

Abstract
Endothelin-1 is a vasoactive peptide that activates both the endothelin A (ETA) and endothelin B (ETB) receptors, and is secreted in high concentrations in many different cancer environments. While ETA receptor activation has an established nociceptive effect in cancer models, the role of ETB receptors on cancer pain is controversial. EDNRB, the gene encoding the ETB receptor, has been shown to be hypermethylated and transcriptionally silenced in many different cancers. In this study we demonstrate that EDNRB is heavily methylated in human oral SCC lesions, which are painful, but not methylated in human oral dysplasia lesions, which are typically not painful. ETB mRNA expression is reduced in the human oral SCC lesions as a consequence of EDNRB hypermethylation. Using a mouse cancer pain model we show that ETB receptor re-expression attenuates cancer-induced pain. These findings identify EDNRB methylation as a novel regulatory mechanism in cancer-induced pain and suggest that demethylation therapy targeted at the cancer microenvironment has the potential to thwart pain-producing mechanisms at the source, thus freeing patients of systemic analgesic toxicity.
Keywords: methylation, cancer pain, EDNRB, Endothelin B receptor, oral squamous cell carcinoma
INTRODUCTION
For almost all oral cancer patients, pain is rated as the worst symptom over all other symptoms. Oral cancer patients suffer severe pain for months and even years, preventing them from functioning normally [5; 6; 11; 12; 19]. The intensity of oral cancer pain is higher than that of other cancers [23; 24]. Oral cancer pain escalates with disease progression, so that terminal patients experience excruciating pain during their final months of life. Most oral cancer patients with severe pain are terminal patients. Approximately 50% of oral cancer patients will not be cured with surgery, chemotherapy or radiation therapy [36]. In the USA this group, which consists of approximately 40,000 new cases per year, is larger than those dying from melanoma, cervical cancer, or ovarian cancer [27].
Cancer pain, including oral cancer pain, is due to the production of nociceptive mediators, such as endothelin-1 (ET-1) into the cancer microenvironment [30; 35]. ET-1 is a vasoactive peptide that is produced and secreted into the cancer microenvironment in high concentration by many cancers including prostate, ovarian, breast, renal, bladder, cervical, bone and oral [2; 4; 13; 28; 30; 32–35; 37]. Oral squamous cell carcinoma (SCC), in particular, expresses extremely high levels of ET-1, compared to other cancers [29]. ET-1 binds two receptors, the endothelin A (ETA) and the endothelin B (ETB) receptors. ETA receptors have an established role in neuropathic and inflammatory pain [1; 7; 14]. Activation of ETA receptors also contributes to cancer pain. We have confirmed that it is the ETA receptors on the primary afferent nerves within the cancer microenvironment that leads to significant pain [30; 32; 35].
Unlike ETA receptor activation, evidence suggests that ETB receptor activation has an anti-nociceptive effect in both non-cancer and cancer models [17; 28]. Khodorova and colleagues demonstrated that ETB receptor activation on keratinocytes leads to an analgesic effect. Oral SCC consists of malignant keratinocytes. However, EDNRB, the gene encoding ETB receptors, is frequently methylated in cancer. EDNRB methylation results in transcriptional silencing and has been demonstrated in cancers of many types, including lung, prostate, esophageal and nasopharyngeal cancer [15; 18; 21; 39].
In this study, we hypothesize that EDNRB is silenced through promoter methylation in oral SCC and that hypermethylation of this gene contributes to cancer induced pain. To investigate the role of EDNRB methylation in oral SCC pain we use a two-pronged approach. Firstly, to establish the clinical significance of EDNRB silencing on oral SCC pain, we quantify EDNRB promoter methylation in the biopsied tissues of oral dysplasia patients, and tumor and contralateral normal tissue from oral SCC patients. Secondly, we use a mouse oral cancer pain model to determine the behavioral effect of EDNRB re-expression in vivo.
MATERIALS AND METHODS
Tissue collection
All procedures were approved by the University of California, San Francisco Committee on Human Research. We enrolled oral SCC patients with the following inclusion criteria: 1) biopsy-proven oral SCC and 2) no history of prior surgical, chemotherapeutic, or radiation treatment for head and neck SCC. We collected tissue at time of surgery from the primary cancer site and contralateral normal epithelium as control. Samples were flash frozen in liquid nitrogen and stored in −80°C. Demographic and health information were recorded for each patient. Cancer patients were staged according to the American Joint Commission on Cancer tumor-node-metastasis (TNM) staging system [10]. Oral dysplasia tissue was obtained from paraffin-embedded tissues archived from excisional biopsies. Patient demographics and tumor characteristics are shown in Table 1.
Table 1.
| Patient ID | Sex | Age | Tumor Location | Staging | Differentiation Staging |
|---|---|---|---|---|---|
| ORAL SCC PATIENTS | |||||
| 1 | M | 62 | mandible | T4aN0Mx | Well |
| 2 | F | 70 | maxilla | T1N0Mx | Well |
| 3 | F | 62 | tongue | T1N0Mx | Moderate |
| 4 | M | 75 | tongue | T1N0Mx | Well |
| 5 | M | 59 | buccal mucosa | T2N1Mx | Well |
| 6 | M | 60 | mandible | T1N2bMx | Moderate |
| 7 | M | 49 | floor of mouth | T1N2cMx | Moderate |
| 8 | F | 84 | mandible | T3N0Mx | Moderate |
| 9 | M | 56 | tongue | T1N0Mx | Moderate |
| 10 | M | 51 | mandible | T4aN0Mx | Well |
| 11 | M | 59 | mandible | T4aN2bMx | Moderate |
| 12 | F | 84 | maxilla | T2N0Mx | Moderate |
| 13 | F | 76 | mandible | T3N2bMx | Well-Moderate |
| 14 | F | 59 | mandible | T1N1Mx | Poor |
| 15 | F | 93 | maxilla | T4aN0Mx | Well |
| 16 | F | 60 | mandible | T3N2bMx | Moderate |
| 17 | F | 60 | tongue | T2N2bMx | Well |
| 18 | M | 62 | maxilla | T4aN0Mx | Well |
| 19 | F | 66 | tongue | T2N0Mx | Well |
| 20 | F | 71 | tongue | T1N1Mx | Moderate |
| ORAL DYSPLASIA PATIENTS | |||||
| 1 | M | 82 | floor of mouth | Severe | |
| 2 | M | 70 | tongue | Carcinoma in situ | |
| 3 | M | 69 | tongue | Severe | |
| 4 | F | 89 | maxilla | Mild | |
| 5 | M | 50 | tongue | Carcinoma in situ | |
| 6 | M | 59 | retromolar trigone | Carcinoma in situ | |
| 7 | M | 51 | tongue | Carcinoma in situ | |
| 8 | F | 75 | tongue | Mild | |
RNA and DNA extraction
Thirty mg of each tissue collection sample was homogenized with a Mini Beadbeater-1 (BioSpec Products, Bartesville, OK) and subject to RNA/DNA extraction with AllPrep DNA/RNA Kit (Qiagen, Valencia, CA). RNA was eluted in a total volume of 50 μl and DNA was eluted in a total volume of 100 μl. RNA and DNA yield and quality were assessed with spectrometry (Nanodrop Technologies, Wilmington, DE).
