

Abstract
The live yellow fever vaccine (YF-17D) offers a unique opportunity to study memory CD8+ T cell differentiation in humans following an acute viral infection. We have performed a comprehensive analysis of the virus-specific CD8+ T cell response using overlapping peptides spanning the entire viral genome. Our results showed that the YF-17D vaccine induces a broad CD8+ T cell response targeting several epitopes within each viral protein. We identified a dominant HLA-A2-restricted epitope in the NS4B protein and used tetramers specific for this epitope to track the CD8+ T cell response over a 2 year period. This longitudinal analysis showed the following. 1) Memory CD8+ T cells appear to pass through an effector phase and then gradually down-regulate expression of activation markers and effector molecules. 2) This effector phase was characterized by down-regulation of CD127, Bcl-2, CCR7, and CD45RA and was followed by a substantial contraction resulting in a pool of memory T cells that re-expressed CD127, Bcl-2, and CD45RA. 3) These memory cells were polyfunctional in terms of degranulation and production of the cytokines IFN-γ, TNF-α, IL-2, and MIP-1β. 4) The YF-17D-specific memory CD8+ T cells had a phenotype (CCR7−CD45RA+) that is typically associated with terminally differentiated cells with limited proliferative capacity (TEMRA). However, these cells exhibited robust proliferative potential showing that expression of CD45RA may not always associate with terminal differentiation and, in fact, may be an indicator of highly functional memory CD8+ T cells generated after acute viral infections.
Immune memory forms the basis of vaccine-induced protection, and memory CD8+ T cells form an important cellular component of this immunity (1). Our understanding of CD8+ T cell memory has advanced a great deal through exhaustive studies in acute viral infections that cause protective, long-lasting memory (2-5). We know from these studies that Ag-driven clonal expansion results in a dynamic antiviral CD8+ T cell response initially manifested as pathogen clearance via cytotoxic molecules and effector cytokines and later by the presence of a small population of memory cells that can be rapidly recruited to blunt subsequent infections. We have also begun to understand the differences between the properties of poor and high quality memory CD8+ T cells based on their cytotoxicity, cytokine production, homeostatic turnover, and proliferative potential (6-17).
The yellow fever vaccine strain 17D (YF-17D),3 a live attenuated form of the wild-type virus, has not only been extremely effective in controlling yellow fever but is also one of the safest vaccines in young adults (18, 19). A single immunization confers protection, and virus-neutralizing Abs can be detected for up to 30 years post-vaccination (20). The live viral nature of the vaccine combined with its efficacy is useful for studying how humans generate functional immunity in the context of an acute viral infection and also as a potential expression vector for recombinant vaccines (21, 22). Some of the studies that address the complex interactions between the virus and the immune system suggest that the ability of YF-17D to infect dendritic cells and activate multiple TLRs may be crucial for generating the robust adaptive immune response seen after vaccination (23-25). The immune memory developed as a consequence comprises the ability to rapidly produce neutralizing Abs as well as the CD8+ T cell effectors (26-28) that are likely required for killing cells infected with the virus that escape the humoral response. This cellular memory has been of interest to our group because it presents a rare opportunity to study primary CD8+ T cell responses in humans. CD8+ T cells that recognize HLA-B35-restricted epitopes in E, NS1, NS2B, and NS3 proteins have been reported in vaccinees (26). In addition, we demonstrated recently that the magnitude of the total effector CD8+ T cells response against YF-17D (or the smallpox live viral vaccine) could be measured using the transient Ki67+Bcl-2lowHLA-DR+CD38+ phenotype of effector T cells (28, 29). However, there are gaps in our knowledge regarding the breadth of the response, the differentiation of virus-specific effector and memory CD8+ T cells, and the functional qualities of these cells.
To address the above questions, we used a peptide library encompassing the entire YF-17D genome for estimating the magnitude of the T cells targeting each viral protein and the range of epitopes recognized. Using a strategy involving direct ex vivo stimulation of cells with overlapping peptides and detection of cytokine production (30), we identified an immunodominant class I-restricted epitope that permitted longitudinal tracking of YF-17D-specific CD8+ T cells in individual vaccinees using MHC class I tetramers. This analysis offers novel perspectives for two reasons. First, our study has the advantage that the subjects were primary vaccinees from a geographical area that is not endemic for yellow fever, i.e., the United States (19). Thus, an accurate determination of the breadth and magnitude of the primary YF-17D-specific response could be made while avoiding the possible cross-reactivity introduced by prior exposure to either the yellow fever virus or other closely related flaviviruses. Second, we performed a longitudinal tetramer-based analysis and followed YF-17D-specific CD8+ T cells from the time they first appeared in circulation until 2 years later, in the same group of vaccinees. Hence, we were able to study various stages of memory CD8+ T cell differentiation longitudinally without the confounding effects introduced by a cross-sectional analysis. This analysis will be valuable in understanding the basic tenets of human memory T cell differentiation after acute viral infections.
Materials and Methods
Study subjects and blood samples
Healthy volunteers (18–40 years of age) were recruited in the study after informed consent. Approval for all procedures was obtained from the Emory University Institutional Review Board (Atlanta, GA). A single dose (0.5 ml containing at least 105 PFU) of 17D live-attenuated yellow fever vaccine strain was administered subcutaneously. The recommendations established by the Advisory Committee on Immunization Practices (Department of Health and Services, Centers for Disease Control and Prevention, Atlanta, GA) were followed for selection and vaccination of individuals in the study. In addition, individuals with a previous history of vaccination with YF-17D or exposure to flaviviruses as evidenced by serology or a history of travel to endemic areas were excluded from the study. Blood samples were analyzed before and at various times postvaccination as indicated in the text. Seroconversion after vaccination was confirmed by assaying the neutralizing Ab titers for YF-17D (data not shown). PBMC were purified from cell preparation tubes (BD Biosciences), and EDTA blood samples were used to quantify viral RNA by real-time PCR as described elsewhere (28).
YF-17D peptide library
To map T cell epitopes, a library of overlapping peptides spanning the entire YF-17D polyprotein was made. All 851 peptides comprising the library were 15-aa long (Synpep) and nonamidylated, with neighboring peptides overlapping by 11 aa. The peptides were organized into 60 pools based on a matrix such that each pool had 24–30 peptides, each at a concentration of 10 μg/ml, and any two pools had no more than one peptide in common (30, 31).
Cell preparation, stimulation, and intracellular cytokine (ICC) staining
PBMC were purified from cell preparation tubes (BD Biosciences) according to standard protocols and cryopreserved in 90% FCS plus 10% DMSO. For mapping T cell epitopes, freshly isolated PBMC were stimulated with each of 60 peptide pools. Assays for confirming epitopes in the single peptides identified using the pools were done using cryopreserved PBMC that were revived, rested overnight at 37°C, and stimulated with 10 μg/ml peptides. Fresh or revived PBMC were stimulated for 6 h in 96-well round-bottom plates in the presence of brefeldin A (1 μl/ml) and anti-CD28/CD49d (10 μl/ml) and then stained for relevant T cell markers and cytokines. Culture medium for all cellular assays was RPMI 1640 containing 10% FCS, 2 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin (RPMI 1640 plus 10% FCS).
Staining and flow cytometry analysis
All mAbs except anti-granzyme B (Caltag), programmed death (PD)-1 (provided by Dr. G. J. Freeman, Dana Farber Cancer Institute, Boston, MA), and CCR7 (R&D Systems) were obtained from BD Biosciences. A2-NS4B 214–222 tetramers were made in-house. For ICC after in vitro stimulation, cells were stained for T cell markers by incubation with the relevant Abs at room temperature for 30 min, washed with PBS, and then stained for cytokines using anti-IFN-γ, TNF-α, IL-2 and Mip-1β Abs after cell permeabilization with the Cytofix/Cytoperm kit (BD Biosciences). Degranulation in stimulated cells was measured by including anti-CD107a-PE (10 μl/well) in the initial culture medium. For phenotypic analysis of A2-NS4B+ CD8+ T cells, 100–200 μl of unprocessed whole blood was incubated at room temperature first with tetramer for 10 min and then for a further 30 min with Abs for surface markers. This was followed by a 10-min lysis of RBC using FACS lysing solution (BD Biosciences), washing with PBS, and fixing in 1% p-formaldehyde. For staining intracellular proteins like Bcl-2, Ki-67, granzyme B, and perforin, cells were permeabilized and stained using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. Data were acquired on a FACSCalibur (BD Biosciences) or LSR-II (BD Biosciences) flow cytometer and analyzed using Flow Jo (Tree Star) software. In addition, the programs SPICE (version 4.1.5) and PESTLE (version 1.5.2) (both from M. Roederer, National Institutes of Health, Vaccine Research Center, Bethesda, MD) were used to quantify CD8+ T cells positive for the various combinations of functions assayed. Statistical analysis and graphical representation of data was performed using GraphPad Prism software.
