

Abstract
The palladium-catalyzed cross-coupling of chiral propargyl acetates and allyl boronates delivers chiral 1,5-enynes with excellent levels of chirality transfer and applied across a broad range of substrates.
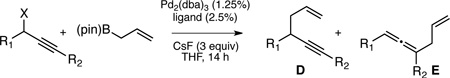
1,5-Enynes are important and versatile synthetic intermediates. In addition to offering differentiated π-systems for selective functionalization, 1,5-enynes can be transformed into a diverse array of cyclic structures.1 A current challenge to reaction methodology surrounding 1,5-enynes lies in the preparation of these structures in an enantiomerically enriched fashion. Synthesis of 1,5-enynes is commonly accomplished by allylation of propargylic electrophiles, employing either stoichiometric or catalytic Lewis acid activation.2 While high levels of regiocontrol have been observed in these processes, they appear to proceed through an achiral carbocation intermediate and this feature precludes the transfer of chirality from enantiomerically-enriched starting materials to 1,5-enyne products (Scheme 1, eq. 1).3,4 Transition metal catalysis could provide a solution to this limitation: palladium undergoes stereospecific anti SN2´ oxidative addition with propargylic electrophiles.5 This reaction delivers an η1-(allenyl)palladium complex (A, Scheme 1) whose configuration reflects that of the starting material. While (allenyl)palladium complexes can undergo isomerization to η1-(propargyl)palladium species (A→B), this transformation is also stereospecific.5,6 With appropriately substituted substrates, both the propargyl (B) and the allenyl (A) palladium complexes are chiral and the fact that they are configurationally stable enables stereospecific cross-couplings.7 However, the Pd-catalyzed cross-coupling of organometallic reagents and branched propargylic electrophiles generally favors the allene as opposed to the 1,5 enyne product.7,8 This regioselectivity likely arises from steric effects; complex A is less hindered than complex B and this leads to allene products on reductive elimination. This reaction manifold renders traditional palladium catalysis ineffective for construction of chiral alkyne-containing compounds from chiral propargylic electrophiles.
Scheme 1.

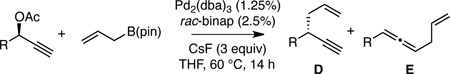
In contrast to the cross-coupling of alkyl, aryl and vinyl metal reagents, cross-couplings of allyl metal reagents may occur with allyl migration. To exploit this feature, our lab has studied cross-couplings of allyl metal reagents and allylic electrophiles and has found that these reactions appear to occur by an inner sphere 3,3’-reductive elimination.9,10,11 In the presence of appropriately selected ligands (bidentate, small bite-angle diphosphines), allyl-allyl cross-couplings occur with excellent levels of regio- and stereocontrol. Along these lines, we considered that allyl-propargyl cross-couplings might also occur by 3,3’-elimination and, if the elimination occurred from an allenyl(allyl)Pd complex (C, Scheme 2), this might provide a method for the stereocontrolled construction of non-racemic 1,5-enynes from readily available enantiomerically-enriched propargylic alcohols.12
Scheme 2.

To initiate these studies, cross-couplings of allylB(pin) and propargylic chlorides were examined in the presence of Pd2(dba)3 and CsF. With dppf as the ligand, the reaction furnished the 1,5-enyne product regioselectively for both aromatic (entry 1) and aliphatic (entry 2) substrates. More conveniently prepared propargylic acetates were also found to participate in the coupling, and while the regioselectivity was moderate with dppf (entries 3 and 4), a marked improvement was observed when rac-binap was employed. These reactions occurred with high levels of selectivity for the 1,5-enyne product and in exceptional yields (entries 5 and 6). In light of prior studies on the impact of ligand structure on regioselectivity,9a it was not surprising that a monodentate ligand such as triphenyl phosphine (entries 7 and 8) favored allene products. Lastly, internal alkynes appear to react with diminished levels of selectivity (entry 9), perhaps because the added substituent at C1 of the allenyl palladium intermediate (C, Scheme 2) suffers an interaction with the ligand framework. Fortunately, this synthetic limitation is easily addressed: C-alkylation of the terminal alkyne-derived products (D, R2=H, Table 1) provides ready access to the corresponding internal alkyne allylation products.
Table 1.
