

Abstract
How the brain determines which memories are selected for long-term storage is critical for a full understanding of memory. One possibility is that memories are selected based on the history of activity and current state of neurons within a given memory circuit. Many in vitro studies have demonstrated metaplasticity-like effects whereby prior neuronal activity can affect the ability of cells to express synaptic plasticity in the future; however, the significance of these findings to memory is less clear. Here we show in rats that a single pairing of a light with shock, insufficient to support either short- or long-term fear memory, primes future learning such that another trial delivered within a circumscribed time window lasting from ∼60 min to 3 d results in the formation of a long-lasting and robust fear memory. Two adequately spaced training trials support long-term fear memory only if the two trials are signaled by the same cue. Furthermore, although a single training trial does not support formation of an observable fear memory, it does result in the phosphorylation of several targets of protein kinase A (PKA) in the amygdala. Accordingly, blocking PKA signaling in the amygdala before the first training trial completely prevents the ability of that trial to facilitate the formation of long-term fear memory when a second trial is delivered 24 h later. These findings may provide insight into how memories are selected for long-term storage.
Introduction
The ability to successfully predict dangerous or rewarding situations is critical for survival and depends on the ability to select out associations in the environment that are consequential for the animal from those that are not important. Information is more likely to be retained the more often it is encountered, particularly if these encounters are spaced over time (Ebbinghaus, 1885; Carew et al., 1972; Josselyn et al., 2001; Detert et al., 2008). However, laboratory conditions typically used to study learning and memory are much more predictable (i.e., many trials, regular spacing) than they are under natural settings where it is unlikely that the same stimulus will predict the same outcome over and over again within a short time period. Thus, there must be a neurobiological mechanism that endows animals with the ability to select out important information for long-term memory with only limited experience.
One attractive possibility is that memories are selected for based on the current neural state of cells within dedicated memory circuits. This idea comes from work on metaplasticity showing that the ability of neurons to undergo long-term synaptic changes can be influenced by their history of neuronal stimulation (Abraham and Bear, 1996). For example, long-term potentiation (LTP) can be facilitated or inhibited by various electrophysiological and pharmacological treatments applied before the induction of LTP, which themselves do not elicit LTP (Huang et al., 1992, Cohen and Abraham, 1996). Thus, prior events alter the state of neural circuits such that they can influence future bouts of synaptic plasticity. Although a considerable amount of work has gone into characterizing the mechanisms of metaplasticity-like findings in vitro (Abraham, 2008), the significance of these results to learning and memory is less clear.
We reasoned that if a metaplasticity-like mechanism is important in determining how fear memories are selected for storage then weak training, insufficient to support long-term fear memory, should prime future learning. We trained rats with a single training trial, or two trials separated by various intertrial intervals. A single training trial did not support the formation of an overt fear memory, but if another trial was delivered within a time window lasting from 60 min to 3 d a robust and long-lasting fear memory was formed. Furthermore, a single training trial activated several targets of PKA in the amygdala, and the priming of future learning induced by this single training trial could be blocked by inhibiting PKA. We believe our results shed new light on how memories are selected for long-term storage.
Materials and Methods
Subjects
Three hundred forty-two male Sprague Dawley rats weighing 325–450 g at the time of testing and obtained from Charles River served as subjects. All rats were ∼2 months old upon arrival and were housed 4 to a cage in a vivarium maintained on a 12 h light/dark cycle. Behavioral testing began within a month after arrival and took place during the light portion of the cycle. Food and water were freely available. All procedures were performed with approval of the Emory University Institutional Animal Care and Use Committee.
Surgery
Before surgery animals were anesthetized with IP injections of dexdomitor (0.5 mg/kg) and ketamine (75 mg/kg). Each rat received a subcutaneous injection of metacam (1 mg/kg) on the day of surgery. Rats were implanted with 22-gauge bilateral cannulae aimed at the amygdala (AP = −2.8/L = ±5.2/V = −8.0). The cannulae were anchored to the skull using stainless steel screws and acrylic cement. Stainless steel obdurators, cut to be flush with the end of the guide cannulae, were inserted to prevent blockage.
Drug preparation and infusion
The PKA inhibitor Rp-cAMPS (Tocris Bioscience) was diluted with ACSF to a concentration of 36 μg/μl (Schafe and LeDoux, 2000). In all cases rats received bilateral infusions of Rp-cAMPS or ACSF into the amygdala 30 min before the first training trial. The total volume of the infusion (0.5 μl/side) was given over 2 min and the injection cannula remained in place for an additional 90 s to ensure diffusion away from the tip of the injection cannulae, which were cut to extend 1 mm beyond the guide cannulae.
Apparatus
As described previously (Parsons and Davis, 2011), rats were trained and tested in two identical 9 × 14 × 10 cm (depth × width × height; internal dimensions) Plexiglas and wire-mesh cages. Each cage was suspended between compression springs within a steel frame and located within a custom-designed 60 × 79.5 × 59.5 cm sound-attenuating chamber lined with 6.3 mm thick Plexiglas. The floor of each cage consisted of four 6.0 mm diameter stainless steel bars spaced 18 mm apart. Affixed to the bottom of each cage was an Endevco accelerometer (Model 2217E). Cage movement resulted in displacement of the accelerometer that produced a voltage output proportional to the velocity of cage movement. The accelerometer's output was amplified by an Endevco Model 104 amplifier and digitized on a scale of 0–2500 U by an InstruNET device (GW Instruments, Model 100B) interfaced to a Macintosh G3 computer. Startle amplitude was defined as the maximal peak-to-peak voltage that occurred during the first 300 ms after onset of the startle-eliciting noise burst.