CFSE labeling and in vitro expansion of CD8+ T cells
PBMC from vaccinated subjects were labeled for 5 min with 1 μM CFSE (Molecular Probes) in PBS at room temperature; cold FCS was then added and cells were washed extensively with RPMI 1640 plus 10% FCS. CFSE-labeled cells were incubated with or without the NS4B 214–222 peptide (10 μg/ml) for 6 days, at the end of which flow cytometry and analysis were performed as described above. Responding CD8+ T cells were identified either by tetramer staining or by ICC staining after a 6-h recall with peptide.
Results
YF-17D elicits a broad diversity of memory CD8+ T cells
To define the breadth of the primary antiviral CD8+ T cell response, we created a library of overlapping peptides encompassing the entire viral polyprotein. This library was organized into 60 pools of multiple peptides based on a 24 × 36 matrix such that each peptide was present in precisely two pools (30). Responses to all of these pools were studied in nine healthy subjects vaccinated 2 mo previously with a single s.c. injection of YF-17D. PBMC isolated from vaccinees were stimulated with individual pools followed by ICC staining assays, and responding virus-specific CD8+ T cells were identified by their ability to produce IFN-γ. Of 60 peptide pools tested with this assay, between eight and 25 pools were capable of eliciting IFN-γ production in different vaccinees. Many pools contained peptides from adjacent sequences in the viral polyprotein and thus had epitopes primarily from one protein. IFN-γ production stimulated by these pools was used to estimate the contribution of individual viral proteins toward eliciting a CD8+ T cell response (Fig. 1). Overall, each vaccinee had CD8+ T cells targeting multiple proteins and several epitopes within each protein, resulting in broad cellular immunity. Despite variation in their magnitude, CD8+ T cells specific for each of the 10 YF-17D proteins were detected, indicating that every viral protein was immunogenic. IFN-γ+ CD8+ T cells specific for E, NS3, and NS5 were elicited in all vaccinees with an average of 10.7% (SD 8.0), 16.7% (SD 7.0), and 27.7% (SD 14.0) of the total responding CD8+ T cells specific for E, NS3, and NS5 respectively. NS1 also elicited frequent (in 8/9 vaccinees) but lower numbers (mean 7.2%; SD 3.7) of CD8+ T cells, whereas the magnitude of C-, M-, NS2A-, and NS2B-reactive CD8+ T cells was low (<3%) when detected. Strikingly, in five vaccinees the NS4B component was dominant and accounted for ~42.1% (SD 13.5) of the total IFN-γ+ CD8+ T cells (Fig. 1).
FIGURE 1.
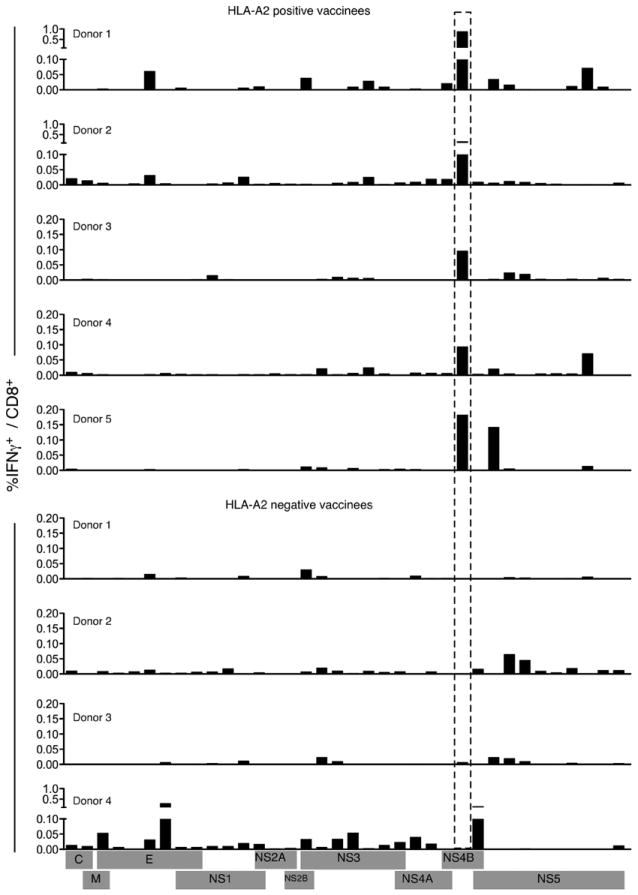
YF-17D elicits a broad diversity of memory CD8+ T cells. PBMC from YF-17D-vaccinated individuals were stimulated with peptide pools derived from YF-17D proteins and IFN-γ production was assayed by ICC. The percentage of IFN-γ+ CD8+ T cells for individual vaccinees is shown.
YF-17D harbors a dominant HLA-A2-restricted epitope in the NS4B protein
Although vaccinees had CD8+ T cells recognizing multiple epitopes, IFN-γ production in some pools containing NS4B-derived peptides consistently dominated the CD8+ T cell response in five vaccinees. These pools had either the peptide NS4B 209–223 or NS4B 213–227 in common. Both peptides were individually capable of stimulating CD8+ T cells from the same five vaccinees, confirming that these peptides contained MHC class I-restricted epitopes (Fig. 2, A and B). Notably, all the above vaccinees were positive for the HLA-A2 serotype (data not shown), suggesting that the response was HLA-A2 restricted. An epitope prediction algorithm (32) was used to identify putative HLA-A0201-restricted epitopes in the 15-mer NS4B 209–223 and NS4B 213–227 peptides. Of the several nonamer epitopes predicted, three epitopes with the highest scores (and hence the best binding motifs), NS4B 213–221, NS4B 214–222, and NS4B 215–223 were tested by ICC staining for their ability to stimulate IFN-γ production. Only one peptide, NS4B 214–222 (amino acid sequence LLWNGPMAV) generated IFN-γ-producing CD8+ T cells in HLA-A2+ vaccinees, identifying it as the HLA-A2-restricted CD8+ T cell epitope (Fig. 2, A and B). In addition, control peptides that had a single amino acid substitution (either L214 to V214 or L215 to E215) did not stimulate IFN-γ production, thus validating the specificity of the NS4B 214–222 epitope. MHC-peptide tetramers containing HLA-A0201 and the NS4B 214–222 peptide were prepared and tested in individuals. Tetramer-stained CD8+ cells were seen in vaccinated HLA-A2+ but not HLA-A2− individuals, confirming the specificity of the epitope (Fig. 2C). This tool enabled us to perform a longitudinal analysis of YF-17D-specific CD8+ T cells.
FIGURE 2.