Selectivity in Allyl-Propargyl Coupling.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R1 | R2 | X | ligand | temp (°C) |
D:E | yield (%) |
| 1 | Ph | H | Cl | dppf | rt | 83:17 | 85 |
| 2 | pentyl | H | Cl | dppf | rt | 95:5 | 75 |
| 3 | Ph | H | OAc | dppf | 60 | 68:32 | 79 |
| 4 | pentyl | H | OAc | dppf | 60 | 84:16 | 74 |
| 5 | Ph | H | OAc | rac-binap | 60 | 93:7 | 91 |
| 6 | pentyl | H | OAc | rac-binap | 60 | 98:2 | 96 |
| 7 | Ph | H | OAc | PPh3 | 60 | <1:99 | - |
| 8 | pentyl | H | OAc | PPh3 | 60 | <1:99 | - |
| 9 | pentyl | Bu | OAc | rac-binap | 60 | 27:73 | 68 |
Reactions employed 1.2 equiv of allylB(pin). Regioselectivity was determined by 1H NMR analysis; yield refers to isolated yield of the regioisomer mixture.
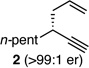
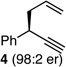
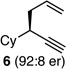
With an effective protocol for enyne-selective coupling, the capacity for chirality transfer from enantiomerically-enriched propargylic acetate substrates was explored. As depicted in Table 2, the reaction showed excellent conservation of enantiomeric enrichment (>99% cee) with aliphatic substrates (entries 1, 3 and 4). While the aromatic substrate in entry 2 suffered some loss of optical purity with rac-binap (77% cee),13 high levels of chirality transfer (>99% cee) were obtained when (R)-methoxy(furyl)biphep14 was employed as the ligand (entry 2). Of special interest was the ability to utilize tertiary acetates to access enantiomerically-enriched 1,5-enynes bearing all-carbon quaternary centers (entry 4). Despite slightly diminished regioselection, the ability to establish quaternary centers with high cee (>99%) and yield could prove especially useful in the construction of complex products.
Table 2.
Conservation of Enantiomeric Enrichment in Allyl-Propargyl Couplings.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | substrate | product | cee (%) | D:E | yield (%) |
||
| 1 | 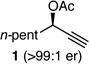 |
 |
>99 | 98:2 | 93 | ||
| 2b | 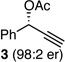 |
 |
>99 | 98:2 | 90 | ||
| 3 | 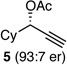 |
 |
96 | 98:2 | 87 | ||
| 4 | 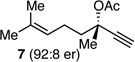 |
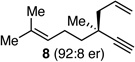 |
>99 | 89:11 | 96 | ||
Reactions employed 1.2 equiv of the allylboronate. Yield refers to isolated yield of the regioisomer mixture. Ratio of D:E was determined by 1H NMR analysis; enantiomer ratios were determined by GC analysis on a chiral stationary phase.
For entry 2, (R)-methoxy(furyl)biphep (2.5 %) used as the ligand; rac-binap gave a product er of 87:13, 77% cee, 93:7 D:E, and 80% yield.
In addition to the substrates in Table 2, a broad array of other propargylic acetates were found to participate in the regioselective allyl-propargyl coupling and generally delivered 1,5-enyne products in good to excellent yield. As depicted in Table 3, substrates can possess silyl and benzyl ethers, remote alkenes, indoles and aryl chloride groups. Of particular note is reaction product 13, which results from regioselective reductive elimination from a conjugated (enallenyl)palladium intermediate.
Table 3.
