydroxyestrone. J Endocrinol. 2004;183(1):91–99. doi: 10.1677/joe.1.05802. [DOI] [PubMed] [Google Scholar]
- 21.Ju J, Hao X, Lee MJ, et al. A gamma-tocopherol-rich mixture of tocopherols inhibits colon inflammation and carcinogenesis in azoxymethane and dextran sulfate sodium-treated mice. Cancer Prev Res (Phila Pa) 2009;2(2):143–152. doi: 10.1158/1940-6207.CAPR-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Y, Lee MJ, Cheung C, et al. Analysis of multiple metabolites of tocopherols and tocotrienols in mice and humans. J Agric Food Chem. 2010;58(8):4844–4852. doi: 10.1021/jf904464u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jan YH, Mishin V, Busch CM, Thomas PE. Generation of specific antibodies and their use to characterize sex differences in four rat P450 3A enzymes following vehicle and pregnenolone 16alpha-carbonitrile treatment. Arch Biochem Biophys. 2006;446(2):101–110. doi: 10.1016/j.abb.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 24.Lee HJ, Liu H, Goodman C, et al. Gene expression profiling changes induced by a novel Gemini Vitamin D derivative during the progression of breast cancer. Biochem Pharmacol. 2006;72(3):332–343. doi: 10.1016/j.bcp.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 25.Ravoori S, Vadhanam MV, Sahoo S, Srinivasan C, Gupta RC. Mammary tumor induction in ACI rats exposed to low levels of 17beta-estradiol. Int J Oncol. 2007;31(1):113–120. [PubMed] [Google Scholar]
- 26.Kurz SG, Hansen KK, McLaughlin MT, et al. Tissue-specific actions of the Ept1, Ept2, Ept6, and Ept9 genetic determinants of responsiveness to estrogens in the female rat. Endocrinology. 2008;149(8):3850–3859. doi: 10.1210/en.2008-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li GX, Lee MJ, Liu AB, et al. delta-tocopherol is more active than alpha - or gamma -tocopherol in inhibiting lung tumorigenesis in vivo. Cancer Prev Res (Phila) 2011;4(3):404–413. doi: 10.1158/1940-6207.CAPR-10-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sontag TJ, Parker RS. Cytochrome P450 omega-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J Biol Chem. 2002;277(28):25290–25296. doi: 10.1074/jbc.M201466200. [DOI] [PubMed] [Google Scholar]
- 29.Mense SM, Remotti F, Bhan A, et al. Estrogen-induced breast cancer: alterations in breast morphology and oxidative stress as a function of estrogen exposure. Toxicol Appl Pharmacol. 2008;232(1):78–85. doi: 10.1016/j.taap.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chamras H, Barsky SH, Ardashian A, Navasartian D, Heber D, Glaspy JA. Novel interactions of vitamin E and estrogen in breast cancer. Nutr Cancer. 2005;52(1):43–48. doi: 10.1207/s15327914nc5201_6. [DOI] [PubMed] [Google Scholar]
- 31.Bonofiglio D, Gabriele S, Aquila S, et al. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin Cancer Res. 2005;11(17):6139–6147. doi: 10.1158/1078-0432.CCR-04-2453. [DOI] [PubMed] [Google Scholar]
- 32.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4(1):61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 33.Campbell SE, Musich PR, Whaley SG, et al. Gamma tocopherol upregulates the expression of 15-S-HETE and induces growth arrest through a PPAR gamma-dependent mechanism in PC-3 human prostate cancer cells. Nutr Cancer. 2009;61(5):649–662. doi: 10.1080/01635580902825654. [DOI] [PubMed] [Google Scholar]
- 34.Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor gamma in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 2009;8(7):6–15. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- 35.Schulz TJ, Thierbach R, Voigt A, et al. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J Biol Chem. 2006;281(2):977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- 36.Jarrar MH, Baranova A. PPARgamma activation by thiazolidinediones (TZDs) may modulate breast carcinoma outcome: the importance of interplay with TGFbeta signalling. J Cell Mol Med. 2007;11(1):71–87. doi: 10.1111/j.1582-4934.2007.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frohlich DA, McCabe MT, Arnold RS, Day ML. The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene. 2008;27(31):4353–4362. doi: 10.1038/onc.2008.79. [DOI] [PubMed] [Google Scholar]
- 38.Barve A, Khor TO, Nair S, et al. Gamma-tocopherol-enriched mixed tocopherol diet inhibits prostate carcinogenesis in TRAMP mice. Int J Cancer. 2009;124(7):1693–1699. doi: 10.1002/ijc.24106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu S, Khor TO, Cheung KL, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS One. 2010;5(1):e8579. doi: 10.1371/journal.pone.0008579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao Y, Brodie AM, Davidson NE, Kensler TW, Zhou Q. Inhibition of estrogen signaling activates the NRF2 pathway in breast cancer. Breast Cancer Res Treat. 2010;124(2):585–591. doi: 10.1007/s10549-010-1023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu G, Xiao H, Li GX, et al. A gamma-tocopherol-rich mixture of tocopherols inhibits chemically induced lung tumorigenesis in A/J mice and xenograft tumor growth. Carcinogenesis. 2010;31(4):687–694. doi: 10.1093/carcin/bgp332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.