Quantitative reverse transcription PCR (RT-PCR) analysis
RNA extracted from fresh frozen paired oral SCC and normal tissues from the 20 patients was converted to cDNA. mRNA was reverse transcribed with Random Hexamers. An 8 μl cDNA aliquot was amplified in 25 μl of 2x TaqMan universal master mix and 2.5 μl of 20x Taqman primer and probe mix (Applied Biosystems, Carlsbad, CA) under the following PCR conditions: 2 minutes at 50°C, 10 minutes at 95°C, 50 cycles of 95°C for 15 seconds and 60°C for 1 minute. The Taqman gene expression assay used for EDNRB was Hs00240747_ml and does not detect residual genomic DNA. Human GUSB (product 4326320E) was used as the endogenous control (Applied Biosystems, Carlsbad, CA). Using GUSB as an internal locus control and the 2−ΔΔCt quantitation method on Microsoft Excel, relative EDNRB transcript levels were obtained. Each sample was measured in duplicate.
Quantitative methylation analysis
Quantitative methylation analysis of the EDNRB promoter was performed through the Genome Analysis Core Facility at the University of California San Francisco, using the EpiTYPER assay (Sequenom, San Diego, CA) in conjunction with the MassARRAY system. The target region was located at chrl3:78492226–78493582 on the antisense strand of the human genome on the UCSC Genome Browser. This target region includes a CpG island of 77 CpG sites and spans from −792 to +451 relative to the EDNRB transcription start site (GenBank entry AY275463.1). At least 1 μg of DNA from each sample was treated with sodium bisulfite using the EZ DNA methylation kit (Zymo Research, Orange, CA) and the converted DNA was amplified by PCR. Primer sequences were designed with EpiDesigner software and are listed in Table 2. The PCR product was treated with shrimp alkaline phosphate and served as the template for transcription according to manufacturer’s instructions. The samples were spotted on a 384-pad Spectro-CHIP and analyzed using a MassARRAY analyzer compact MALDI-TOF MS. Methylation calls were analyzed using EpiTyper software v1.0 to produce quantitative results for each CpG unit, which consists of a single CpG site or aggregate of adjacent CpG sites. Fully methylated DNA was used as positive control and water was used as negative control.
Table 2.
| Name | Primer Sequence | 5′ Position | Amplified Strand |
|---|---|---|---|
| EDNRB-1F | aggaagagagTTATTGATTGAATTTTATTTTGGGG | −792 | Antisense |
| EDNRB-1R | cagtaatacgactcactatagggagaaggctTTAAAACACCTAAAAACCAAAAACTT | −425 | |
| EDNRB-2F | aggaagagagTAGAGGAAGGAAGATAGGATATTTGG | −63 | Sense |
| EDNRB-2R | cagtaatacgactcactatagggagaaggctCAAAACTTAACACAAACCCTTAACC | −463 | |
| EDNRB-3F | aggaagagagTAGTTGAGAGGGTATTAGGAAGGAG | −250 | Antisense |
| EDNRB-3R | cagtaatacgactcactatagggagaaggctTTAAAACCCTTAAACCATAAAATCT | 176 | |
| EDNRB-4F | aggaagagagTGTGTAGTAGGTTTTTTAGAGTTAAGTTGG | 151 | Antisense |
| EDNRB-4R | cagtaatacgactcactatagggagaaggctAAAACCTTATAACCCAAAAATTCCA | 451 |
Recombinant adenovirus and in vitro transduction
A pCMV6-AC-GFP plasmid with GFP-tagged ORF clone of human EDNRB, transcript variant 1, was purchased from Origene (Rockville, MD). Subcloning of the plasmid and viral particle purification were completed through Viraquest (North Liberty, IA). Adenovirus containing only GFP was also obtained from Viraquest for use as a transduction control. The human head and neck cancer cell line derived from human tongue SCC, HSC-3 (ATCC, Manassas, VA), was transduced with recombinant adenovirus (Ad-EDRN or Ad-GFP) at increasing multiplicities of infection (MOI; number of viral particles per cell), at 50, 100, and 200. Transduction was performed in Dulbecco’s Modified Eagle Medium (DMEM) with 4.5 g/L glucose, l-glutamine and sodium pyruvate, supplemented with 2% fetal bovine serum (FBS), 25 μg/mL fungizone, 100 μg/mL streptomycin sulfate, and 100 U/mL penicillin G. Twenty-four hours following transduction, cell media was changed to DMEM containing 10% FBS and the supplements mentioned above. mRNA quantification of transduced cells was performed using RT-PCR.
Immunofluorescence
Immunofluorescence was performed to evaluate the expression of the transgenes. HSC-3 cells were transduced with recombinant adenovirus at increasing multiplicities of infection. Twenty-four hours after transduction they were trypsinized and grown overnight at 37°C on 12-mm glass cover slips, stabilized in a 6-well plate with DMEM containing 10% fetal bovine serum and the aforementioned supplements. The cells were washed twice with PBS, fixed in ice-cold acetone for 5 minutes at room temperature (RT), permeabilized with 0.2% Triton X-100 for 15 minutes, washed three times in PBS then non-specifically blocked with 3% bovine serum albumin (BSA) for 2 hours. Incubation with primary rabbit polyclonal ETB receptor antibody (Abeam Inc., Cambridge, MA) diluted 1:500 in 3% SA was performed at RT for 2 hours followed by incubation with goat anti-rabbit Texas Red-conjugated IgG secondary antibody (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) diluted 1:200 in 3% BSA for 1 hour at RT. Nuclei were stained with 1:500 Hoechst stain (Invitrogen, Carlsbad, CA). Cover slips were washed and mounted on slides in Gel/Mount mounting medium (Biomeda Crop., Foster City, CA) and visualized on the Zeiss Axiolmager 2. Fluorescence quantification of captured images was performed with CellProfiler software. Data were manipulated according to the developer’s instructions.