Substrate Scope of Propargyl-Allyl Cross-Coupling.a
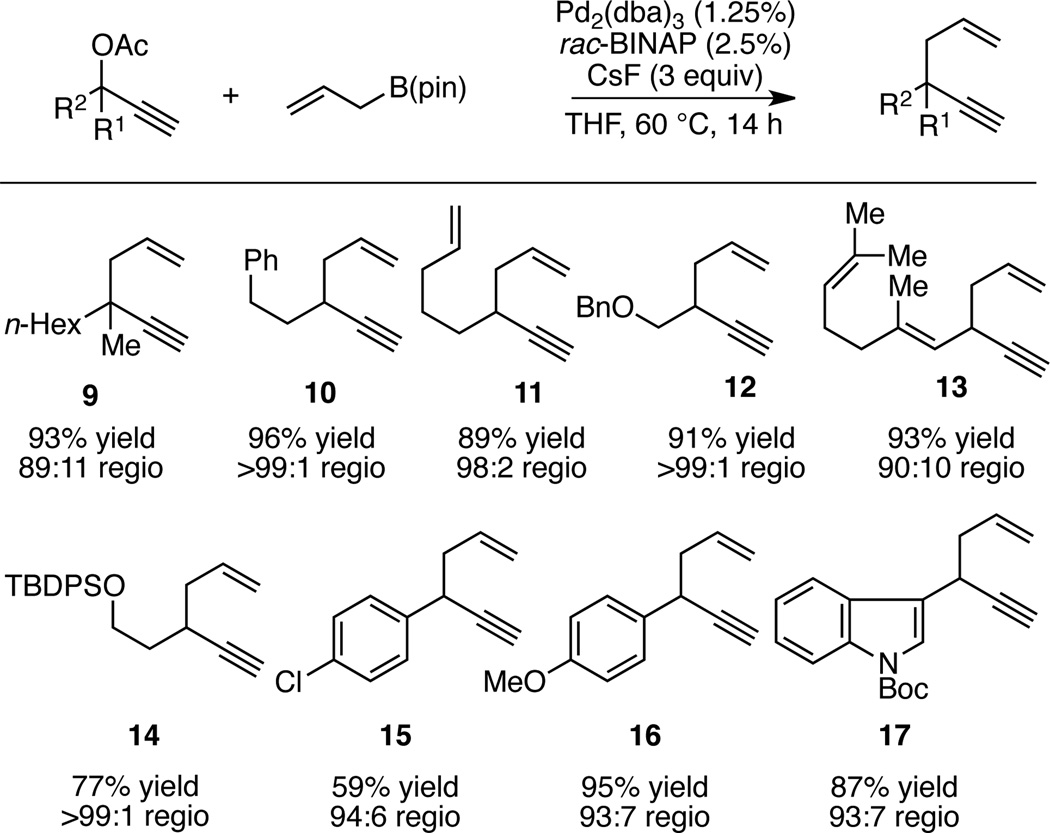 |
Reactions were conducted for 14 h at 60 °C with 1:1 ligand:Pd(0) and employed 1.2 equiv of the allylboronate relative to the propargyl acetate. Yield refers to isolated yield of the regioisomeric mixture. Regioselectivity was determined by 1H NMR analysis.
Aspects of the allyl-propargyl coupling reactions described above merit mention. First the high level of chirality transfer observed with rac-binap suggests that the catalyst configuration may have little impact on the product stereochemistry. To examine this feature in more detail, both enantiomers of ligand were employed with enantiomerically enriched substrate 1. As shown in Scheme 3 (eq. 3), there is a slight level of double diastereodifferentiation but both reactions deliver the product with net inversion of configuration at carbon as the predominant outcome. These experiments indicate that the product configuration is dictated almost exclusively by the starting material structure. To learn more about the innate basis for the product regioselectivity, the experiment in equation 4 was conducted. In this experiment, the reaction of allenyl B(pin) and cinnamyl acetate was found to deliver 1,5-enyne 20 as the predominant reaction product. In this experiment, the presumed reactive intermediate bears an interconverting allyl/propargyl ligand without a steric bias between the two isomers (F and G). This reaction delivers the 1,5 enyne selectively, an outcome that is most consistent with 3,3' reductive elimination from allenyl complex G. Thus, even in the absence of a steric bias, the innate preference appears to be for reductive elimination through the allenyl palladium intermediate.
Scheme 3.

Allylboronates substituted at the β and γ positions are readily available by catalytic hydroboration of dienes and by catalytic borylation of allylic electrophiles.15 To learn about the broader generality of the allyl-propargyl cross-coupling, the utility of these substituted nucleophiles was examined. As depicted in Table 4, β–substituted allyl boronates maintain high levels of regioselectivity in coupling with propargyl acetates (products 21 and 22). γ–Substituted allyl boronates can also exhibit high levels of reactivity and selectivity; however, they require modifications to the reaction. To produce compound 23 in good yield required use enantiomerically enriched substrate (R)-3 and (R)-methoxy(furyl)biphep. With these conditions, the reaction was efficient and, while both crotyl-(H) and cis-2-butenyl derivatives (J) were generated, the reaction exhibited a strong preference for the alkyne product. Similarly, efficient production of 24 and 25 could be accomplished with the use of the (R)-propargylic acetate and (S,S)-QuinoxP*. In these reactions, when the opposite ligand enantiomer was employed diminished levels of selectivity were observed. A rationale for the observed matched and mismatched interactions is presented in Figure 1 and is based on a stereochemical model for related allyl-allyl couplings. As depicted, the matched pairing appears to minimize non-bonded interactions between the pseudoequatorial furyl ring and the allyl/allenyl ligands as the later couple to form a staggered C-C bond.