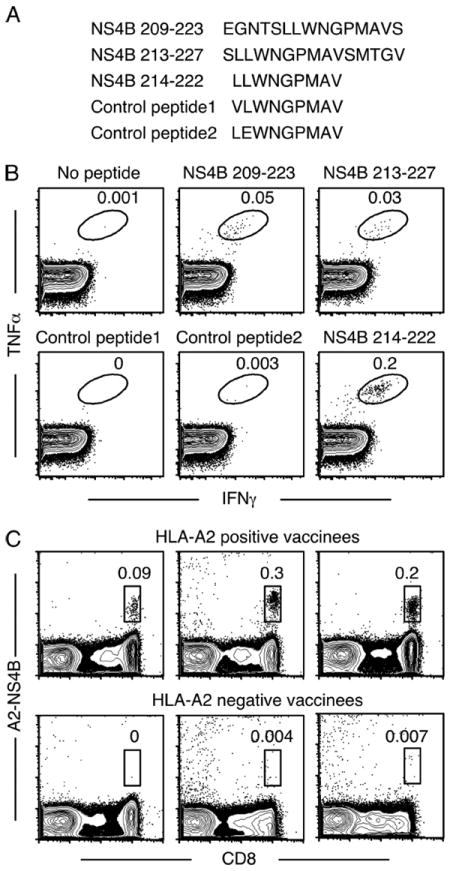
YF-17D harbors a dominant HLA-A2 restricted epitope in the NS4B protein. A, Sequences of the peptides used to confirm and identify the immunogenic nonamer epitope in NS4B. B, PBMC from HLA-A2+ vaccinees were cultured in the presence or absence of 15-mer peptides (NS4B 209–223 or NS4B213–227) and ICC staining was performed (upper panels). To confirm the nonamer epitope in these peptides, PBMC were stimulated with either the NS4B 214–222 peptide or with control peptides that differed by one amino acid (lower panels). Plots gated on CD8+ T cells for one representative donor are shown. C, Identification of YF-17D-specific CD8+ T cells using MHC class I tetramers. Peripheral blood from HLA-A2+ or HLA-A2− individuals vaccinated with YF-17D 2 wk previously was stained with the A2-NS4B tetramer. Plots are gated on all lymphocytes and numbers indicate the percentage of tetramer+ cells from total lymphocytes.
Kinetics of the YF-17D-specific primary CD8+ T cell response tracked by MHC class I tetramers
We analyzed the dynamics of viral replication after the vaccination of HLA-A2+ individuals who did not have a history of exposure to flaviviruses (Fig. 3). Most vaccinees did not have detectable viral RNA in the first 2 days following vaccination; on day 3, several vaccinees tested positive for viral RNA (mean 114 copies/ml; SD 50), and the number of positives increased further such that all vaccinees had YF-17D genomes in the plasma by day 5. The highest level of viral RNA in circulation was seen on day 5 for most vaccinees (mean 3598 copies/ml; SD 1813) and on day 7 for others (mean 1443 copies/ml; SD 613), after which it declined quickly. By day 11 all except two vaccinees had no viral RNA in the plasma, and by day 14 viral genomes were absent.
FIGURE 3.
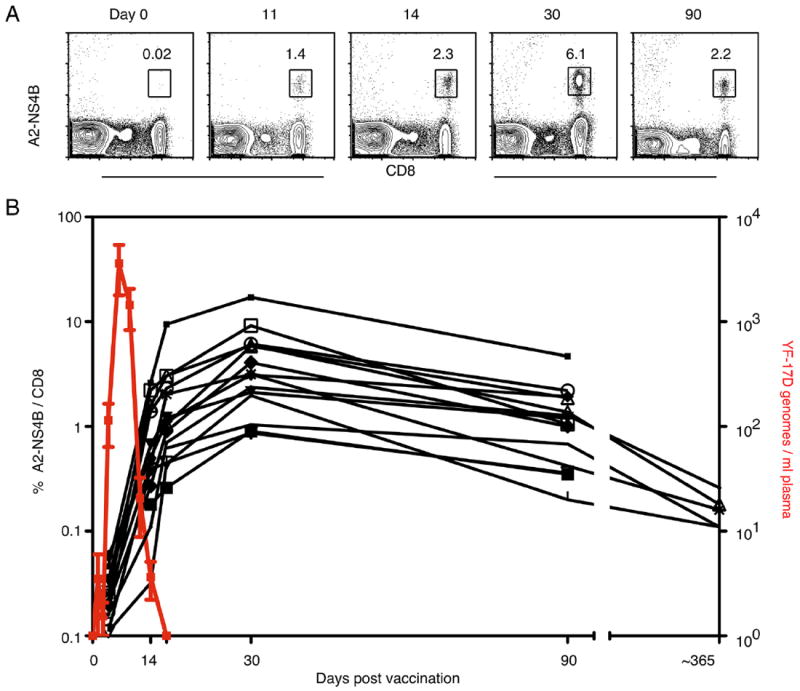
Kinetics of the YF-17D-specific primary CD8+ T cell response tracked by MHC class I tetramers. A, Tetramer kinetics in one representative vaccinee is shown. Plots show all lymphocytes but the numbers indicate the percentage of A2-NS4B+ cells from total CD8+ T cells. B, YF-17D genomes in the plasma (red line; mean ± SD) and the percentage of A2-NS4B+ (black lines; each for one vaccinee) in YF-17D vaccinees over 1 year is shown.
Ag dictates the size of the effector CD8+ T cell response and the kinetics of initial expansion (33). We used A2-NS4B 214–222 tetramers to quantify the size and examine the kinetics of the YF-17D-driven primary CD8+ T cell response in vaccinees. Strikingly, A2-NS4B tetramer+ CD8+ T cells were seen in nearly all HLA-A2+ vaccinees (19 of 21). The A2-NS4B+ CD8+ cells were often detectable as early as 11 days after vaccination and definitely after 14 days in all responding individuals, coincident with the decline in plasma viral RNA. Tetramer+ cells continued to expand till 30 days postvaccination, when the peak tetramer frequency ranging from 0.5 to 17% of CD8+ T cells was seen in the vaccinees; it diminished by ~3.6-fold by day 90. The size of the A2-NS4B+ pool at this time was roughly proportional to that seen on day 30; vaccinees with high peak frequencies retained high numbers of memory cells. We were able to track these cells for an additional time in five vaccinees from the same cohort and found that tetramer+ cells were readily detectable even 1 year after vaccination (Fig. 3B). In conclusion, HLA-A2+ vaccinees have a robust and long-lived response to the NS4B 214–222 epitope, showing expansion, contraction, and memory kinetics typical of CD8+ T cells elicited in a primary acute viral infection.
Effector and memory differentiation of YF-17D-specific CD8+ T cells
To understand the differentiation of effector and memory CD8+ T cells and to gain an insight into the generation of YF-17D-specific CD8+ memory, we performed a detailed longitudinal analysis of tetramer-stained CD8+ T cells in 15 HLA-2+ vaccinees (Fig. 4). Because rapid clonal expansion is the hallmark of a virus-driven CD8+ T response, we assessed proliferation of YF-17D-specific CD8+ T cells using Ki-67, a marker that is tightly associated with cycling CD8+ T cells (34). Compared with naive CD8+ T cells that were Ki-67 negative, >95% of the A2-NS4B+ cells detected early on (days 11 to 14) were Ki-67 positive. The Ki-67 staining declined significantly (present in <5% of the cells) by day 30 and decreased to baseline by day 90. The proliferation seen early on was coupled with decreased expression of the antiapoptotic protein Bcl-2, suggesting that a majority of these cells were destined to undergo apoptosis. Progressive reexpression of Bcl-2 occurred concomitantly with loss of Ki-67 and differentiation to a memory phenotype. Expression of the activation markers HLA-DR and CD38 mirrored the kinetics seen with Ki-67. Activated but not naive CD8+ T cells also expressed the chemokine receptor CCR5. Although we observed that the CCR5 expression kinetics was similar to those of the proliferation and activation markers, unlike the other markers CCR5 was retained in 40–50% of A2-NS4B+ memory cells. Potent effector properties were evidenced by high levels of granzyme B (Fig. 4), granzyme A, and perforin (data not shown) expression until as late as 30 days after YF-17D vaccination, and tetramer+ cells continued to express granzyme B later, although at lower levels.
FIGURE 4.