ELISA measurement of HSC-3 supernatant
ET-1 levels were quantified in the supernatant of nontransduced HSC-3 cells and Ad-EDNRB transduced HSC-3 cells. The samples were prepared as follows. Cells were grown to 60% confluency in 24-well plates and transduced in DMEM with 2% FBS. Twenty-four hours following transduction, transduction media was replaced with 1 ml DMEM containing 10% FBS and supplements. Control nontransduced HSC-3 cells were treated similarly. Cells were incubated for 4 hours at 37°C. Cell supernatant was collected and 10 μl Halt Protease Inhibitor (Thermo Scientific, Rockford, IL) added. Samples were added to the ET-1 ELISA plate (Assay Designs, Plymouth Meeting, PA) and ELISA was performed according to manufacturer’s instructions. To perform the beta-endorphin ELISA, cells were incubated for 1 hour at 37°C. Cell supernatant was collected similarly and added to the beta-endorphin ELISA plate (MD Biosciences, St. Paul, MN). Samples were run in duplicate and measured using a spectrophotometer at recommended wavelengths.
Cancer pain mouse model
The cancer pain mouse model was produced as previously described [29]. Experiments were performed on 4 week-old adult female BALB/c, athymic, immunocompromised mice weighing 16–20 g at the time of human oral SCC inoculation. The mice were housed in a temperature-controlled room on a 12:12 h light cycle (0700–1900 h light), with unrestricted access to food and water. All the procedures were approved by the University of California, San Francisco Committee on Animal Research. Researchers were trained under the Animal Welfare Assurance Program. The mice were divided into three inoculation groups: (1) Ad-EDNRB transduced in HSC-3 (n=6), (2) Ad-GFP transduced in HSC-3 (n=6), or (3) HSC-3 (n=5). Inoculation of HSC-3 cells was performed 48 hours after adenovirus transduction, as preliminary characterization of the adenoviruses showed maximal ETB receptor or GFP expression by 24 hours. Approximately 5 × 106 cells from each group were suspended in a mixture of 35 μl of Matrigel (Becton Dickinson & Co., Franklin Lakes, NJ) and 15 μL DMEM and inoculated into the plantar surface of the right hind paw under isofluorane inhalational anesthesia.
Paw withdrawal and paw volume measurement
Paw withdrawal testing was performed as described previously [29]. Testing was performed by an observer blinded to the experimental groups between 0900 and 1200 h. Mice were placed in a plastic cage with a wire mesh floor which allowed access to the paws. Fifteen minutes were allowed for acclimation prior to testing. The probe was applied to the mid-plantar right hind paw. Paw withdrawal thresholds were determined in response to pressure from an electronic von Frey anesthesiometer (2390 series, IITC Life Sciences, Woodland Hills, CA). The amount of pressure (g) needed to produce a paw withdrawal response was measured six times on each paw separated by 3 min intervals to allow resolution of previous stimuli. The results of the six values were averaged for each paw for that day. Paw volume measurements were performed with a plethysmometer (IITC Life Sciences, Woodland Hills). The paw was inserted into a water cell of which pressure is changed due to the immersion. The pressure change was calibrated in milliliters and shown on an electronic monitor. The measurements were accurate by .001 ml. Triplicate measurements were taken for each mouse. Paw withdrawal and paw volume measurements were made at −2, 0, 4, 7, 9, 11, 13, 15, 18 and 21 days relative to inoculation of HSC-3 cells.
RESULTS
Oral dysplasia and SCC patient demographics
A total of 20 patients with biopsy-proven oral SCC were enrolled in the study. Patient demographics and tumor characteristics are described in Table 1. Nine men and eleven women were enrolled, with an age range of 49–93 and a median age 62. Pathology reports for all patients were reviewed to confirm the diagnosis of oral SCC. Tumor staging was also confirmed. None of the oral SCC patients had a cancer of another histologic type during the period of the study. A total of eight patients with biopsy-proven oral dysplasia were enrolled, with an age range of 50–89 and a median age of 69.5.
Higher EDNRB methylation in human oral SCC than normal tissue
A CpG island of 77 CpG sites in the EDNRB promoter was identified on UCSC Genome Browser. Primers were designed using EpiDesigner software. The primer sets are detailed in Table 2. Taken together, the four primer pairs span the region −792 to 451 relative to the EDNRB transcription start site (GenBank entry AY275463.1). The PCR products of the second and third primer sets overlapped each other by 188 bases. Due to the proximity of many CpG sites, not all CpG sites could be quantified. The four primer sets had the ability to quantify methylation at 61 out of the total 77 CpG sites, resulting in a total coverage of about 80%. The location of quantified sites is detailed in Figure 1.
Figure 1. Heat map of EDNRB methylation.

The heat map signifies quantified methylation values. Within each cell is the methylation value of a patient sample at a particular CpG unit. The values are color coded using a green-red scale, where green is low methylation and red is high methylation. The top heat map shows methylation values of oral SCC tissue. The bottom heat map shows methylation values of contralateral normal tissue. The EDNRB PCR product in which the CpG unit is located is indicated in the gene map. Each PCR product is shown in relation to the EDNRB promoter region and exon 1. Start and end bases for each product are noted; bases are numbered relative to the transcription start site (TSS). PCR product EDNRB 1 surveys the extreme 5′ end of the CpG island. EDNRB 2 and 3 are located proximal to the TSS. They overlap each other by 188 bases. EDNRB 4 is located in exon 1 of the gene. Green circles denote the 61 out of 77 CpG sites whose methylation frequencies were quantified. Grey circles denote the 16 CpG sites that were not quantified.
Methylation was quantified using the MassARRAY System. The result was a methylation value for a CpG unit, which consisted of a single CpG site or aggregate of adjacent CpG sites. Methylation values ranged from 0 to 1, with 1 signifying 100% methylation of the CpG unit in the sample. Figure 1 shows heat maps of methylation values obtained for the CpG units within each EDNRB PCR product. Normal tissue has relatively low baseline methylation. Comparing the different EDNRB PCR products, EDNRB 4 has a higher baseline methylation level in normal tissues than any of the other products. EDNRB 4 is also the only product to be entirely located downstream of the transcription start site, in exon 1.