Table 4.
Propargyl-Allyl Cross-Coupling with Substituted Allylboronates.a
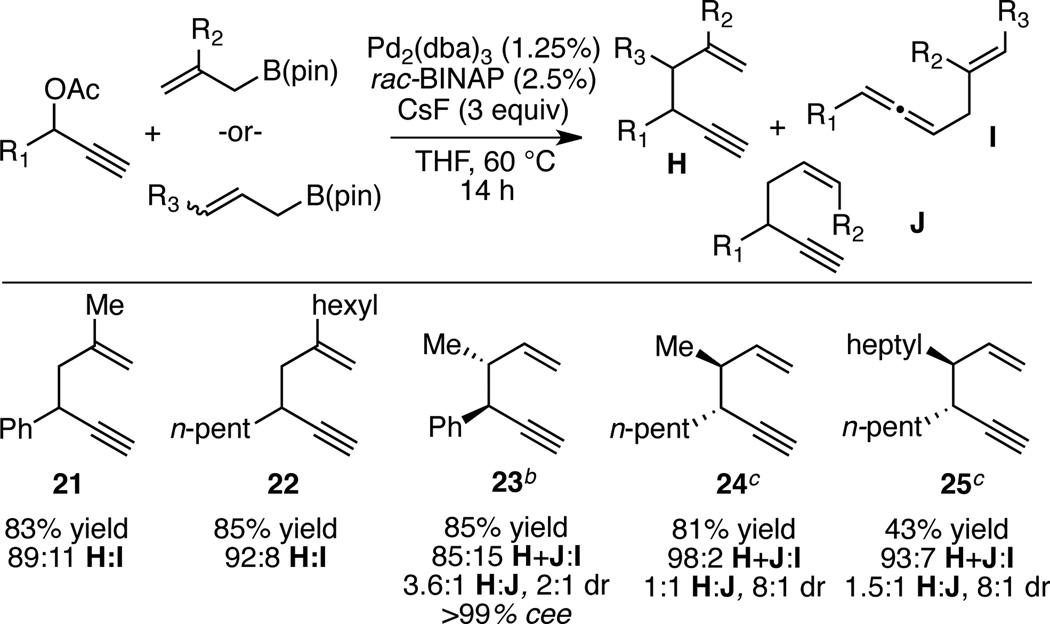 |
Reactions were conducted for 14 h at 60 °C with 1:1 ligand:Pd(0) and employed 1.2 equiv of the allylboronate relative to the propargyl acetate. Yield refers to isolated yield of the regioisomeric mixture. Regioselectivity was determined by 1H NMR analysis.
(R)-methoxyfurylbiphep employed as the ligand and 10 equiv. CsF employed.
(S,S)-QuinoxP* employed as the ligand and 10 equiv. CsF employed.
Figure 1.

Matched and Mismatched Catalyst Substrate Interactions in Propargyl-Allyl Couplings.
Synthetically, the allyl-propargyl coupling serves as a strategically useful link between methodologies for synthesis of enantiomerically-enriched propargyl alcohols and methods for the transformation of enyne substrates. For example, Kozmin and co-workers have described a straightforward protocol for the synthesis of substituted cyclohexenones through cyclization of siloxy alkynes derived from 1,5-enynes.16 Using the allyl-propargyl-coupling described above, the requisite substrates can be prepared in an enantiomerically-enriched fashion thereby facilitating the use of the Kozmin transformation in asymmetric synthesis; as depicted in Scheme 4, coupling of methallylB(pin) converts 1 to 26 with excellent stereocontrol. From 26, disubstituted cyclohexenone 27 can be easily produced in high yield and without racemization. With a general procedure to access a broad range of highly enantiomerically-enriched enynes bearing aliphatic, aromatic and all carbon quaternary substitution, the allyl-propargyl coupling should serve as a useful new route to a myriad of other optically enriched synthetic intermediates.
Scheme 4.

In conclusion, the development of a new Pd(0) catalyzed allyl-propargyl coupling has opened a general method for construction of enantiomerically-enriched 1,5-enynes. These motifs are often used in a racemic fashion, and this strategy now provides a simple method for their construction in enantiomerically enriched form.