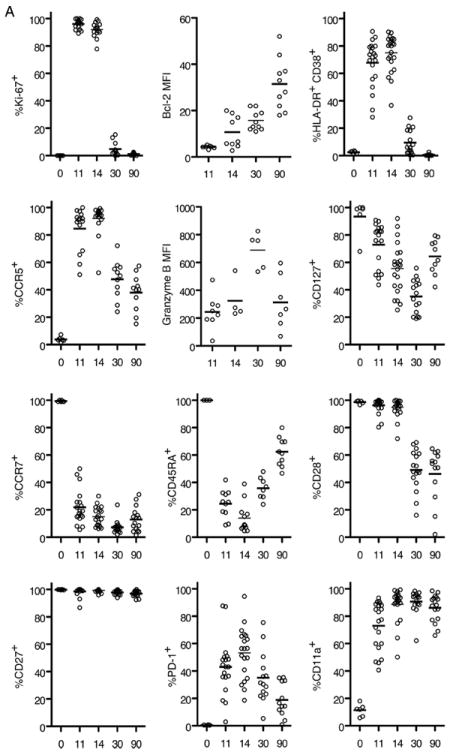
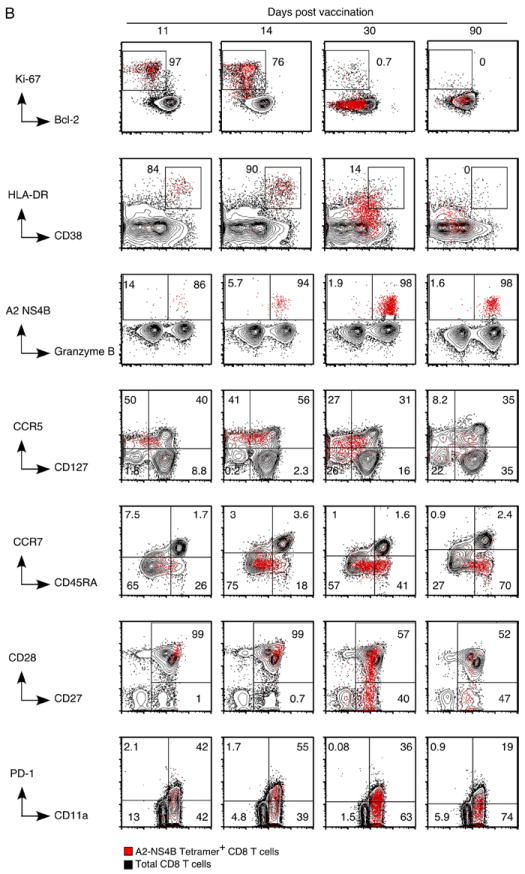
Effector and memory differentiation of YF-17D-specific CD8+ T cells. A, Results from the longitudinal phenotypic analysis of A2-NS4B+ cells present in blood of 10 to 15 vaccinees are summarized. The day 11, 14, 30, and 90 data are from the same group of vaccinees. The day 0 data are from a separate group and represent expression of the relevant marker on naive (CD45RA+CCR7+) CD8+ T cells. The percentage of A2-NS4B+ CD8+ T cells expressing the relevant marker for each individual (open circles) and the group mean (horizontal line) are shown. MFI, Mean fluorescence intensity. B, Flow plots representing the phenotype of YF-17D-specific CD8+ T cells. Plots are gated on total CD8+ T cells (black background) or YF-17D-specific (A2-NS4B+) CD8+ T cells (red dots). Numbers show the percentage of A2-NS4B+ CD8+ T cells in the gate.
We analyzed the expression of markers associated with other relevant functions of CD8+ T cells (Fig. 4). CD127 (IL-7Rα) has been shown earlier to be an important marker that is down-regulated on effectors and selectively reexpressed on a subset destined to form precursors of the memory pool (10, 35). We observed substantial but not complete down-regulation of CD127 on A2-NS4B+ CD8+ T cells over the 30 days following vaccination, and a gradual reexpression thereafter. Coexpression of CCR7, a homing marker, and CD45RA, a transmembrane tyrosine phosphatase, is characteristic of naive cells (36). Both markers were down-regulated in the YF-17D-specific CD8+ T cells during effector differentiation. However in contrast to the lack of CCR7 on a majority of memory cells, CD45RA was reexpressed over time. We next examined the profiles of signaling molecules that regulate T cell activation. PD-1 is a regulatory receptor of the CD28 family that is transiently expressed on activated T cells during acute viral infections (37). Its up-regulation by CD8+ T cells (transient in acute and long-term in chronic viral infections) inhibits the activation, expansion, and acquisition of effector CD8+ T cell functions (38). A2-NS4B+ cells transiently up-regulated PD-1 between 11 and 14 days after YF-17D vaccination, similar to reports with other acute viral infections in mice (38). To assess the ability to respond to costimulatory signals at the different stages of differentiation, we evaluated the expression of two critical signaling proteins, CD27 and CD28. CD27 was uniformly expressed at high levels during all stages of the differentiating virus specific CD8+ T cells, whereas CD28 expression was uniform in effectors (day 11–14) and heterogeneous on memory cells. CD11a, a component of the adhesion molecule LFA-1 (39), was expressed by YF-17D-experienced but not naive CD8+ T cells as described in studies with other infections (40).
To summarize, the primary antiviral response was characterized by a pronounced activation and expansion phase that led to increasing frequencies of A2-NS4B+ cells by day 30. At this stage, YF-17D-specific cells were at the end of the expansion phase and showed a phenotype intermediate to that of effector and memory CD8+ T cells. Substantial contraction resulted in a pool of memory cells that had lost activation and proliferation markers, uniformly expressed CD45RA and CD27, and had heterogeneous expression with respect to proteins such as CCR5, CD28, and CD127.
The YF-17D-specific CD8+ T cell memory is fine tuned over time
Memory differentiation is a gradual and continuous process resulting in subtle changes in the memory pool that may not be obvious over a short period of time. We followed A2-NS4B+ memory cells at 3 mo and 2 years after vaccination in the same three individuals to determine any such gradual changes (Fig. 5). After 2 years, the frequency of A2-NS4B+ memory cells in the blood of these individuals had decreased from 1.3, 0.43, and 0.36% at 3 mo to 0.13, 0.12, and 0.06%, respectively (data not shown). Phenotypic analysis revealed that strikingly, >95% of the A2-NS4B+ cells continued to express CD45RA even 2 years later, verifying that reexpression of this molecule was not a transient phase of memory differentiation (Fig. 5A). In addition, uniform CD27 expression and heterogeneous CD28 expression were observed at both early (3 mo) and late (2 years) memory stages. However, we did observe a subtle increase over time with respect to CCR7 and CD127, two markers associated with high-quality immune memory (Fig. 5A). The expression of CD45RA has been associated with senescence in several studies (41), whereas CD127 confers the ability to respond to IL-7 mediated survival signals (4). To further dissect the issue of whether these memory cells were still capable of differentiation, we examined two markers, CD56 and CD57, associated with terminal effectors (Fig. 6). A2-NS4B+ CD8+ T cells did not express either CD56, which is thought to identify CD8+ T cells with direct cytolytic activity (42), or CD57, which is associated with replicative senescence (43), suggesting that YF-17D-specific memory cells were not terminally differentiated even though they expressed CD45RA and were mostly CCR7 negative, a phenotype often associated with terminal effectors.
FIGURE 5.
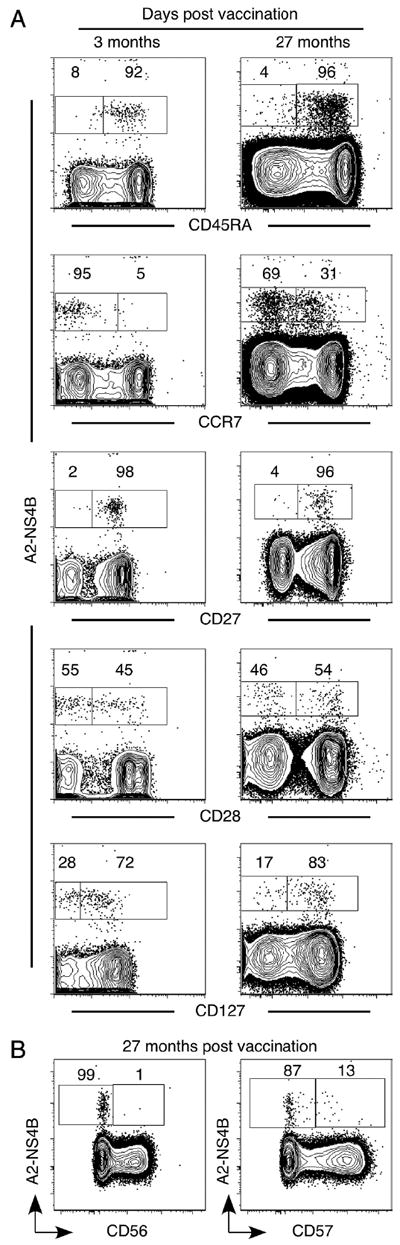
The YF-17D-specific CD8+ T cell memory is fine-tuned over 2 years. A, Comparison of the phenotype YF-17D-specific memory CD8+ T cells 3 mo and 2 years after vaccination. Representative flow plots (gated on total CD8+ T cells) from one of three vaccinees are shown. B, Representative flow plots (gated on total CD8+ T cells) show expression of the late differentiation markers CD56 and CD57 on YF-17D-specific memory CD8+ T cells in an individual vaccinated 2 years earlier.