Comparing the paired oral SCC to normal tissue, oral SCC tissue clearly has higher methylation than contralateral-matched normal tissue in all patients, at almost all CpG units. Out of the 61 CpG sites quantified, the average increase in methylation in oral SCC tissue compared to the corresponding normal tissue was at least 10% (methylation value change 0.1) in 57 sites (93% of sites). The four sites with a methylation value change of lower than 0.1 were CpG 1, 2, 3, and 6 in PCR product EDNRB 4, which starts 151 downstream of the transcription start site. Table 3 summarizes the mean methylation difference between oral SCC and normal tissue at each CpG unit, and the corresponding p-value from the Student’s t-test. Figure 2a shows box plots representing methylation difference values between oral SCC and normal tissue, for each of the four EDNRB PCR products. The difference values were averaged across all patients for each CpG site. The median, 10th, 25th, 75th and 90th percentiles were calculated for each group of CpG sites within a PCR product. PCR product EDNRB 2 had the highest methylation increase from normal to oral SCC tissue, with a median increase of .34 (34%). PCR products EDNRB 1 and 3 had a median increase in methylation of 0.25 (25%). Due to already higher baseline methylation levels of EDNRB 4 in normal tissues, the median increase of methylation in oral SCC tissue was 0.13 (13%). The Mann Whitney Rank Sum Test demonstrated that there was a significant difference between methylation values of oral SCC tissue and the matched normal tissue. Methylation values for each sample were averaged across all CpG sites within each PCR product and oral SCC tissue was compared to normal tissue. There was a significant difference (p<.001) in methylation between cancer and normal tissue in all four PCR products, indicating significant increase in methylation spanning the whole EDNRB promoter in oral SCC.
Table 3.
| EDNRB PCR Product | CpG Site | Mean Methylation Difference | p-value of Student t test |
|---|---|---|---|
| EDNRB 1 | CpG 4 | 0.15 | <.0001 |
| CpG 5 | 0.24 | <.0001 | |
| CpG 6–7 | 0.30 | <.0001 | |
| CpG 8–9 | 0.29 | <.0001 | |
| CpG 10–11–12 | 0.29 | <.0001 | |
| CpG 13 | 0.24 | <.0001 | |
| CpG 14–15 | 0.27 | <.0001 | |
| CpG 16 | 0.17 | <.0001 | |
| CpG 17–18 | 0.20 | <.0001 | |
| CpG 19 | 0.30 | <.0001 | |
| EDNRB 2 | CpG 1–2 | 0.22 | <.0001 |
| CpG 3–4 | 0.51 | <.0001 | |
| CpG 5 | 0.32 | <.0001 | |
| CpG 8 | 0.34 | <.0001 | |
| CpG 9–10 | 0.19 | .0001 | |
| CpG 13–14–15 | 0.36 | <.0001 | |
| CpG 16 | 0.32 | <.0001 | |
| CpG 17 | 0.51 | <.0001 | |
| CpG 18–19 | 0.55 | <.0001 | |
| CpG 20 | 0.36 | <.0001 | |
| CpG 21 | 0.56 | <.0001 | |
| CpG 22–23 | 0.43 | <.0001 | |
| CpG 24 | 0.36 | <.0001 | |
| CpG 25 | 0.39 | <.0001 | |
| CpG 26 | 0.39 | <.0001 | |
| EDNRB 3 | CpG 1 | 0.23 | .0001 |
| CpG 2–3–4 | 0.27 | <.0001 | |
| CpG 5 | 0.21 | <.0001 | |
| CpG 6–7 | 0.37 | <.0001 | |
| CpG 8 | 0.32 | <.0001 | |
| CpG 9 | 0.26 | <.0001 | |
| CpG 10–11 | 0.33 | <.0001 | |
| CpG 12 | 0.28 | <.0001 | |
| CpG 14 | 0.14 | .0002 | |
| CpG 15 | 0.26 | <.0001 | |
| CpG 16 | 0.26 | <.0001 | |
| CpG 18 | 0.19 | <.0001 | |
| CpG 19 | 0.22 | <.0001 | |
| CpG 20 | 0.32 | <.0001 | |
| CpG 27 | 0.19 | <.0001 | |
| CpG 28 | 0.31 | <.0001 | |
| EDNRB 4 | CpG 1 | −0.03 | .4728 |
| CpG 2–3 | 0.02 | .3934 | |
| CpG 4 | 0.10 | .0557 | |
| CpG 6 | 0.16 | .0011 | |
| CpG 7 | 0.08 | .0084 | |
| CpG 8 | 0.10 | .0082 | |
| CpG 9–10 | 0.27 | <.0001 | |
| CpG 12 | 0.32 | <.0001 | |
| CpG 14–15 | 0.26 | <.0001 | |
| CpG 16 | 0.20 | .0003 |
Figure 2.
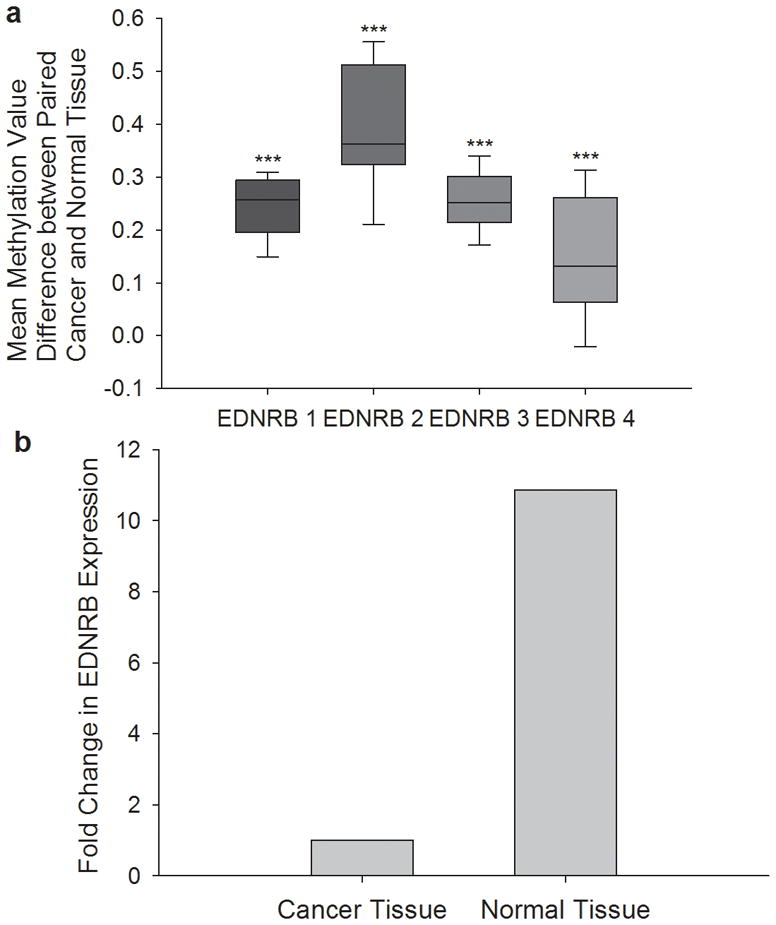
(a) CpG sites have increased methylation in oral SCC tissue compared to normal tissue The box plots represent methylation difference values between oral SCC and normal tissue from the same patient. The difference values are averaged from all patients within each CpG site. All CpG sites within a PCR product are grouped together; the median, 10th, 25th, 75th, and 90th percentiles are shown for each group. PCR product EDNRB 2 has the highest increase in methylation in oral SCC tissue (median = 0.34, 34%). EDNRB 1 and 3 have a median increase of 0.25 (25%). EDNRB 4 has the smallest increase of 0.13 (13%). Methylation values for each sample are averaged across all CpG sites within each PCR product and oral SCC tissue is compared to normal tissue using the Mann Whitney Rank Sum Test (***p<.001). (b) EDNRB mRNA expression is lower in oral SCC than normal tissue. EDNRB mRNA expression in normal and oral SCC tissue was quantified using RT-PCR. GUSB was used as the internal locus control. The relative EDNRB mRNA levels was obtained using delta-delta Ct. When compared to oral SCC tissue, normal contralateral tissue has on average 10.9 times more EDNRB mRNA than oral SCC tissue (Student’s t test, p > .001).