Supplementary Material
ACKNOWLEDGMENT
The NIH (GM-064451) is acknowledged for financial support; Frontier Scientific is acknowledged for a generous donation of allylB(pin). We are grateful to Ms. Angelika Neitzel for preliminary experimental assistance.
Footnotes
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1. Nishida M, Adachi N, Onozuka K, Matsumura H, Mori M. J. Org Chem. 1998;63:9158. Rhode O, Hoffmann HMR. Tetrahedron. 2000;56:6479. Luzung MR, Markham JP, Toste FD. J. Am. Chem. Soc. 2004;126:10858. doi: 10.1021/ja046248w. Pianowski Z, Rupnicki L, Cmoch P, Staliński K. Synlett. 2005:900. Debleds O, Campagne J-M. J. Am. Chem. Soc. 2008;130:1562. doi: 10.1021/ja0780986. Graham TJA, Gray EE, Burgess JM, Goess BC. J. Org. Chem. 2010;75:226. doi: 10.1021/jo9020375. (g) Review: Zhang L, Sun J, Kozmin SA. Adv. Synth. Catal. 2006;348:2271.
- 2.(a) Kuninobu Y, Ishii E, Takai K. Angew. Chem. Int. Ed. 2007;46:3296. doi: 10.1002/anie.200700183. [DOI] [PubMed] [Google Scholar]; Schwier T, Rubin M, Gevorgyan V. Org. Lett. 2004;6:1999. doi: 10.1021/ol0494055. [DOI] [PubMed] [Google Scholar]; (b) Dau-Schmidt J-P, Mayr H. Chem. Ber. 1994;127:205. [Google Scholar]; (c) Hayashi M, Inubushi A, Mukaiyama T. Chem. Lett. 1987:1975. [Google Scholar]; (d) Ishikawa T, Okano M, Aikawa T, Saito S. J. Org. Chem. 2001;66:4635. doi: 10.1021/jo010157p. [DOI] [PubMed] [Google Scholar]; (e) Kabalka GW, Yao M-L, Borella S. J. Am. Chem. Soc. 2006;128:11320. doi: 10.1021/ja061379d. [DOI] [PubMed] [Google Scholar]; (f) Zhan Z-P, Yu J-L, Liu H-J, Cui Y-Y, Yang R-F, Yang W-Z, Li J-P. J. Org. Chem. 2006;71:8298. doi: 10.1021/jo061234p. [DOI] [PubMed] [Google Scholar]; (g) Sanz R, Martínez A, Miguel D, Álvarez-Gutiérrez JM, Rodríguez F. Synthesis. 2007:3252. [Google Scholar]
- 3.(a) Georgy M, Boucard V, Campagne J-M. J. Am. Chem. Soc. 2005;127:14180. doi: 10.1021/ja0534147. [DOI] [PubMed] [Google Scholar]; (b) Georgy M, Boucard V, Debleds O, Dal Zotto C, Campagne J-M. Tetrahedron. 2009;65:1758. [Google Scholar]; (c) Sanz R, Martínez A, Álvarez-Gutiérrez JM, Rodríguez F. Eur. J. Org. Chem. 2006:1383. [Google Scholar]
- 4.For stereoselective propargyl/allyl coupling using chiral allyl silanes, see: Luzung MR, Toste FD. J. Am. Chem. Soc. 2003;125:15760. doi: 10.1021/ja039124c.
- 5.Elsevier CJ, Kleijn H, Boersma J, Vermeer P. Organometallics. 1986;5:716. [Google Scholar]
- 6.For characterization of relevant (η3-allenyl)Pd compounds, see: Ogoshi S, Tsutsumi K, Kurosawa H. J. Organomet. Chem. 1995;493:C19. Tsutsumi K, Kawase T, Kakiuchi K, Ogoshi S, Okada Y, Kurosawa H. Bull. Chem. Soc. Jpn. 1999;72:2687. Baize MW, Blosser PW, Plantevin V, Schimpff DG, Gallucci JC, Wojcicki A. Organometallics. 1996;15:164.