FIGURE 6.
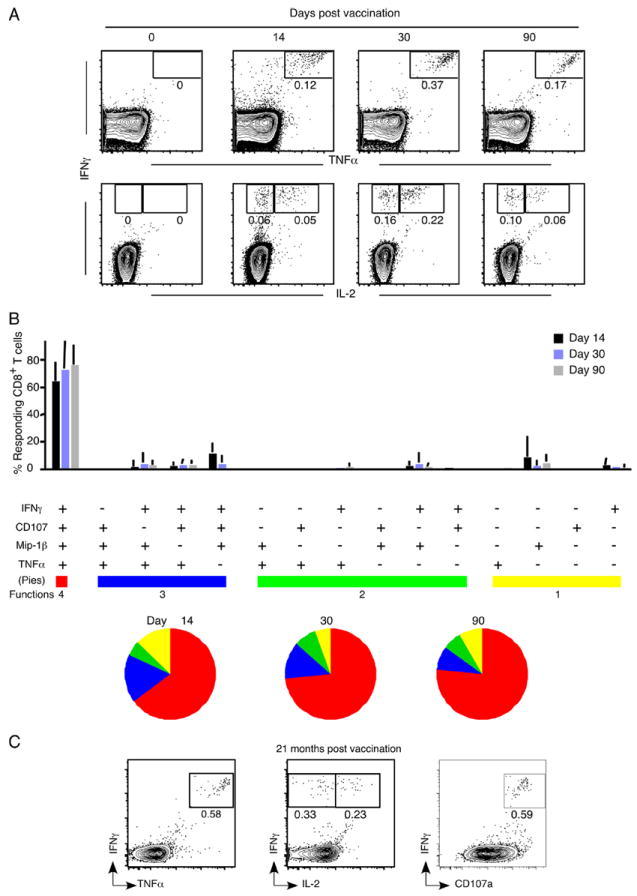
YF-17D elicits polyfunctional, long-lived memory CD8+ T cells. A, Kinetics of cytokine-producing CD8+ T cells in individuals immunized with YF-17D. PBMC were isolated from vaccinees and responses to the NS4B 214–222 peptide were measured by ICC staining. Representative flow plots (gated on total CD8+ T cells) from one vaccinee are shown. B, Functional profile of the YF-17D-specific CD8+ T cell response. All possible combinations of four functions (degranulation by CD107a staining and secretion of IFN-γ, TNF-α, and Mip-1β) are shown on the x-axis. Bars indicate the percentage of the total response contributed by CD8+ T cells with the combination of responses on the x-axis. Responses are grouped according to the number of functions and the data summarized by the pie charts. Each slice of the pie represents the fraction of the total response that consists of CD8+ T cells positive for a given number of functions. Mean data from four YF-17D-immunized individuals are shown. C, Functional profile of the YF-17D-specific memory CD8+ T cells 2 years after vaccination. Plots gated on CD8+ T cells show the cytokines produced in response to the stimulation of PBMC with the NS4B 214–222 peptide. Data from one representative vaccinee are shown.
YF-17D elicits polyfunctional, long-lived memory CD8+ T cells
CD8+ T cells have a spectrum of functions to achieve viral control. We evaluated five functions (degranulation and secretion of IFN-γ, TNF-α, Mip1β, and IL-2) of CD8+ T cells recognizing the A2-NS4B epitope by stimulation of PBMC in vitro with the NS4B 214–222 peptide followed by ICC staining and multicolor flow cytometry. The kinetics of IFN-γ-producing CD8+ T cells (Fig. 6A) matched that seen using tetramer-based analysis (Fig. 3), with highest frequencies seen 30 days post-vaccination. Upon assessing the composition of the cytokine-producing population, we found that at every time point analyzed the cells that simultaneously produced IFN-γ and TNF-α dominated the response, and a prominent fraction of these cells additionally produced IL-2 (Fig. 6A). Surface mobilization of CD107a (lysosome-associated membrane protein (LAMP)-1) after peptide stimulation indicates the ability to release cytolytic granules (44). Independent and simultaneous measurement of four functions showed that the majority of virus-specific CD8+ T cells from days 11 through 90 were CD107a+ as well as capable of IFN-γ, TNF-α, and Mip-1β production at the same time (Fig. 6B). Notably, this polyfunctional CD8+ T cell memory was retained even 2 years after vaccination (Fig. 6C). Successful pathogen clearance depends on both quantity and quality of CD8+ T cells, and our findings show that highly polyfunctional memory CD8+ T cells are elicited and maintained after YF-17D vaccination.
A cardinal property of memory CD8+ T cells is their ability to undergo rapid proliferation upon reencountering Ag (5). This is an important component of protective immunity; a higher proliferative potential implies a larger pool of secondary effectors. To examine the proliferative potential of A2-NS4B-specific CD8+ T cells, CFSE-labeled PBMC from vaccinees were stimulated in vitro with the relevant peptide and the dilution of CFSE was assayed as a measure of cell division (Fig. 7A). Expansion of A2-NS4B+ cells at day 90 was much higher (7.2-fold) compared with day 14 (2.3-fold). This proliferative potential was preserved even when >95% of tetramer+ cells were CD45RA+ (supplemental Fig. 1).4 Thus, YF-17D-specific CD8+ T cells progressively acquire proliferative potential over the course of time. The quality of memory CD8+ T cells generated in chronic viral infections is inferior to that in response to acute viral infections (9). Ag-driven proliferation in particular is reported to be lower (45, 46). We were able to make a direct comparison in this regard between YF-17D, an acute virus, and CMV, a chronic virus, in an individual vaccinated 1 year previously with YF-17D. This vaccinee had CD8+ T cells specific for A2-NS4B as well as for a CMV pp65-derived epitope (A2-NLV). A2-NS4B+ CD8+ T cells expanded to higher levels (8.6-fold) than A2-NLV-specific CD8+ T cells (1.45-fold) upon stimulation with the corresponding peptides, indicative of the superior proliferation capability in YF-17D memory CD8+ T cells (Fig. 7B). Strikingly, A2-NS4B+ cells that diluted CFSE could be detected in an individual immunized 10 years previously (Fig. 7C). This vaccinee had no history of recent travel to a yellow fever endemic area, strongly suggesting that long-term persistence of YF-17D-specific CD8+ T cell memory does not require antigenic stimulation.
FIGURE 7.