Startle responses were evoked by 50 ms 95-dB white noise bursts (5 ms rise-decay time, 0–22 kHz) generated by a Macintosh G3 computer sound file, amplified by a Radio Shack amplifier (Model MPA-200), and delivered through high-frequency speakers (Radio Shack Supertweeter) located in front of each cage. Background noise (60-dB wideband) was produced by an ACO Pacific white noise generator (Model 3024) and was delivered through the same speakers as those used to provide white noise bursts. Sound level measurements were made with a Brüel and Kjaer model 2235 sound level meter (A scale; random input) with the microphone (Type 4176) located 10 cm from the center of the speaker, which approximates the distance of the rat's ear from the speaker during testing.
The unconditioned stimulus for all experiments was a 0.5 s, 0.4 mA scrambled shock delivered through the floor bars. Shock intensity was measured with a 1 kOhm resistor across a differential channel of an oscilloscope in series with a 100 kOhm resistor connected between adjacent floor bars within each cage. Current was defined as the root-mean-square voltage across the 1 kOhm resistor where mA = 0.707 × 0.5 × peak-to-peak voltage. Shocks were produced by LeHigh Valley shock generators (SGS 004). The presentation and sequencing of all stimuli was under the control of the Macintosh G3 computer using custom-designed software (The Experimenter; Glassbeads).
Behavioral procedures
Baseline startle.
On two consecutive days, rats were placed into the test cages and presented with 30 95-dB startle-eliciting noise bursts (30 s interstimulus interval, ISI) after a 5 min stimulus-free period. Rats were matched into different treatment groups on the basis of the baseline startle.
Pretest.
All rats were placed into the startle chamber and presented with 50 startle stimuli (30 s ISI). Of the last 20 startle trials, every fourth startle trial was in the presence of the light, resulting in a total of five light pretest trials.
Training.
The next day rats were returned to the same chamber and received a single 3.7 s light (82 lux)-shock (0.4 mA/ 0.5 s) pairing (n = 16) or two pairings separated by 4 min (n = 18), 45 min (n = 10), 60 min (n = 30), 3 h (n = 13), 6 h (n = 8), 9 h (n = 10), 24 h (n = 17), 3 d (n = 12), 7 d (n = 10), or 30 d (n = 8). Each pairing occurred 5 min after placement into the chamber and the 0.5 s shock was delivered 3.2 s after onset of the 3.7 s light cue. To control for the amount of time in the training chamber, separate groups of rats were trained with a single trial or two trials separated by 4 or 60 min. Animals in these separate groups were in the chamber for either 12 (Fig. 1D) or 67 min (Fig. 1C). All other groups were removed from the testing chamber in between trials. For the experiment in Figure 3A,B, training involved presenting a light paired with shock, a white noise (82 dB/3.7 s) with a shock, a shock alone, or a light alone. Various configurations of these stimuli were presented to different groups of rats (see Fig. 3A). During training for this experiment all rats were in the chamber for a total of 67 min. The data for the “light-shock/noise-shock” and “noise-shock/light-shock” groups were no different and were combined for statistical purposes. Shock reactivity was measured in a manner very similar to the startle responses. Any cage movement resulted in displacement of the accelerometer that produced a voltage output proportional to the velocity of cage movement. Shock reactivity was defined as the maximal peak-to-peak voltage that occurred during the 500 ms shock.
Figure 1.

Optimal trial-spacing permits long-term memory formation using two training trials. A, Rats received a single pairing of light that predicted shock (n = 16) or two trials separated by intertrial intervals ranging from 4 min to 30 d (n's = 8–30/group) and memory was tested 2 d following the last trial. B, Spacing the trials by 60 min to 3 d resulted in a very robust memory. C, D, Superior memory in rats trained with a 60 min interval versus those trained with a single trial or two trials spaced by 4 min is still observed when time in the training chamber is equated. E, Levels of contextual fear did not differ between groups in rats left in during the ITI. F, Rats trained with a 60 min ITI and removed during the ITI showed higher levels of contextual fear compared with the group trained with a single trial. G, Rats were trained with a single trial and tested for short term memory 1 h later and long-term memory 2 d later. Another group was only tested for long term memory. H, Rats showed no evidence of short or long term memory. I, Rats pre-exposed to the light showed similar levels of fear as those not pre-exposed to the light cue (*p < 0.05 vs 1 trial; #p < 0.05 vs 4 min). In all graphs, error bars indicate ±SEM.
Figure 3.
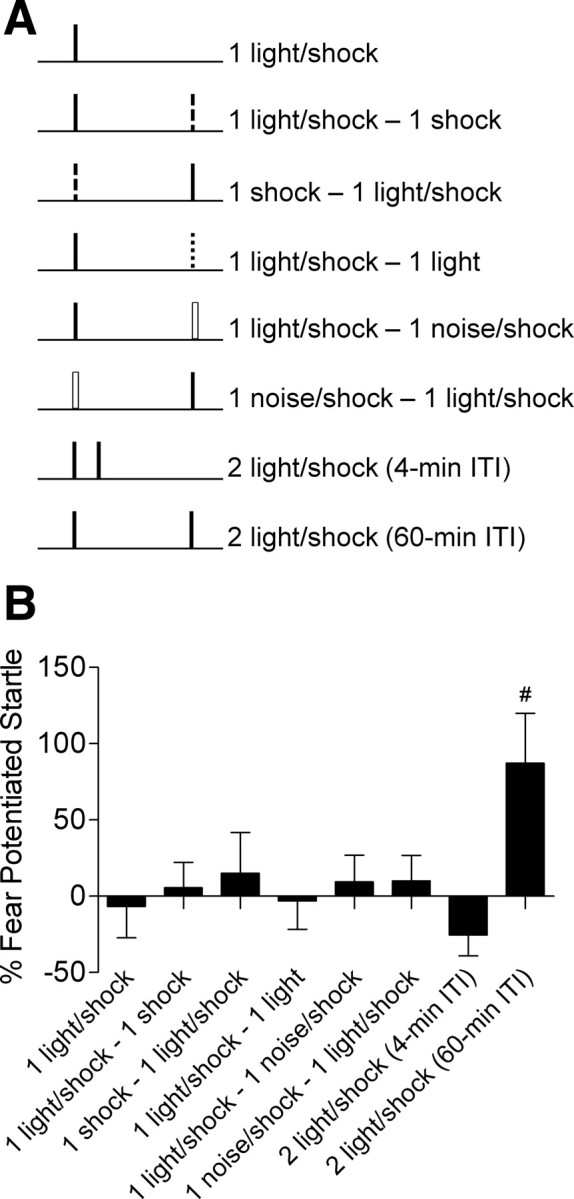
Fear memory in rats trained with two trials is cue specific. A, Separate groups of rats (n's = 6–13/group) were trained with one of the listed sets of stimuli. B, Only rats trained with two trials at a 60 min intertrial interval using the same cue show a long-term memory (#p values <0.05 vs all groups).