- 7.(a) Elsevier CJ, Stehouwer PM, Westmijze H, Vermeer P. J. Org. Chem. 1983;48:1103. [Google Scholar]; (b) Elsevier CJ, Mooiweer HH, Kleijn H, Vermeer P. Tetrahedreon Lett. 1984;48:5571. [Google Scholar]; (c) Elsevier CJ, Vermeer P. J. Org. Chem. 1985;50:3042. [Google Scholar]; (d) Konno T, Tanikawa M, Ishihara T, Yamanaka H. Chem. Lett. 2000:1360. [Google Scholar]; (e) Dixneuf PH, Guyot T, Ness MD, Roberts SM. Chem. Commun. 1997:2083. [Google Scholar]; (f) Yoshida M, Gotou T, Ihara M. Tetrahedron Lett. 2004;45:5573. [Google Scholar]; (g) Molander GA, Sommers EM, Baker SR. J. Org. Chem. 2006;71:1563. doi: 10.1021/jo052201x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yoshida M, Ueda H, Ihara M. Tetrahedron Lett. 2005;46:6705. [Google Scholar]
- 8. Jeffrey-Luong T, Linstumelle G. Tetrahedron Lett. 1980;21:5019. Ruitenberg K, Kleijn H, Elsevier CJ, Meijer J, Vermeer P. Tetrahedron Lett. 1981;22:1451. Ma S-M, Zhang A-B. Pure Appl. Chem. 2001;73:337. Moriya T, Miyaura N, Suzuki A. Synlett. 1994:149. Reviews: Ma S-M. Eur. J. Org. Chem. 2004:1175. Tsuji J, Mandai T. Angew. Chem. Int. Ed. Engl. 1995;34:2589.
- 9.(a) Zhang P, Brozek LA, Morken JP. J. Am. Chem. Soc. 2010;132:10686. doi: 10.1021/ja105161f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang P, Le Hai, Kyne RE, Morken JP. J. Am. Chem. Soc. 2011;133:9716. doi: 10.1021/ja2039248. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brozek LA, Ardolino MJ, Morken JP. J. Am. Chem. Soc. 2011;133:16778. doi: 10.1021/ja2075967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For other recent allyl-allyl couplings, see: Flegeau EF, Schneider U, Kobayashi S. Chem. Eur. J. 2009;15:12247. doi: 10.1002/chem.200902221. Jiménez-Aquino A, Flegeau EF, Schneider U, Kobayashi S. Chem. Commun. 2011;47:9456. doi: 10.1039/c1cc13348a.
- 11. Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem. Eur. J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P. Cárdenas DJ, Echavarren AM. New J. Chem. 2004;28:338. Perez-Rodrguez M, Braga AAC, de Lera AR, Maseras F, Alvarez R, Espinet P. Organometallics. 2010;29:4983.
- 12.Reviews of asymmetric carbonyl alkynylation: Trost BM, Weiss AH. Adv. Synth. Catal. 2009;351:963. doi: 10.1002/adsc.200800776. Pu L. Tetrahedron. 2003;59:9873. Frantz DE, Fässler R, Tomooka CS, Carreira EM. Acc. Chem. Res. 2000;33:373. doi: 10.1021/ar990078o. For enantioselective catalytic ynone reduction, see: Matsumura K, Hashiguchi S, Ikariya T, Noyori R. J. Am. Chem. Soc. 1997;119:8738. Helal CJ, Magriotis PA, Corey EJ. J. Am. Chem. Soc. 1996;118:10938. Parker KA, Ledeboer MW. J. Org. Chem. 1996;61:3214. doi: 10.1021/jo951712o.
- 13.Possible racemization mechanisms include redox transmetallation, see: Ogoshi S, Nishida T, Fukunishi Y, Tsutsumi K, Kurosawa H. J. Organometallic Chem. 2001;620:190. Banerjee M, Roy S. Chem. Comm. 2003:534. and references therin.
- 14.Broger EA, Foricher J, Heiser B, Schmid R. 5274125. U.S. Patent. 1993
- 15.a) Wu JY, Moreau B, Ritter T. J. Am. Chem. Soc. 2009;131:12915. doi: 10.1021/ja9048493. [DOI] [PubMed] [Google Scholar]; b) Ely RJ, Morken JP. J. Am. Chem. Soc. 2010;132:2534. doi: 10.1021/ja910750b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ishiyama T, Ahiko T, Miyaura N. Tetrahedron Lett. 1996;37:6889. [Google Scholar]; d) Zhang P, Roundtree IA, Morken JP. Org. Lett. 2012;14:1416. doi: 10.1021/ol3001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Zhang L, Kozmin SA. J. Am. Chem. Soc. 2004;126:10204. doi: 10.1021/ja046586x. [DOI] [PubMed] [Google Scholar]; b) Zhang L, Sun J, Kozmin SA. Tetrahedron. 2006;62:11371. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.