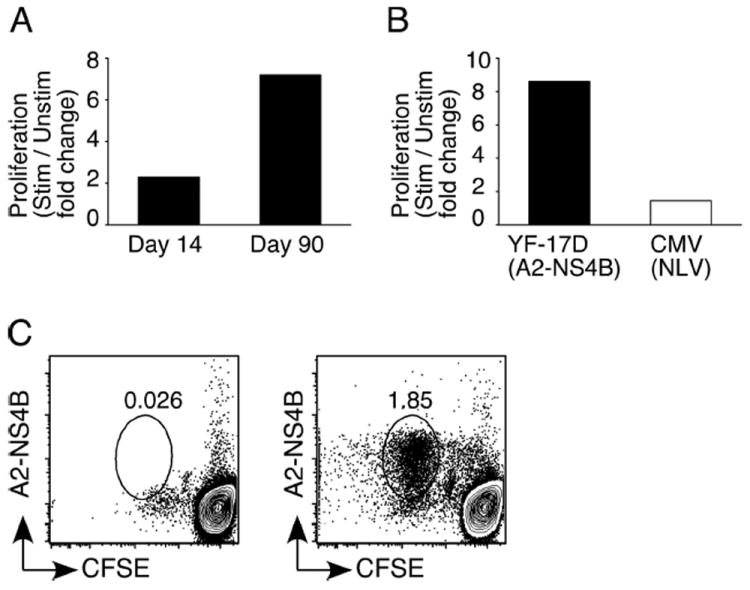
YF-17D elicits memory CD8+ T cells with a robust proliferative potential. A, Proliferative potential of YF-17D-specific effector (day 14) and memory (day 90) CD8+ T cells. PBMC from vaccinated individuals were labeled with CFSE and stimulated (Stim) or not (Unstim) with the A2-NS4B peptide for 6 days. CFSE dilution and staining with the A2-NS4B tetramer was used to identify divided cells. The percentage of CD8+ T cells that are A2-NS4B+ cells is shown as the fold increase above the percentage in blood. B, Expansion of YF-17D-specific memory CD8+ T cells compared with the expansion of CMV-specific CD8+ T cells from the same individual. CFSE-labeled PBMC were stimulated with either A2-NS4B or the CMV-NLV peptide for 6 days. The percentage of CD8+ T cells that are tetramer+ is shown as the fold increase above the percentage in blood. C, Retention of proliferation potential by YF-17D-specific memory CD8+ T cells. PBMC from an individual vaccinated with YF-17D 10 years previously were labeled and stimulated as described above and A2-NS4B specific responses were measured. Plots are gated on CD8+ T cells.
To summarize, YF-17D generates a CD8+ T cell response that is high in magnitude, broadly diverse, and polyfunctional. This results in memory CD8+ T cells that progressively acquire potent proliferative potential and retain it long after the virus is cleared.
Discussion
Live viral vaccines like YF-17D offer a rare opportunity to study the progressive differentiation of human Ag-specific CD8+ T cells that are generated as part of a protective antiviral immune response. We have studied the magnitude, breadth, and dynamics of the YF-17D-specific CD8+ T cell response as a model to study the generation and maintenance of CD8+ T cell memory in the context of acute viral infections. We used a set of overlapping peptides spanning the entire virus to analyze the total antiviral CD8+ T cells and report several interesting findings. Firstly, each vaccinee responded to multiple proteins, and although some proteins such as E, NS3, and NS5 were targeted more commonly, most viral proteins contained CD8+ T cell epitopes. Secondly, the magnitude of the NS4B-specific response was particularly high in HLA-A2+ and not HLA-A2− vaccinees. We identified an immunodominant, HLA-A2-restricted epitope (NS4B 214–222) responsible for this. MHC class I tetramers specific for this epitope were used for longitudinal analysis of A2-NS4B+ CD8+ T cells in a cohort of HLA-A2+ vaccinees. A2-NS4B+ CD8+ T cells had an activated Ki67+Bcl-2lowHLA-DR+ CD38+ phenotype on day 14 that was progressively lost by day 90. This differentiation was also accompanied by the acquisition of proliferation potential. Notably, although the earliest detectable A2-NS4B+ cells were heterogeneous for many markers, nearly all had the activated Ki67+Bcl-2lowHLA-DR+ CD38+ phenotype, indicating a prominent phase of activation and expansion. Lastly, A2-NS4B+ memory CD8+ T cells were detected in circulation 2 years after vaccination. Interestingly, a majority of these cells were CD45RA+CCR7−, a phenotype that has been associated with terminal differentiation and lack of proliferative potential in many studies (6, 41, 47). However, they retained high proliferation potential 5–10 years after vaccination and produced multiple cytokines upon antigenic recall in vitro. Overall, these characteristics may be used to understand the qualities of antiviral CD8+ T cell memory in healthy individuals.
Using an immunodominant epitope identified in YF-17D to quantify virus-specific CD8+ T cells over time, we observed expansion, contraction, and long-term maintenance typical of a primary CD8+ T cell response. Tetramer+ CD8+ T cells were first detected 11 days after vaccination, reached highest frequency at 30 days, diminished to ~30% of the peak by day 90, and only 5–10% of the peak cell numbers were seen 1 year later. However, it is evident that YF-17D-specific memory persists even longer, because A2-NS4B+ cells were detectable in subjects vaccinated 5–10 years previously. Significantly, these individuals had no history of recent exposure to the virus, strongly suggesting that the persistence of YF-17D CD8+ memory is Ag independent.
Based on an estimated 1–3 × 1011 total CD8+ T cells in humans (48) and the percentage of tetramer+ cells observed in the current study (0.5–17% at the peak), the approximate number of A2-NS4B+ CD8+ T cells is between 5 × 108 and 2 × 109, comparable to the number of total YF-17D-specific CD8+ T cells (1.5–9 × 109) approximated on the basis of IFN-γ production in our earlier study (28). We found that at day 14 A2-NS4B+ cells formed a subset of total responding CD8+ T cells; both populations had the Ki67+Bcl-2lowHLA-DR+ CD38+ activated phenotype (28). However, we saw discordance between the times at which these two populations peaked. The highest frequency of total activated cells was seen 2 wk after vaccination, whereas the frequency of A2-NS4B+ cells continued to increase for an additional 2 wk. This suggested that the YF-17D-specific CD8+ T cells continued to expand beyond day 14. In keeping with this, we could detect Ki-67+ CD8+ T cells at day 21 with a frequency that was 70–75% lower than that seen at day 14 (data not shown). Because naive CD8+ T cell expansion is Ag driven, this indicated that YF-17D could persist for slightly longer (possibly in tissues other than blood) than indicated by the viral genomes in circulation that are undetectable by day 14.
Elaboration of the breadth of YF-17D epitopes recognized showed that vaccinees had CD8+ T cells specific for multiple proteins with variation among donors in the size of the response to each protein. However, the hierarchy of CD8+ T cell reactivity established early on was retained through the memory stage as evidenced in stimulations with recombinant vesicular stomatitis virus expressing YF-17D proteins (data not shown). Interestingly, specificity of CD8+ T cells in HLA-A2+ vaccinees was highly skewed toward NS4B, where we later identified an epitope. In some vaccinees up to 86.6% of the proliferating (Ki-67+) CD8+ T cells recognized the NS4B 214–222 epitope and the frequency was as high as 17% of total CD8+ T cells at the peak of expansion, suggesting that the NS4B 214–222 epitope was highly immunogenic. Binding affinity of the peptide for MHC class I is an important factor contributing to the immunogenicity of CD8+ T cell epitopes, with most known peptide epitopes having high (IC50 < 50 nM) or intermediate (IC50 of 50–500 nM) binding affinities. We speculate that the high binding affinity of the NSB 214–222 peptide predicted for HLA-A2 alleles (IC50 < 10 nM; Ref. 49) is instrumental for the immunogenicity of the NS4B 214–222 peptide. Also, the response to NS4B was much more vigorous compared with that directed toward the more frequently recognized E, NS3, and NS5 proteins. A similar observation has been reported recently in a murine vaccinia infection where Oseroff and colleagues find that the most frequently recognized epitopes differ from the immunodominant ones (50).
Studies of total CD8+ T cell populations in healthy individuals have provided useful T cell differentiation models by categorizing CD8+ T cells into subsets based on their distinct phenotypes and finding functions unique to each subset. In one widely accepted model, Ag-experienced CD8+ T cells are classified into central memory cells (CD45RA−CCR7+), effector memory cells (CD45RA−CCR7−), or CD45RA+ effector memory cells (TEMRA; CD45RA+CCR7−) (13, 36). Unexpectedly, YF-17D-specific memory cells shared the CD45RA+CCR7− phenotype of TEMRA cells. Cells with this phenotype are reported to have very little proliferative capacity and to be sensitive to apoptosis and associated with senescence. However, in contrast to the “bulk” TEMRA population, A2-NS4B+ cells had a high proliferative potential and did not express other markers of terminal differentiation (CD56 and CD57). Recent reports showed that memory CD8+ T cells for two acute viruses, vaccinia (7, 28) and B19 (51), were CD45RA+. Together with our findings, this strongly suggests that CD45RA can be reexpressed in the absence of antigenic stimulation. Thus, the presence of CD45RA in Ag-experienced cells may not always be associated with the lack of proliferative potential and in fact may be an indicator of highly functional memory CD8+ T cells generated after acute viral infections.