Memory test.
Testing occurred in the same chamber as was used in the previous sessions. After 5 min, 30 95-dB startle-eliciting noise bursts were presented, all separated by 30 s. Rats then received 40 test trials consisting of 10 light-startle test trials each followed by 3 startle-alone test trials. For test trials, the 95-dB noise burst was presented 3.5 s after onset of the cue. For startle-alone test trials, the 95-dB noise burst was presented alone. The ISI for all stimuli was 30 s. As noted in the text and figures, the test sessions took place 2, 14, or 30 d after the last training trial.
To quantify fear potentiated startle, each rat's mean startle amplitude during the baseline startle session and on light test trials was determined and a percentage change score was calculated. Percentage change scores were used (i.e., vs absolute difference scores) because previous work from our lab found that they remain stable across variations in baseline startle amplitude (Walker and Davis, 2002). Fear potentiated startle to the context was measured by taking the mean startle amplitude values during the 2 d of baseline startle testing and the mean of the 30 startle-alone trials that preceded presentation of the light during testing. Percentage change scores were derived from this comparison. These scores were then analyzed using ANOVA, Welch F test, or Student's t tests to examine differences between groups. Fisher's LSD post hoc tests were used when appropriate, and the criterion for significance was p < 0.05.
Retraining and retesting.
Two days after testing, a subset of rats (n = 13) from the experiment in Figure 5 were returned to the same chamber and received five acetophenone (10% in propylene glycol)-shock pairings. The first pairing occurred 5 min after placement into the startle chamber and successive shocks occurred every 4 min. For each pairing, the 0.5 s shock (0.4 mA) was delivered 3.5 s after onset of the 4.0 s odor cue. Five days later rats were given a test session with 10 trials of the odor cue, but otherwise identical to the testing procedure described above.
Figure 5.
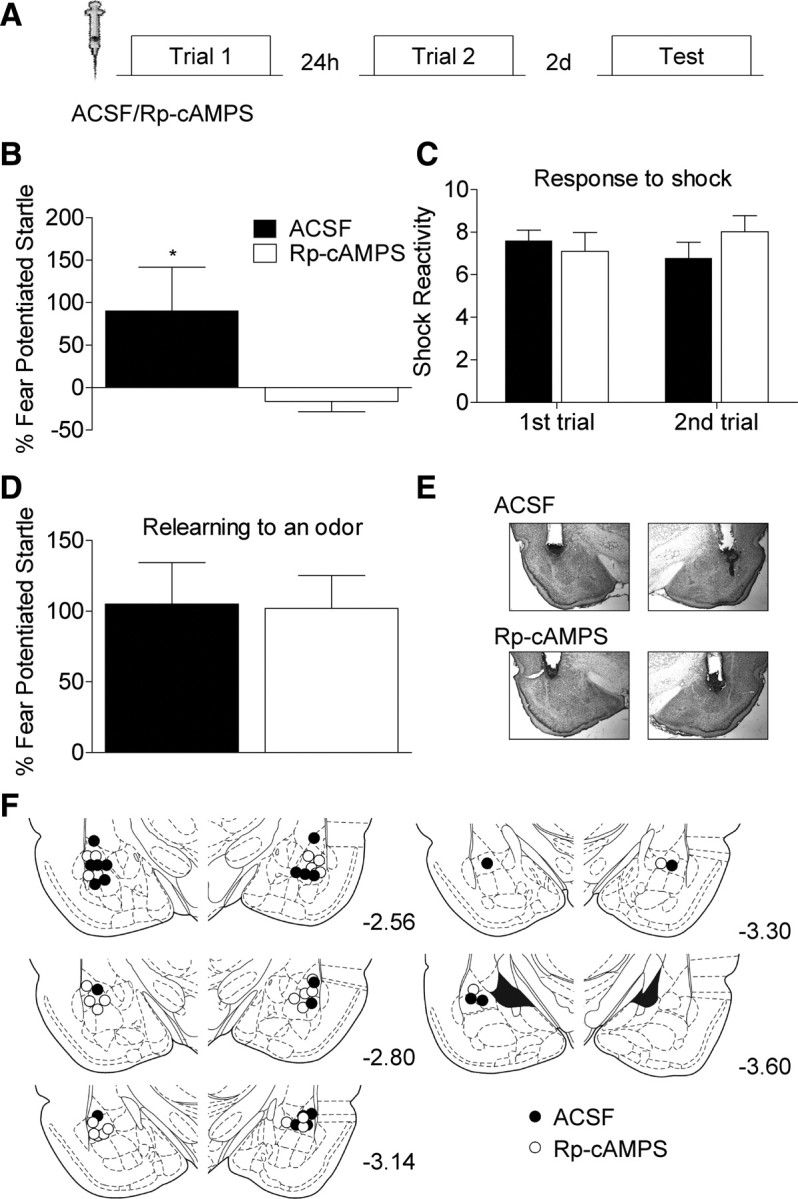
Blocking PKA activity in the amygdala before trial 1 prevents the priming of future learning. A, ACSF (n = 12) or Rp-cAMPS (n = 14) was infused into the amygdala (18 μg/0.5 μl/hemisphere) 30 min before the first light shock trial. Rats were administered another trial 24 h later. B, Rats infused with Rp-cAMPS showed no evidence of memory when tested 2 d after the final trial. C, Reactivity to the footshock was not affected by the PKA inhibitor. D, Both groups show intact fear memory when retrained with an odor that signaled shock (*p < 0.05). E, Representative Nissl-stained images in rats infused with ACSF or Rp-cAMPS into the amygdala. F, Cannulae placements for all rats in this experiment.