These results show that there are consistently higher methylation levels in oral SCC tissue in 93% of the queried CpG sites in 100% of sample pairs. Furthermore, there is a larger increase in methylation in the first three PCR products, EDRNB 1, 2 and 3, which are located upstream of the transcription start site. There is a modest increase in methylation of CpG sites of EDNRB 4, which is located downstream of the transcription start site. These results suggest that increase in methylation upstream, and not downstream, of the EDNRB transcription start site, has a more significant contribution to carcinogenesis.
Oral SCC patients demonstrate higher EDNRB methylation than oral dysplasia patients
Methylation quantification of EDNRB was performed on paraffin-embedded tissue of eight biopsy-proven oral dysplasia patients. Methylation results were then averaged and compared to the average methylation of oral SCC tissue. EDNRB 2 PCR product was too long and could not be reliably amplified using DNA purified from the paraffin embedded tissue of dysplasia patients. Oral SCC tissue demonstrated higher methylation than oral dysplasia tissues at all queried CpG sites, and were on average 20% higher (range of 6–39%) (Figure 3). There was a significant difference between the methylation values at almost all CpG sites (Mann Whitney Rank Sum Test).
Figure 3. Oral SCC patients demonstrate higher EDNRB methylation than oral dysplasia patients.

The three plots show methylation values of CpG sites from PCR products EDNRB 1,3, and 4, respectively, in oral SCC and oral dysplasia tissues. Oral SCC tissues exhibit an average of 20% higher methylation than oral dysplasia tissues at all CpG sites. The Mann Whitney Rank Sum Test is used to demonstrate significant differences in methylation between oral SCC and dysplasia tissues (*p < .05, **p < .01, ***p < .001).
EDNRB promoter methylation levels are higher in patients with nodal metastasis
We also classified our patients by their N staging in their TNM classification and compared EDNRB methylation among the different N stages. There were eleven patients with N staging of 0, three patients with N staging of 1, and six patients with N staging of 2. Figure 4 is a line and scatter plot graphing the difference in methylation between the oral SCC tissue of a patient and the normal tissue. The patients are categorized into the three possible N staging groups; each point on the graph represents the average of difference in methylation of oral SCC versus normal tissue, of all samples in the group. The plot shows average methylation difference in CpG sites comprised in PCR product EDNRB 1. These sites are the extreme 5′ end of the CpG island in the EDNRB promoter, and are located −792 to −425 upstream of the transcription start site. The plot shows that methylation difference is lowest in the N=0 group, and is highest in the N=2 group. There was a significant difference between the three groups in seven of the ten CpG units, equating to 11 or 16 CpG sites (ANOVA). Furthermore, the N=2 group as significantly different from the N=0 group in all seven CpG units, and the N=l as significantly different from the N=0 group at one CpG unit (p < .05, Holm-Sidak).
Figure 4. EDNRB promoter methylation levels are higher in patients with nodal metastasis.
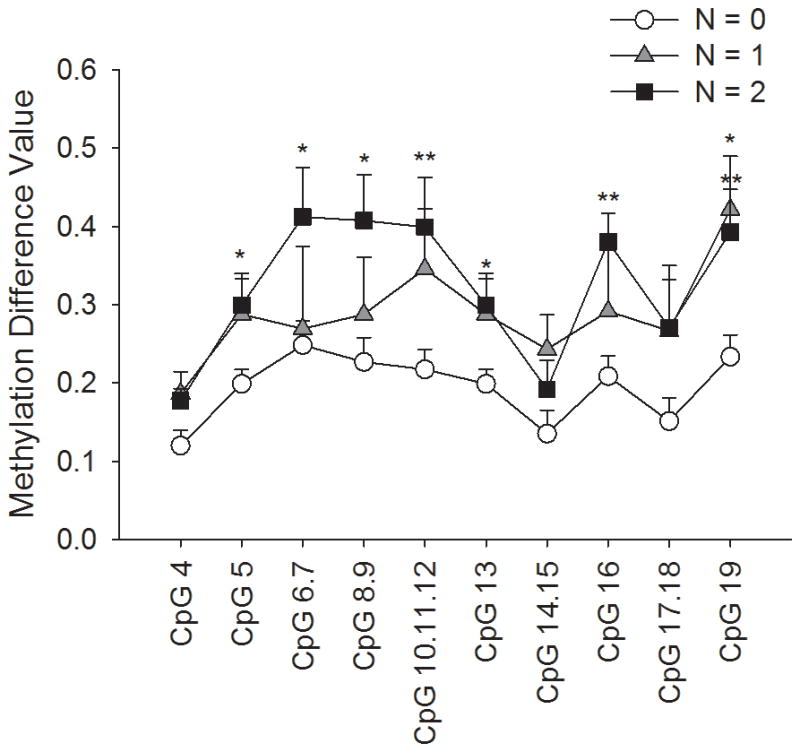
The graph shows three plots of different nodal metastasis levels. Patients were classified by their TNM staging as N=0, N=l, or N=2. The N=0 group had 11 patients, the N=l group had 3 patients, and the N=2 group had 6 patients. The plots represent methylation difference values (methylation value of normal tissue subtracted from that of oral SCC tissue) at each CpG unit in the EDNRB 1 product, which is the extreme 5′ end of the CpG island in the EDNRB promoter. The N=0 group has the lowest methylation value differences out of the three groups. The N=1 has intermediate differences, and the N=2 group has the highest methylation value differences. Using ANOVA and the Holm-Sidak test, we showed that the N=2 group is significantly higher than the N=0 group at 7 of 10 CpG units, and that the N=1 group is significantly different from the N=0 group at one CpG unit (*p < .05, **p <.01).