The smallpox vaccine (Dryvax) is another highly efficacious live viral vaccine that offers some interesting comparisons with the yellow fever vaccine. Possibly due to factors like the route of vaccination used and the complexity and tropism of each virus, there are some differences between the CD8+ T cell responses elicited by them. Firstly, Dryvax elicits a CD8+ T cell response 4-to 10-fold larger than that seen for YF-17D (28). Secondly, the contraction of YF-17D-specific CD8+ T cells seems more protracted (4-fold contraction by day 90) than that of Dryvax-specific CD8+ T cells (10- to 20-fold contraction by day 90) (28). Lastly, in Dryvax vaccinees the peak of tetramer+ cells occurs at the same time as the effector (Ki-67+ Bcl-2low) peak, unlike the discordance seen for YF-17D vaccinees. Overall however, both vaccines elicit a brisk, broadly targeting, polyfunctional CD8+ T cell response (7, 28, 52) with similarities in the phenotype of virus specific-CD8+ T cells, such as the reexpression of CD45RA on memory cells.
Given the remarkable safety and efficacy of YF-17D, it has recently been exploited to develop vaccine candidates against other flaviviral diseases by substitution of the YF-17D M and E genes with those of the target virus. YF-17D-based West Nile, Dengue, and Japanese encephalitis chimeric vaccines have already been evaluated in several phase I–III human trials (53-55). This strategy is also being used to test vaccines against some pathogens unrelated to YF-17D, such as the malaria parasites of the plasmodium genus (56-58). The current study may provide a useful benchmark for evaluation of the responses to these vaccines that use YFV-17D as a vector. In addition, the YF-17D epitope identified may be useful in a simultaneous comparison of CD8+ responses to vector and recombinant proteins in chimeric vaccines, which may be useful for ruling out vector-dominant responses as reported in a study with an modified vaccinia Ankara recombinant vaccine (59).
We define the attributes of a human CD8+ T cell response that generates high-quality immune memory by performing a comprehensive analysis of the CD8+ T cells elicited after vaccination with the efficacious yellow fever live virus vaccine. The YF-17D-specific CD8+ T cells displayed broad specificity, high magnitude, multiple functions, robust proliferative potential, and long-term persistence, all characteristics of protective cellular immunity. We saw an interesting discordance between the “terminal effector-like” phenotype of yellow fever memory CD8+ T cells and their robust recall potential. This observation restates the importance of multiparameter analysis for evaluating an immune response. Recently available systems biology approaches to analyzing changes in global gene expression profiles induced by the vaccine have proved useful for examining the YF-17D-specific innate immune response in depth (29). The ability to track YF-17D-specific CD8+ T cells using tetramers raises the possibility of defining the signature of high quality CD8+ T cell memory in humans.
Supplementary Material
Proliferative potential of CD45RA+ CCR7- CD8+ T cells. Cells from one individual vaccinated two years previously were analyzed for CD45RA expression ex-vivo (left) and proliferative potential in vitro (right). For the in vitro proliferation, PBMC from this individual were labeled with CFSE and stimulated with the A2-NS4B peptide for 6 days and the end of which a further peptide stimulation for 6 hours was done followed by ICC to identify responding cells. The histogram shows CFSE intensity by the IFNγ+ cells compared to the cells that have not divided
Acknowledgments
We thank K. Araki and R. Aubert for input and critical reading of the manuscript and A. Popkowski for technical assistance. We also thank Mario Roederer, National Institutes of Health, Vaccine Research Center, for providing the PESTLE and SPICE software.
Footnotes
This work was supported by National Institutes of Health (NIH) U19 Grant AI057266 (to R.A.) and in part by Sanofi-Pasteur, Lyon, France. C.M.R. receives support from the Greenberg Medical Research Institute, the Starr Foundation, the Foundation for the National Institutes of Health through the Grand Challenges in Global Health initiative (grant identification nos. 334 and 574), General Clinical Research Center Grant M01-RR00102 (to Rockefeller University Hospital), and Center for Translational Science Award Grant 1UL1 RR024143-01 (to Rockefeller University Hospital) from the NIH National Center for Research Resources.
Abbreviations used in this paper: YF-17D, yellow fever virus strain 17D; ICC, intracellular cytokine; PD-1, programmed death-1; TEMRA, effector memory RA T cell.
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 2.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 3.Kalia V, Sarkar S, Gourley TS, Rouse BT, Ahmed R. Differentiation of memory B and T cells. Curr Opin Immunol. 2006;18:255–264. doi: 10.1016/j.coi.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 4.Williams MA, Holmes BJ, Sun JC, Bevan MJ. Developing and maintaining protective CD8+ memory T cells. Immunol Rev. 2006;211:146–153. doi: 10.1111/j.0105-2896.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 5.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, Appay V, Rizzardi GP, Fleury S, Lipp M, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 2001;410:106–111. doi: 10.1038/35065118. [DOI] [PubMed] [Google Scholar]
- 7.Precopio ML, Betts MR, Parrino J, Price DA, Gostick E, Ambrozak DR, Asher TE, Douek DC, Harari A, Pantaleo G, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8+ T cell responses. J Exp Med. 2007;204:1405–1416. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, Busch DH. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc Natl Acad Sci USA. 2004;101:5610–5615. doi: 10.1073/pnas.0308054101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wherry EJ, Barber DL, Kaech SM, Blattman JN, Ahmed R. Antigen-independent memory CD8 T cells do not develop during chronic viral infection. Proc Natl Acad Sci USA. 2004;101:16004–16009. doi: 10.1073/pnas.0407192101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, Ahmed R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Appay V, Rowland-Jones SL. Lessons from the study of T-cell differentiation in persistent human virus infection. Semin Immunol. 2004;16:205–212. doi: 10.1016/j.smim.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 14.de Bree GJ, Heidema J, van Leeuwen EM, van Bleek GM, Jonkers RE, Jansen HM, van Lier RA, Out TA. Respiratory syncytial virus-specific CD8+ memory T cell responses in elderly persons. J Infect Dis. 2005;191:1710–1718. doi: 10.1086/429695. [DOI] [PubMed] [Google Scholar]
- 15.He XS, Mahmood K, Maecker HT, Holmes TH, Kemble GW, Arvin AM, Greenberg HB. Analysis of the frequencies and of the memory T cell phenotypes of human CD8+ T cells specific for influenza A viruses. J Infect Dis. 2003;187:1075–1084. doi: 10.1086/368218. [DOI] [PubMed] [Google Scholar]
- 16.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 17.Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang J, Zuckerman J, Clarke P, Barrett P, Kirkpatrick C, Blondeau C. Comparison of the immunogenicity and safety of two 17D yellow fever vaccines. Am J Trop Med Hyg. 1999;60:1045–1050. doi: 10.4269/ajtmh.1999.60.1045. [DOI] [PubMed] [Google Scholar]
- 19.Barnett ED. Yellow fever: epidemiology and prevention. Clin Infect Dis. 2007;44:850–856. doi: 10.1086/511869. [DOI] [PubMed] [Google Scholar]
- 20.Poland JD, Calisher CH, Monath TP, Downs WG, Murphy K. Persistence of neutralizing antibody 30–35 years after immunization with 17D yellow fever vaccine. Bull World Health Organ. 1981;59:895–900. [PMC free article] [PubMed] [Google Scholar]