Latent inhibition experiment.
Baseline startle levels were measured as described previously. The next day, during the latent inhibition phase, one group was given 30 presentations of the light cue which later served as the conditioned stimulus. Another group of rats were exposed to the same chamber for an equivalent period of time. All rats were trained the following day with 2 light-shock trials (60 min ITI), and were tested 2 d later as described before.
Summation experiment.
Baseline startle levels were measured as described previously. For the pretest, all rats were placed into the startle chamber and presented with 70 (30 s ISI) startle stimuli. Of the last 40 startle trials, every fourth startle trial was in the presence of the light or odor cue (10% acetophenone), resulting in a total of 10 pretest trials (5 light, 5 odor). For training, rats received a single light-shock trial followed 60 min later by a single odor-shock trial. Two days later rats were returned to the same chamber and after 5 min, 30 95-dB startle-eliciting noise bursts were presented, all separated by 30 s. Rats then received 60 startle trials consisting of 15 test trials each followed by 3 startle-alone test trials. For test trials, the 95 dB noise burst was presented 3.5 s after onset of the odor, light, or odor-light in compound. The order of the test trials was counterbalanced such that no more than 2 of the same cue or cue combinations were presented consecutively. For startle-alone test trials, the 95 dB noise burst was presented alone. The ISI for all stimuli was 30 s.
Behavioral procedures for Western blot experiments.
All rats were put through the Baseline Startle and Pretest sessions as described above. In the first experiment, groups of rats were given a single light-shock trial and then killed 4 min, 60 min, or 24 h later. Home cage rats were transported to the testing area, left in the holding chamber, and killed throughout the day. In the second experiment, rats were either given a single trial and killed 60 min later, or received two light-shock pairings separated by 60 min and were killed 1 h later. Rats in the “Box” group were given two exposures to the training context separated by 60 min, but received no lights or shocks, and were killed 1 h later. On the same day, a group of home cage rats was killed as described above.
Histology
Rats were killed by overdose of chloral hydrate and perfused intracardially with 0.9% saline followed by 10% buffered formalin. The brains were removed and immersed in a 30% sucrose-formalin solution for several days. The tissue was frozen and 50 μm coronal sections were cut through the area of interest using a cryostat. Cannulae placements were determined with the aid of a rat brain atlas (Paxinos and Watson, 2007) and were judged by a scorer blind to the animal's group assignment. To be considered an accurate placement, cannulae tips needed to be within or no further than 0.5 mm from the basolateral amygdala and medial to the external capsule.
Western blot procedure
To kill the animals, rats were removed from their home cages and placed in a chamber that had isoflurane vapors in it. Approximately 30 s later, the rats were decapitated and their brains were rapidly removed and immediately placed on dry ice. The brains stayed frozen at −80°C until they were dissected. Both sides of the amygdala were dissected out by making coronal cuts to reach the anterior and posterior extent of the amygdala. Punches of the amygdala were taken using a 2 mm punch tool from this section and were immediately placed in homogenization buffer (all in 100 ml of DDH20; tris-HCl 0.605 g; sodium deoxycholate 0.25 g; NaCl 0.876 g; EDTA 0.038 g; 0.0042 g NaF; PMSF 1 μg/ml; leupeptin 1 μg/ml; aprotinin 1 μg/ml; 10 ml of 10% SDS, 1 mm sodium orthovanadate) and briefly sonicated until no traces of solid matter were visible. The homogenates were then immediately returned to dry ice.
Samples were stored at −80°C until they were thawed and centrifuged at 4k rpm for 20 min. The supernatant was taken and protein levels were measured using a Micro BCA protein assay kit (Thermo Scientific). Samples were normalized and 25 μg of protein was loaded on 15-well precast 7.5% SDS/PAGE gels (Bio-Rad). Proteins were then transferred from the gel to a nitrocellulose membrane using a wet transfer apparatus (Bio-Rad). Membranes were blocked (1% nonfat dry milk) for 2 h and then overnight at 4°C in primary antibody. Membranes were probed with antibodies against phospho-MAPK (1:1000), total MAPK (1:5000), phospho-CREB (1:1000), total-CREB (1:1000; all from Cell Signaling Technology), phospho-GLUR1 (SER 845; 1:1000), or total-GLUR1(1:1000; both from Millipore).
Following primary antibody exposure, all membranes were incubated in goat anti-rabbit secondary antibody (1:2500–1:5000; Millipore) for 90 min at room temperature. Membranes were washed and exposed to pico-sensitive chemiluminescence substrate for 5 min (Thermo Scientific). The membranes were then placed between two transparencies and images were taken using a FluorChem SP imager (Alpha Innotech). Following the collection of the images, the membranes were washed several times with PBS-T and exposed to a membrane stripping solution (Restore Plus, Thermo Scientific) for 10 min. The membranes were then reblocked for 1 h at room temperature, exposed (overnight, 4°C) to antibodies for total protein levels corresponding to the particular phospho-protein, and images were taken as described above.
Western blot data were analyzed from the image files generated by the imaging system and densitometry was performed using ImageJ (NIH). A single mean optical density value was determined for each sample by positioning a properly sized sample box over each band and subtracting out the background levels of the image. These values were averaged across the different treatment groups for the phospho-specific and total-protein antibodies. A ratio of phosphorylated to total was calculated and expressed as a percentage of the ratio for home cage rats. To increase the power of the statistical tests, across the two experiments the percentage of home cage values were combined in the groups that received a single trial and were killed 1 h later and in the home cage group. However, the ratios and the percentage values were computed from the home cage rats in the same experiment (i.e., same Western blot development). ANOVA, Student's t tests, and appropriate post hoc comparisons were used to test for differences between groups.