EDNRB mRNA expression is lower in oral SCC than normal tissue
The fold-change in expression of EDNRB in normal tissue compared to oral SCC tissue in the same patient was calculated. A fold change of more than 1 indicated that EDNRB expression was higher in the normal tissue than in the oral SCC tissue. Of the 20 patients, the average fold change of EDNRB expression was 10.9. Nineteen out of 20 patients (95%) had higher EDNRB expression in normal tissue than oral SCC tissue (range in fold change of 1.15–53.0), indicating downregulation of EDNRB in the oral SCC state. Figure 2b compares EDNRB expression in oral SCC to normal tissue. EDNRB expression in normal tissue is significantly higher than oral SCC tissue (p<0.001, Student’s t-test).
Oral SCC cells transduced with Ad-EDNRB express high levels of EDNRB mRNA and ETB receptor
The EDNRB promoter in human oral cancer tissue is heavily methylated, whereas the EDNRB promoter in normal and dysplastic oral tissues has low levels of methylation. Furthermore, EDNRB mRNA expression was considerably reduced in cancer tissue compared to normal tissues. We wanted to explore the biological role of silenced EDNRB expression in the cancer microenvironment. To do so, we used adenoviruses to overexpress EDNRB in vitro. To overexpress EDNRB we transduced oral SCC cells (HSC-3) with adenovirus containing the EDNRB gene. HSC-3 has nondetectable levels of EDNRB mRNA and, when methylation levels of the EDNRB promoter in this cell line were quantified using MassARRAY, an average methylation level for all CpG sites was 85%. Our goal was to re-express EDNRB and determine the effect on the cancer microenvironment in vitro.
To confirm overexpression subsequent to transduction, immunofluorescence with EDNRB antibody was performed. ETB receptor expression in oral SCC cells was minimal, while transduction of Ad-EDNRB resulted in a marked increase in ETB receptor membrane expression. Figure 5 shows images from cells transduced at 200 MOI, as 50 MOI and 100 MOI show similar trends.
Figure 5. Immunofluorescence images of Ad-EDNRB transduced and nontransduced oral SCC cells.
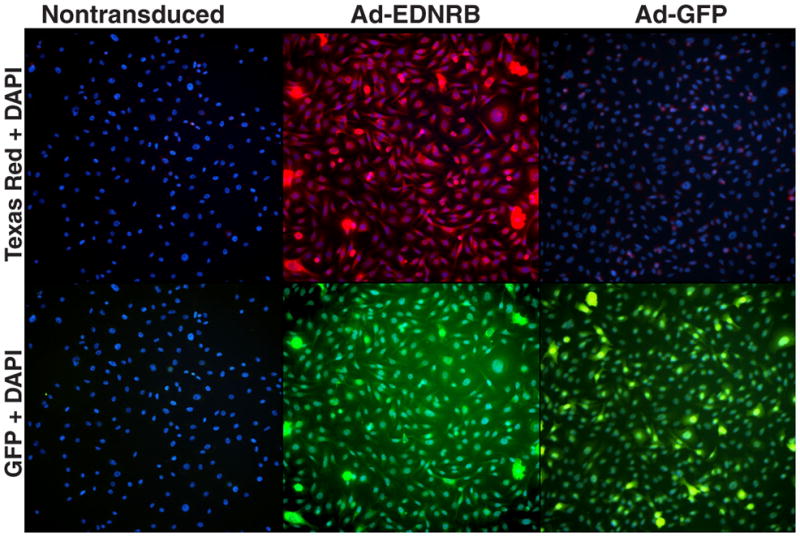
Images are shown at 20X magnification. Images are taken from a single exposure setting. The top row of images shows Texas Red and Hoechst staining. The bottom row of images shows GFP and Hoechst staining. ETB receptor antibody was secondarily tagged with Texas Red and cell nuclei were counterstained with Hoechst. Nontransduced and Ad-GFP transduced HSC-3 cells display minimal ETB receptor expression, whereas Ad-EDNRB transduced HSC-3 cells exhibit strong ETB receptor expression. Ad-GFP transduced HSC-3 cells serve as the positive control for transduction. Ad-GFP transduced HSC-3 cells show strong GFP expression. Ad-EDNRB transduced HSC-3 cells also show GFP expression due to a C-terminal GFP tag on the ETB receptor protein.
RT-PCR was performed on mRNA extracted from nontransduced oral SCC cells and oral SCC cells transduced with Ad-EDNRB at increasing MOI. RT-PCR results shown in figure 6a illustrate a dose-dependent increase in EDNRB mRNA expression with increasing MOI. Oral SCC cells transduced with Ad-EDNRB at 200 MOI express 3 × 107 more mRNA than nontransduced oral SCC.
Figure 6.
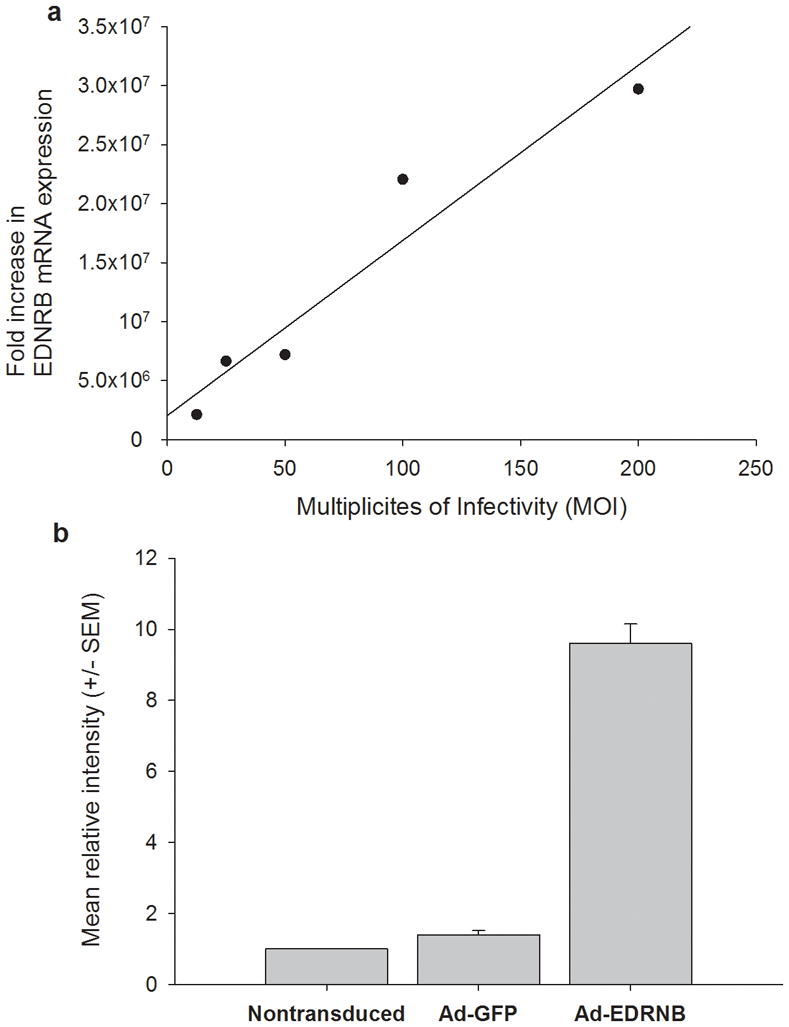
(a) Oral SCC cells show dose-dependent increase in EDNRB mRNA expression in response to transduction with Ad-EDNRB HSC-3 cells are transduced at 12.5, 25, 50, 100, and 200 MOI. RT-PCR performed on each group reveals that there is a dose-dependent increase in EDNRB mRNA expression with increasing MOI. Fold change is calculated relative to nontransduced HSC-3. (b) Ad-EDNRB transduced oral SCC cells express high levels of ETB receptor compared to control oral SCC cells.