- 21.McAllister A, Arbetman AE, Mandl S, Pena-Rossi C, Andino R. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine experimental solid tumors and pulmonary metastases. J Virol. 2000;74:9197–9205. doi: 10.1128/jvi.74.19.9197-9205.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaucher D, Therrien R, Kettaf N, Angermann BR, Boucher G, filai-Mouhim A, Moser JM, Mehta RS, Drake DR, III, Castro E, et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J Exp Med. 2008;205:3119–3131. doi: 10.1084/jem.20082292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barba-Spaeth G, Longman RS, Albert ML, Rice CM. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J Exp Med. 2005;202:1179–1184. doi: 10.1084/jem.20051352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–424. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins MA, Silva ML, Marciano AP, Peruhype-Magalhaes V, Eloi-Santos SM, Ribeiro GL, Correa-Oliveira R, Homma A, et al. Activation/modulation of adaptive immunity emerges simultaneously after 17DD yellow fever first-time vaccination: is this the key to prevent severe adverse reactions following immunization? Clin Exp Immunol. 2007;148:90–100. doi: 10.1111/j.1365-2249.2006.03317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Co MD, Terajima M, Cruz J, Ennis FA, Rothman AL. Human cytotoxic T lymphocyte responses to live attenuated 17D yellow fever vaccine: identification of HLA-B35-restricted CTL epitopes on nonstructural proteins NS1, NS2b, NS3, and the structural protein E. Virology. 2002;293:151–163. doi: 10.1006/viro.2001.1255. [DOI] [PubMed] [Google Scholar]
- 27.Santos AP, Matos DC, Bertho AL, Mendonca SC, Marcovistz R. Detection of TH1/TH2 cytokine signatures in yellow fever 17DD first-time vaccinees through ELISpot assay. Cytokine. 2008;42:152–155. doi: 10.1016/j.cyto.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 28.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 29.Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol. 2009;10:116–125. doi: 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kern F, I, Surel P, Brock C, Freistedt B, Radtke H, Scheffold A, Blasczyk R, Reinke P, Schneider-Mergener J, Radbruch A, et al. T-cell epitope mapping by flow cytometry. Nat Med. 1998;4:975–978. doi: 10.1038/nm0898-975. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmeister B, Kiecker F, Tesfa L, Volk HD, Picker LJ, Kern F. Mapping T cell epitopes by flow cytometry. Methods. 2003;29:270–281. doi: 10.1016/s1046-2023(02)00349-3. [DOI] [PubMed] [Google Scholar]
- 32.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 33.Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 34.Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O’Callaghan CA, Steven N, McMichael AJ, Rickinson AB. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J Exp Med. 1998;187:1395–1402. doi: 10.1084/jem.187.9.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 36.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73:975–983. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 37.Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. 2007;19:309–314. doi: 10.1016/j.coi.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 39.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 40.Wills MR, Okecha G, Weekes MP, Gandhi MK, Sissons PJ, Carmichael AJ. Identification of naive or antigen-experienced human CD8+ T cells by expression of costimulation and chemokine receptors: analysis of the human cytomegalovirus-specific CD8+ T cell response. J Immunol. 2002;168:5455–5464. doi: 10.4049/jimmunol.168.11.5455. [DOI] [PubMed] [Google Scholar]
- 41.Hamann D, Kostense S, Wolthers KC, Otto SA, Baars PA, Miedema F, van Lier RA. Evidence that human CD8+CD45RA+CD27− cells are induced by antigen and evolve through extensive rounds of division. Int Immunol. 1999;11:1027–1033. doi: 10.1093/intimm/11.7.1027. [DOI] [PubMed] [Google Scholar]
- 42.Pittet MJ, Speiser DE, Valmori D, Cerottini JC, Romero P. Cutting edge: cytolytic effector function in human circulating CD8+ T cells closely correlates with CD56 surface expression. J Immunol. 2000;164:1148–1152. doi: 10.4049/jimmunol.164.3.1148. [DOI] [PubMed] [Google Scholar]
- 43.Brenchley JM, Karandikar NJ, Betts MR, Ambrozak DR, Hill BJ, Crotty LE, Casazza JP, Kuruppu J, Migueles SA, Connors M, et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 2003;101:2711–2720. doi: 10.1182/blood-2002-07-2103. [DOI] [PubMed] [Google Scholar]
- 44.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 45.Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol. 2002;3:1061–1068. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- 46.Wherry EJ, Blattman JN, Ahmed R. Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination. J Virol. 2005;79:8960–8968. doi: 10.1128/JVI.79.14.8960-8968.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260–4266. doi: 10.1182/blood-2002-11-3577. [DOI] [PubMed] [Google Scholar]
- 48.Ganusov VV, De Boer RJ. Do most lymphocytes in humans really reside in the gut? Trends Immunol. 2007;28:514–518. doi: 10.1016/j.it.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 49.Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, et al. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 2005;3:e91. doi: 10.1371/journal.pbio.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oseroff C, Peters B, Pasquetto V, Moutaftsi M, Sidney J, Panchanathan V, Tscharke DC, Maillere B, Grey H, Sette A. Dissociation between epitope hierarchy and immunoprevalence in CD8 responses to vaccinia virus Western Reserve. J Immunol. 2008;180:7193–7202. doi: 10.4049/jimmunol.180.11.7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isa A, Kasprowicz V, Norbeck O, Loughry A, Jeffery K, Broliden K, Klenerman P, Tolfvenstam T, Bowness P. Prolonged activation of virus-specific CD8+ T cells after acute B19 infection. PLoS Med. 2005;2:e343. doi: 10.1371/journal.pmed.0020343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jing L, Chong TM, McClurkan CL, Huang J, Story BT, Koelle DM. Diversity in the acute CD8 T cell response to vaccinia virus in humans. J Immunol. 2005;175:7550–7559. doi: 10.4049/jimmunol.175.11.7550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guirakhoo F, Kitchener S, Morrison D, Forrat R, McCarthy K, Nichols R, Yoksan S, Duan X, Ermak TH, Kanesa-Thasan N, et al. Live attenuated chimeric yellow fever dengue type 2 (ChimeriVax-DEN2) vaccine: phase I clinical trial for safety and immunogenicity: effect of yellow fever pre-immunity in induction of cross neutralizing antibody responses to all 4 dengue serotypes. Hum Vaccin. 2006;2:60–67. doi: 10.4161/hv.2.2.2555. [DOI] [PubMed] [Google Scholar]
- 54.Monath TP, Guirakhoo F, Nichols R, Yoksan S, Schrader R, Murphy C, Blum P, Woodward S, McCarthy K, Mathis D, et al. Chimeric live, attenuated vaccine against Japanese encephalitis (ChimeriVax-JE): phase 2 clinical trials for safety and immunogenicity, effect of vaccine dose and schedule, and memory response to challenge with inactivated Japanese encephalitis antigen. J Infect Dis. 2003;188:1213–1230. doi: 10.1086/378356. [DOI] [PubMed] [Google Scholar]
- 55.Monath TP, Liu J, Kanesa-Thasan N, Myers GA, Nichols R, Deary A, McCarthy K, Johnson C, Ermak T, Shin S, et al. A live, attenuated recombinant West Nile virus vaccine. Proc Natl Acad Sci USA. 2006;103:6694–6699. doi: 10.1073/pnas.0601932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li S, Locke E, Bruder J, Clarke D, Doolan DL, Havenga MJ, Hill AV, Liljestrom P, Monath TP, Naim HY, et al. Viral vectors for malaria vaccine development. Vaccine. 2007;25:2567–2574. doi: 10.1016/j.vaccine.2006.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao D, Barba-Spaeth G, Rai U, Nussenzweig V, Rice CM, Nussenzweig RS. Yellow fever 17D as a vaccine vector for microbial CTL epitopes: protection in a rodent malaria model. J Exp Med. 2005;201:201–209. doi: 10.1084/jem.20041526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Epps HL. Broadening the horizons for yellow fever: new uses for an old vaccine. J Exp Med. 2005;201:165–168. doi: 10.1084/jem.20042565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith CL, Mirza F, Pasquetto V, Tscharke DC, Palmowski MJ, Dunbar PR, Sette A, Harris AL, Cerundolo V. Immunodominance of poxviral-specific CTL in a human trial of recombinant-modified vaccinia Ankara. J Immunol. 2005;175:8431–8437. doi: 10.4049/jimmunol.175.12.8431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Proliferative potential of CD45RA+ CCR7- CD8+ T cells. Cells from one individual vaccinated two years previously were analyzed for CD45RA expression ex-vivo (left) and proliferative potential in vitro (right). For the in vitro proliferation, PBMC from this individual were labeled with CFSE and stimulated with the A2-NS4B peptide for 6 days and the end of which a further peptide stimulation for 6 hours was done followed by ICC to identify responding cells. The histogram shows CFSE intensity by the IFNγ+ cells compared to the cells that have not divided