Results
A single weak training trial primes future learning
To test whether a weak training trial would facilitate fear memory formation for future events, we presented rats with a single pairing of a 3.7 s light that predicted a mild foot shock (0.4 mA, 0.5 s) or two trials separated in time by various intervals ranging from 4 min to 30 d (Fig. 1A). Fear memory was calculated by testing whether the animals' acoustic startle response was greater in the presence of the cue which predicted shock, versus in its absence (Walker and Davis, 2002). When memory was tested 2 d after training rats given a single training trial, or two trials separated by 4 min, showed no evidence of long-term fear memory (Fig. 1B). Rats with the two trials separated by 60 min to 3 d showed robust memory when tested 2 d later, and rats trained with 7 or 30 d between trials showed intermediate levels of fear. A Welch F test on these data yielded a significant difference between groups, Welch's F(10,47.662) = 6.645, p < 0.001), and post hoc tests showed that rats trained with intervals ranging from 60 min to 7 d were significantly different (p values <0.05) than the group given a single trial. Similarly, groups with training intervals ranging from 60 min to 3 d showed significantly more fear than rats trained with a 4 min interval (p values <0.05). Because background contextual cues can compete with discrete cues for associative strength (Rescorla and Wagner, 1972) it is possible that the rats trained with a single trial or with two trials using the 4 min interval showed poor learning to the cue because the contextual cues outcompete the light cue for associative value during training. However, this does not seem to be a factor in our data because when time in the context during training was equated the rats trained with the 60 min interval still showed superior learning to the light cue whether or not they were left in the box (F(2,29) = 4.323, p < 0.05; Fig. 1C) or removed from the box during the ITI (F(2,29) = 8.271, p < 0.05; Fig. 1D). Post hoc tests in both conditions revealed that the 60 min group showed more fear potentiated startle than both the 1 trial and 4 min groups (p values <0.05). Furthermore, learning to the contextual cues in rats trained with one trial and those trained with the 4 min interval was not superior (Fig. 1E,F), and in fact an ANOVA on data from the rats removed during the during the ITI yielded a significant difference in contextual learning (F(2,29) = 3.801, p < 0.05) with post hoc tests showing that rats trained with a 60 min ITI exhibiting better contextual memory than the rats trained with a single trial (p < 0.05; Fig. 1F).
Other rats were trained with a single trial and tested for short-term memory 1 h later and long term memory 2 d following training (Fig. 1G). The results (Fig. 1H) revealed no evidence of short or long-term fear memory indicating that a single training trial using these weak training conditions was insufficient to produce even an observable short-term fear memory. Finally, to test whether our pretest procedure was producing latent inhibition, rats were presented with 30 trials of the light alone the day before training. A t test on the data from the test session revealed no difference between groups (p > 0.05), demonstrating that under these conditions pre-exposure to the CS does not affect learning. These findings suggest that although a single training trial does not result in the formation of an overt fear memory, it primes future learning via a mechanism that is fully engaged 60 min later and persists for at least 3 d. Hence this single event sets up a mechanism that allows a long-term memory to form if that same event in encountered again within this time window.
The memory formed by two trials is robust and long-lasting
To determine whether the fear memory formed by two optimally spaced training trials is long lasting, we trained rats with some of the same intervals and tested 2 wks or 30 d after training (Fig. 2A). Rats trained with a 60 min or 24 h interval still showed a very robust fear memory 2 wks later (Fig. 2B). An ANOVA showed a significant main effect (F(2,22) = 3.833, p < 0.05), and post hoc comparisons showed that rats trained with a 24 h interval showed significantly higher levels of fear compared with those trained with a 4 min interval (p < 0.05). Since there was only a trend for the 60 min group to show higher levels of fear when tested 2 wks after training, we trained another set of rats and tested them 30 d later. These rats exhibited a very robust fear memory (Fig. 2C), and this is supported by a one-sample t test showing that the group mean is significantly different from zero (t7 = 5.168, p < 0.01). Finally, rats trained with single pairings of a light and shock followed by an odor and shock showed no evidence for summation as there was very little fear when the two cues were presented in compound (Fig. 2E).
Figure 2.

Fear memory in rats trained with a 60 min or 24 h intertrial interval is long lasting. A, Rats were trained with a 4 min (n = 9), 60 min (n = 8) or 24 h (n = 8) training interval and tested 14 d after the second training trial. B, Rats with a 60 min or 24 h intertrial interval showed robust retention. C, Another group of rats (n = 8) trained with a 60 min intertrial interval and tested 30 d later showed very robust fear memory. D, Rats were given a single pairing of light and shock followed by a pairing of odor with shock. Rats were tested 2 d later in the presence of the light, odor, or light and odor in compound. E, There was no evidence of summation between the light and odor, as the rats showed negligible amounts of fear when the cues were presented in compound (*p < 0.05 vs 4 min).
Two-trial memory is cue specific
If our results are indicative of how specific memories are selected for long-term storage, then presenting rats with two dissimilar events should not yield a long-term memory. However, if the facilitation of future learning was the result of the first trial triggering some general arousal process then memory should be facilitated, for example, by an un-signaled shock placed 60 min before a light-shock pairing. We tested this by training rats with various configurations of stimuli (Fig. 3A) and testing for fear to the light 2 d later. Only the rats trained with two light-shock pairings separated by 60 min showed long-term fear memory (Fig. 3B), indicating cue specificity. This conclusion was supported by an ANOVA which showed a significant among the groups (F(6,56) = 2.508, p < 0.05) and post hoc comparisons showed that the group given two trials separated by 60 min was significantly different from all the other groups (p values <0.05). The observation that no memory was formed in any of the other groups, especially those that received a noise-shock trial followed by light-shock trial (or vice versa) or a shock followed by a light-shock pairing (or vice versa), demonstrates this is not a nonspecific effect of arousal caused by the first shock pairing, but is specific to the cue paired with shock. These results indicate that there is a high degree of specificity in how fear memories are selected for long-term storage under these conditions.