Immunofluorescence images are quantified using Cell Profiler software. ETB receptor expression is quantified, and relative expression is calculated based on nontransduced HSC-3 expression. Cells tranduced with Ad-GFP do not express high levels of ETB receptor. However, cells transduced with Ad-EDRN express 9.6 times more ETB receptor than control, nontransduced cells.
Figure 6b illustrates relative ETB receptor expression in the three groups of oral SCC cells: nontransduced, Ad-GFP and Ad-EDNRB transduced at 200 MOI. ETB receptor expression was quantified relative to nontransduced oral SCC cells, which have a relative expression of 1. While Ad-GFP transduced cells do not exhibit a difference in ETB receptor expression, those transduced with Ad-EDNRB express 9.6 times more ETB receptor than control oral SCC cells.
Oral SCC cells transduced with Ad-EDNRB secrete lower levels of ET-1 and higher levels of beta-endorphin
We have previously shown that oral SCC cells secrete very high levels of ET-1 [29]. We wanted to determine the effect of re-expressing ETB receptor on ET-1 levels in oral SCC cells. We transduced oral SCC cells with Ad-EDNRB and then performed ELISA to measure ET-1 in the supernatant. Our results showed that oral SCC cells transduced with Ad-EDNRB secreted lower levels of ET-1 than nontransduced oral SCC. Supernatant of oral SCC cells transduced with Ad-EDNRB at 200 MOI had an average ET-1 concentration of 9.2 pg/l. Supernatant of control oral SCC cells, which were transduced with Ad-GFP, had an average ET-1 concentration of 37.3 pg/l.
Conversely, oral SCC cells transduced with Ad-EDNRB secreted more beta-endorphin than control. Supernatant beta-endorphin concentration of Ad-EDNRB-transduced oral SCC cells was 0.94 ng/ml. Control cells, which were transduced with Ad-GFP, secreted an average of 0.66 ng/ml. Each sample was run in duplicate, so statistical significance was not assessed.
Transduction of Ad-EDNRB decreases nociception without affecting tumor size in a cancer mouse model
Human cancer cell proliferation and cancer-induced mechanical allodynia were assessed in the three orthotopic mouse model groups: (1) Ad-EDNRB-transduced HSC-3 tumors; (2) Ad-GFP-transduced HSC-3 tumors; and (3) non-transduced HSC-3 tumors. The results up to 21 days after inoculation of HSC-3 tumors showed that the three groups had similar paw volume, indicating that overexpressing EDNRB did not significantly alter tumor proliferation (Figure 7a). Overexpression of EDNRB did, however, affect cancer-induced mechanical allodynia. Cancers expressing EDNRB displayed a significantly higher paw withdrawal threshold than either the nontransduced cancer or the cancer expressing GFP. Figure 7b shows that mice with cancers expressing EDNRB have paw withdrawal thresholds that stabilized at 48% below baseline, regardless of the fact that tumor size was increasing. The average paw withdrawal threshold of these mice on day 21 was 2.12 g, approximately 48% of the day 0 threshold of 4.09 g. The Ad-GFP transduced HSC-3 cancers and non-transduced HSC-3 cancer groups showed a 65% (day 0 average = 4.06g; day 21 average = 1.41 g) and 67% (day 0 average = 3.90g; day 21 average = 1.29g) reduction in paw withdrawal threshold, respectively. The baseline paw withdrawal of the three groups was not significantly different from each other. The paw withdrawal threshold of tumors overexpressing EDNRB was significantly higher than the control groups, indicating lower mechanical allodynia (Student’s t test, p-values as indicated in Figure 7b).
Figure 7.

(a) Ad-EDNRB transduction does not alter oral SCC tumor growth The graph shows change in paw volume, which is a measure of tumor growth. The X axis shows the days of the experiment, and the Y axis shows the average change in paw volume. All three study groups show a similar increase in paw volume as the experiment progresses. There is no statistically significant difference in paw volumes among the study groups. Standard error bars are shown, (b) Ad-EDNRB transduction decreases mechanical allodynia. The graph shows change in paw withdrawal threshold, which is a measure of mechanical allodynia. Mice with tumors that have been transduced with Ad-EDNRB exhibit significantly higher paw withdrawal thresholds, indicating lower mechanical allodynia. ANOVA and Holm-Sidak tests are used (*p < .05,**p < .01, ***p< .001). Standard error bars are shown.
DISCUSSION
In this study we demonstrate that methylation of EDNRB is a novel mechanism generating cancer pain and expression of the ETB receptor reverses cancer pain. Evidence supporting this result includes our finding that EDNRB is heavily methylated in painful human oral cancer specimens compared to matched normal controls and oral dysplasia, which is typically not painful. In addition, oral cancer specimens express less EDNRB mRNA than matched normal controls, indicating a correlation between increased EDNRB methylation and decreased mRNA expression. Using a cancer pain mouse model we show that re-expression of ETB receptors using adenoviruses leads to significant attenuation of cancer pain behavior. This study is the first to demonstrate that gene methylation could be a significant mechanism causing cancer-induced pain, and that reversing the gene silencing process with targeted therapy could produce analgesia.