A single weak training trial activates PKA in the amygdala
To begin to understand the mechanisms supporting this priming effect, we asked what cell signaling pathways become engaged after a single fear conditioning trial in the amygdala, given the importance of this brain region in fear learning (LeDoux, 2000; Davis and Whalen, 2001; Josselyn, 2010). Rats were trained with a single pairing of light and shock and killed 4 min, 60 min or 24 h later (Fig. 4A). Another group was given two trials of training spaced by 60 min and killed 60 min later (Fig. 4B). Controls were killed from their home cage or after two exposures to the training chamber with no shock or light presentations. Tissue punches were taken from the amygdala and processed for Western blotting.
Figure 4.

Phosphorylation of PKA targets in the amygdala following training with a single trial or two trials. A, Rats were given a single training trial and killed 4 min (n = 6), 60 min (n = 13), or 24 h (n = 6) later or (B) given two trials separated by 60 min (n = 8) and killed 1 h later. “Box” controls (n = 8) were exposed to the training apparatus twice separated by 60 min but did not experience the light or shock and were killed 1 h later. Home cage rats (n = 13) were transported on the day of training and killed from their home cage. Sixty minutes after a single trial there was a significant increase in the phosphorylation of MAPK (C), GLUR1 at SER845 (D) and CREB at SER 133 (E). One hour after two trials spaced by 60 min there was a significant increase in phosphorylation of GLUR1 (D) and CREB (E) in the amygdala, but no change in MAPK (C). Representative images from the Western blots are shown below each graph (*p < 0.05 vs HC; **p < 0.05 vs 4 min; #p < 0.05 vs 24 HR; ##p < 0.05 vs BOX).
We examined the phosphorylation state of the mitogen-activated protein kinase (MAPK), cyclic adenosine monophosphate response element binding protein (CREB), and the GLUR1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor at serine 845, all of which are targets of PKA (Waltereit and Weller, 2003; Abel and Nguyen, 2008). An ANOVA showed that MAPK (F(3,36) = 5.028, p < 0.01), CREB (F(3,36) = 3.113, p < 0.05), and GLUR1 (SER 845; F(3,36) = 3.032, p < 0.05) all showed a significant change in phosphorylation following a single pairing of light and shock (Fig. 4C–E). Post hoc tests indicate that for MAPK and CREB, the group killed 60 min after training showed significantly higher levels compared with the other three groups (p values <0.05), while phosphorylation of GLUR1 was increased 60 min after training compared with the home cage control (p < 0.05), but was not different from the other two groups. Thus, despite the fact that a single trial does not support observable short or long-term fear memory, it does trigger the phosphorylation of several targets of PKA in the amygdala. The phosphorylation signal engaged by a single trial is transient however, because 24 h later the phosphorylation levels of all three targets were back to basal levels.
We also examined the phosphorylation of these same proteins 60 min following two trials spaced by 1 h. An ANOVA indicated a significant difference in levels of phosphorylated GLUR1 (F(2,27) = 8.864, p < 0.01) and CREB (F(2,27) = 4.146, p < 0.05). Post hoc tests confirmed that both GLUR1 (p values <0.01) and CREB (p values <0.05) were significantly elevated following two trials compared with both the homecage and box controls (Fig. 4D,E). However, there was no change in the levels of phospho-MAPK at this time point (Fig. 4C). The observation that MAPK is not phosphorylated after two trials suggests that the intracellular signaling mechanisms supporting the metaplasticity-like priming of fear learning do not completely overlap with those which ultimately trigger the long-term memory. Although it is possible the time course of MAPK phosphorylation is simply different following two trials, our results are conceptually similar to previous findings that ERK/MAPK is only activated when expectations are violated (Huh et al., 2009) or when novel, but not familiar, cues are used to signal an aversive event (Berman and Dudai, 2001).
PKA is necessary for the priming of future learning induced by one trial
The results of our protein expression studies indicate that PKA signaling is engaged after a single trial. To test whether PKA activity is necessary for the priming of future learning, we infused the PKA inhibitor Rp-cAMPS (18 μg/side in 0.5 μl) into the amygdala 30 min before the first training trial (Fig. 5A). Twenty four hours later, when the effects of the drug should have long worn off, the rats were given a second training trial and memory was tested 2 d later. Rats infused with Rp-cAMPS showed essentially no fear to the light cue during the memory test (Fig. 5B). This assertion is supported by a t test showing that rats infused with Rp-cAMPS showed significantly less fear during testing (t(1,24) = 2.094, p < 0.05). A two-way ANOVA on the shock reactivity data (Fig. 5C) showed that there was no main effect for drug treatment or trials, and no drug by trial interaction (p values >0.05), indicating that the impairment does not reflect a disruption in the ability of Rp-cAMPS infused rats to react to the shock or damage to the amygdala which can reduce reactivity to footshock (Hitchcock et al., 1989). Furthermore, a t test on the retesting data showed no difference between groups (p > 0.05) indicating that the rats showed intact fear memory when retrained with pairings of odor and shock (Fig. 5D), again arguing that amygdala function was not permanently compromised by the drug infusions. These results indicate is that even though the second training trial is what triggers the long-term fear memory, formation of that memory can be prevented by disrupting the priming mechanism engaged by the first trial.
Discussion
This study showed that a single pairing of a light and weak shock, insufficient to support observable short or long term memory, results in the formation of a long-lasting and robust fear memory when a second weak training trial is given over a long circumscribed time window (60 min to 3–7 d) that is stimulus specific. We are not saying this initial weak training trial produced no memory, but rather that it was expressed not in the form of overt fear to the light cue but in a change in the effectiveness of a subsequent training trial to produce a long term memory. This priming of future learning by the first training trial led to a time-dependent phosphorylation of several targets of PKA in the amygdala and blocking PKA signaling in the amygdala before the first training trial prevented this priming effect.
Although several targets of PKA were phosphorylated after the single weak training trial, no fear memory was measurable. This might lead to the conclusion that these phosphorylation events are not sufficient for memory formation. For example, this seems to be contrary to results showing that CREB is necessary and sufficient for memory formation (Sekeres et al., 2010). However, in our study we are looking at endogenous phosphorylation of CREB, whereas other studies have exogenously overexpressed CREB activity using viral vectors in tandem with weak training protocols that either do not yield a long-term memory or result in a weak memory. What we tried to do was to unconfound the role these phosphorylation events play over multiple training trials by looking at their role on the very first trial alone. When we did this we found these phosphorylation events were indeed critical for memory formation, but perhaps in a way that has not been possible to reveal when only multiple training trials are used. Our results indicate that phosphorylation events trigger various changes that alter the impact of subsequent training trials over protracted periods of time. Hence, the function of these phosphorylation events in memory might actually be one of priming in addition to, or rather than, fear memory formation.
While it is apparent that the priming of future learning depends on PKA signaling in the amygdala, it is unclear what cellular processes allow for the priming to occur. One possibility is that a single training trial enhances excitability in the amygdala for a period of time afterward. Several studies have shown that learning can enhance neuronal excitability, typically in the form of reduced afterhyperpolarization and decreased action potential threshold (Alkon et al., 1982; Moyer et al., 1996; Saar et al., 1998; McKay et al., 2009). Other work has shown that PKA and some of its targets are critical for the learning-related changes in excitability (Cohen-Matsliah et al., 2007; Oh et al., 2009). Furthermore, the time course of these alterations in excitability (Moyer et al., 1996; Saar et al., 1998) matches up nicely with time course of our behavioral results. Finally, these learning-related changes do not appear to be a substrate of permanent memory storage because memory persists after the changes in intrinsic excitability have dissipated (Moyer et al., 1996). Our findings are consistent with these observations where a single trial could alter neuronal excitability even though it does not support a behavioral memory. The learning-related changes in excitability might be best conceptualized as being a prerequisite to memory formation, as has been suggested (Disterhoft and Oh, 2006). Using this explanation, a single training trial in our experiments is leaving a set of neurons in the amygdala more excitable for a period of time, so that when a second trial is presented during this period of heightened excitability the formation of long-term fear memory is permitted. What is unknown is whether the fear conditioning procedure results in the same kind of learning-related changes in excitability as has been observed in the hippocampus. However, overexpression of CREB in the amygdala does result in excitability changes similar to those observed after learning (Zhou et al., 2009). A critical question going forward will be to determine whether the training protocol in our experiments results in changes in excitability and what the time course of those changes might be.
Our findings are similar to and inspired by a recent study (Philips et al., 2007) in Aplysia, which examined the result of trial spacing on long-term memory for sensitization using two training trials. They found that 2 trials spaced by 45 min, but not 15 or 60 min, gave rise to long-term memory. The facilitation of learning in this case seemed to be the result of the second trial occurring during the peak of MAPK phosphorylation induced by the first trial. While MAPK is also phosphorylated after a single training trial in our paradigm, the time window during which a second trial is effective was much longer, and our behavioral effects are not paralleled by activation of the PKA signaling. Instead, PKA signaling seems to be a trigger for a much longer lasting form of metaplasticity because the second training trial is still capable of producing a long-term memory well after the phosphorylation events have subsided.
Our results show that fear to the light cue is not facilitated if the first trial is signaled by a noise. These data might seem to run contrary to a study (Zelcer et al., 2006) showing that learning an olfactory discrimination task enhances CA1 excitability and facilitates future learning on the Morris water maze. A number of factors might account for the difference between these sets of findings. First, the Zelcer study used a very different form of learning that relies on a different set of brain structures than does cued fear conditioning. There is evidence that cued fear conditioning relies on a very small subset neurons in the amygdala (Josselyn et al., 2001; Rumpel et al., 2005). It has also been shown that while sensory receptive fields in the amygdala do overlap to some extent, different sensory cues generally activate different parts of the amygdala (Uwano et al., 1995). Hence, it should not be surprising that signaling a shock with a tone does not facilitate subsequent learning when the shock is cued by light, because the noise-shock trial would be activating a small number of neurons many of which would be different from those used in a light-shock association. Another consideration is the Zelcer study used two forms of learning, each of which was strong enough to produce a behaviorally observable memory by itself. In contrast, in our study, we saw no behaviorally observable memory with our single trial alone. Thus, although the two studies share some similarities we believe they probably involve fundamentally different mechanisms.
Our results might also be conceived of in terms of synaptic tagging and capture (Redondo and Morris, 2011). In this instance, the first trial would trigger setting of the synaptic tag and the second trial would result in the production of plasticity-related products which are then captured by the tag. However, it is unclear how this mechanism would explain the ability of a single trial to prime learning that occurs a day or more later given that the lifetime of the putative tag is 3 h or less (Frey and Morris, 1997), and behavioral correlates of synaptic tagging are in accordance with that time scale (Moncada and Viola, 2007). For our results to fit into this framework, the synaptic tag would need to be present for several days, and we are not aware of any data that allows for this possibility. However, it is also possible that the tagging and capture events both occur after the second trial and that the first trial serves to lower the threshold for these events to take place.
Our results indicate that although a single training trial does not yield a fear memory it does prime future learning such that presenting a second trial from an hour to several days later results in the formation of a robust and long lasting memory. We believe our findings might help explain how events are selected out for long-term storage from what is essentially a torrent of information encountered during conscious experience. The long time period following a single trial during which a second trial gives rise to a long-term memory opens up the exciting possibility of being able to study the molecular mechanisms supporting the priming versus those involved in the induction of long-term memories independently. This is not possible with typical training procedures which often involve presenting several trials over a relatively short period of time. In this way we might be able to deconstruct how memories are assembled at the behavioral and molecular level, gaining new insight into how memories are formed.
Footnotes
This research was supported by Grants R37 MH047840 (to M.D.) and F32 MH090700 (to R.G.P.) from the National Institutes of Health. Support was also provided by the Center for Behavioral Neuroscience (National Science Foundation agreement IBN-987675), the National Center for Research Resources P51RR165, and the Office of Research Infrastructure Programs/OD P51OD11132. We thank Dr. Tom Carew, Dr. Mark Bouton, and members of the Davis laboratory for helpful comments.
References
- Abel T, Nguyen PV. Regulation of hippocampus-dependent memory by cyclic AMP-dependent protein kinase. Prog Brain Res. 2008;169:97–115. doi: 10.1016/S0079-6123(07)00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Alkon DL, Lederhendler I, Shoukimas JJ. Primary changes of membrane currents during retention of associative learning. Science. 1982;215:693–695. doi: 10.1126/science.7058334. [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y. Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science. 2001;291:2417–2419. doi: 10.1126/science.1058165. [DOI] [PubMed] [Google Scholar]
- Carew TJ, Pinsker HM, Kandel ER. Long-term habituation of a defensive withdrawal reflex in aplysia. Science. 1972;175:451–454. doi: 10.1126/science.175.4020.451. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Abraham WC. Facilitation of long-term potentiation by prior activation of metabotropic glutamate receptors. J Neurophysiol. 1996;76:953–962. doi: 10.1152/jn.1996.76.2.953. [DOI] [PubMed] [Google Scholar]
- Cohen-Matsliah SI, Brosh I, Rosenblum K, Barkai E. A novel role for extracellular signal-regulated kinase in maintaining long-term memory-relevant excitability changes. J Neurosci. 2007;27:12584–12589. doi: 10.1523/JNEUROSCI.3728-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Detert JA, Kampa ND, Moyer JR., Jr Differential effects of training intertrial interval on acquisition of trace and long-delay fear conditioning in rats. Behav Neurosci. 2008;122:1318–1327. doi: 10.1037/a0013512. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus HE. Memory: a contribution to experimental psychology. New York: Dover; 1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Hitchcock JM, Sananes CB, Davis M. Sensitization of the startle reflex by footshock: blockade by lesions of the central nucleus of the amygdala or its efferent pathway to the brainstem. Behav Neurosci. 1989;103:509–518. doi: 10.1037//0735-7044.103.3.509. [DOI] [PubMed] [Google Scholar]
- Huang YY, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- Huh KH, Guzman YF, Tronson NC, Guedea AL, Gao C, Radulovic J. Hippocampal Erk mechanisms linking prediction error to fear extinction: roles of shock expectancy and contextual aversive valence. Learn Mem. 2009;16:273–278. doi: 10.1101/lm.1240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA. Continuing the search for the engram: examining the mechanism of fear memories. J Psychiatry Neurosci. 2010;35:221–228. doi: 10.1503/jpn.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- McKay BM, Matthews EA, Oliveira FA, Disterhoft JF. Intrinsic neuronal excitability is reversibly altered by a single experience in fear conditioning. J Neurophysiol. 2009;102:2763–2770. doi: 10.1152/jn.00347.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada D, Viola H. Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J Neurosci. 2007;27:7476–7481. doi: 10.1523/JNEUROSCI.1083-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Disterhoft JF. Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci. 1996;16:5536–5546. doi: 10.1523/JNEUROSCI.16-17-05536.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, McKay BM, Power JM, Disterhoft JF. Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A. Proc Natl Acad Sci U S A. 2009;106:1620–1625. doi: 10.1073/pnas.0807708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RG, Davis M. Temporary disruption of fear-potentiated startle following PKMzeta inhibition in the amygdala. Nat Neurosci. 2011;14:295–296. doi: 10.1038/nn.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 2007. [DOI] [PubMed] [Google Scholar]
- Philips GT, Tzvetkova EI, Carew TJ. Transient mitogen-activated protein kinase activation is confined to a narrow temporal window required for the induction of two-trial long-term memory in Aplysia. J Neurosci. 2007;27:13701–13705. doi: 10.1523/JNEUROSCI.4262-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo RL, Morris RG. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci. 2011;12:17–30. doi: 10.1038/nrn2963. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Wagner AR. A theory of pavlovian conditioning: variations in the effectiveness of reinforcement and non-reinforcement. In: Black AH, Prokasky WR, editors. Classical conditioning II: current research and theory. New York: Appleton-Century-Crofts; 1972. pp. 64–99. [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci. 1998;10:1518–1523. doi: 10.1046/j.1460-9568.1998.00149.x. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekeres MJ, Neve RL, Frankland PW, Josselyn SA. Dorsal hippocampal CREB is both necessary and sufficient for spatial memory. Learn Mem. 2010;17:280–283. doi: 10.1101/lm.1785510. [DOI] [PubMed] [Google Scholar]
- Uwano T, Nishijo H, Ono T, Tamura R. Neuronal responsiveness to various sensory stimuli, and associative learning in the rat amygdala. Neuroscience. 1995;68:339–361. doi: 10.1016/0306-4522(95)00125-3. [DOI] [PubMed] [Google Scholar]
- Walker DL, Davis M. Quantifying fear potentiated startle using absolute versus proportional increase scoring methods: implications for the neurocircuitry of fear and anxiety. Psychopharmacology (Berl) 2002;164:318–328. doi: 10.1007/s00213-002-1213-0. [DOI] [PubMed] [Google Scholar]
- Waltereit R, Weller M. Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol Neurobiol. 2003;27:99–106. doi: 10.1385/MN:27:1:99. [DOI] [PubMed] [Google Scholar]
- Zelcer I, Cohen H, Richter-Levin G, Lebiosn T, Grossberger T, Barkai E. A cellular correlate of learning-induced metaplasticity in the hippocampus. Cereb Cortex. 2006;16:460–468. doi: 10.1093/cercor/bhi125. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Won J, Karlsson MG, Zhou M, Rogerson T, Balaji J, Neve R, Poirazi P, Silva AJ. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]