Oral cancer is notoriously painful. The pain is consistent with mechanical allodynia and severely limits function [6; 19]. We used resected cancer specimens and anatomically matched normal oral tissue from the same patients to evaluate EDNRB methylation. This approach allows for an accurate analysis of methylation levels associated with carcinogenesis. We quantified methylation in the promoter region of EDNRB spanning 77 CpG sites and 1.2 kilobases, which to date represents the most comprehensive methylation panel of EDNRB in normal, dysplastic, and cancer tissue. Oral cancer patients demonstrated higher methylation of EDNRB than oral dysplasia patients at all queried CpG sites. In our recent study we quantified pain in our cohort of oral dysplasia and oral cancer patients using the validated UCSF Oral Cancer Pain Questionnaire [20]. We demonstrated that only oral cancer patients, and not dysplasia patients, reported significant levels of spontaneous and function-related pain, with function-related pain being markedly pronounced. Additionally, when we compared methylation frequency in patients with and without nodal metastasis, patients with an N staging of 2 showed significantly higher methylation than patients with an N staging of 0 in the 5′ end of the EDNRB promoter. These results correlate with our previous finding that oral cancers that are metastatic are more painful [6].
Next, we wanted to understand the biologic significance of EDNRB expression on cancer-induced pain. Cancer pain has been hypothesized to result from either a tumor-mass effect or the activation of primary afferent nociceptors by mediators liberated by the cancer [3; 8; 9; 22; 25]. The tumor-mass effect is unlikely because even small carcinomas are painful [6]. We hypothesized that cancer pain results from an imbalance in pain-producing and pain-relieving mediators within the cancer microenvironment. The endothelin axis provides a compelling example of this imbalance. We have previously shown that for the human oral SCC cell line, the mRNA for ET-1 is nearly doubled, while mRNA for ETB receptors is down-regulated compared to normal oral keratinocytes [33]. Similarly in lung cancer, ETB receptors are down-regulated via promoter methylation [18]. The ETB receptors could mediate analgesia, attenuating the acute pain generated by ET-1 [16; 31]. This counterbalancing, pain-relieving effect could be lost in cancers because ETB receptors are not expressed. We therefore proposed that if the promoter for ETB receptors could be re-expressed then the receptor could be activated by ET-1. To test this hypothesis we developed a cancer mouse model in which ETB receptors were re-expressed. Since there is currently no reliable method to demethylate a specific gene and activate its transcription, adenovirus transduction was the most feasible method of re-expression. Our results showed that mice with oral SCC tumors expressing EDNRB had significantly lower levels of mechanical allodynia than mice with oral SCC tumors that did not express EDNRB.
ETB receptor expression leads to antinociception through two likely mechanisms: 1) activation of ETB receptor, which activates an endogenous analgesic mechanism, and 2) reduced ET-1 secretion by the carcinoma. The endogenous analgesic mechanism by ETB receptor activation was first demonstrated by Khodorova et al., who also showed that the analgesic cascade involved release of beta-endorphin [17]. We demonstrated in this study that re-expression of ETB receptor resulted in increased secretion of beta-endorphin into the oral SCC supernatant. These results correspond to our earlier results where treatment of oral SCC cells with an ETB receptor agonist caused increased secretion of beta-endorphin compared to control cells [33]. Support for the second analgesic mechanism comes from our finding that ET-1 levels in the cell supernatant were significantly lower in oral SCC re-expressing the ETB receptor. ETB receptors have a known role in ET-1 clearance and inhibiting ET-1 secretion in keratinocytes [26; 38]. Our previous studies in a mouse oral SCC model have shown that ET-1 protein and mRNA concentrations are markedly elevated in oral SCC tumors. These elevated ET-1 levels caused increased pain by activation of the ETA receptor. Not only is ET-1 elevated in numerous cancer types, our studies on salivary biomarkers have shown that ET-1 is significantly elevated in saliva of patients with oral SCC compared to normal subjects [30]. High ET-1 concentration activates ETA and downstream networks resulting in nociception. We also demonstrated that melanoma tumors of equal size to SCC tumors produced significantly less pain, since they had a lower ET-1 concentration. Therefore, cancer pain intensity depends more on ET-1 concentration in that tumor than tumor size itself [29]. From these cumulative findings we propose that increased concentration of ET-1 activates ETA and subsequent nociceptive pathways, and re-expression of the ETB receptor produces an analgesic effect by counter-regulating ET-1/ETA nociceptive mechanisms.
For most cancer patients, uncontrollable pain creates a poor quality of life [5; 6; 11; 12]. Because improved chemotherapy and radiotherapy prolong survival, chronic cancer pain is a major public health problem. To date there is no effective treatment for chronic cancer pain. The role of gene methylation in cancer pain has not been previously demonstrated. The findings from this study establish gene methylation as a novel regulatory mechanism of cancer-induced pain. Furthermore, the findings suggest that targeted gene demethylation could serve as a novel analgesic therapy to chronic cancer pain. Such therapy targeted to the cancer microenvironment like we had demonstrated in this study could antagonize pain while minimizing systemic drug toxicity and solve the current public health problem of chronic cancer pain refractory to traditional therapeutics.
Acknowledgments
This work was supported by National Institutes of Health grant R21 DE018561.
Footnotes
Conflict of Interest Statement: The authors have no conflict of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baamonde A, Lastra A, Villazon M, Bordallo J, Hidalgo A, Menendez L. Involvement of endogenous endothelins in thermal and mechanical inflammatory hyperalgesia in mice. Naunyn Schmiedebergs Arch Pharmacol. 2004;369(2):245–251. doi: 10.1007/s00210-003-0841-1. [DOI] [PubMed] [Google Scholar]
- 2.Bagnato A, Tecce R, Di Castro V, Catt KJ. Activation of mitogenic signaling by endothelin 1 in ovarian carcinoma cells. Cancer Res. 1997;57(7):1306–1311. [PubMed] [Google Scholar]
- 3.Bjordal K, Kaasa S. Psychological distress in head and neck cancer patients 7–11 years after curative treatment. Br J Cancer. 1995;71 (3):592–597. doi: 10.1038/bjc.1995.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carducci MA, Jimeno A. Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin Cancer Res. 2006;12(20 Pt 2):6296s–6300s. doi: 10.1158/1078-0432.CCR-06-0929. [DOI] [PubMed] [Google Scholar]
- 5.Chaplin JM, Morton RP. A prospective, longitudinal study of pain in head and neck cancer patients. Head Neck. 1999;21 (6):531–537. doi: 10.1002/(sici)1097-0347(199909)21:6<531::aid-hed6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 6.Connelly ST, Schmidt L. Evaluation of pain in patients with oral squamous cell carcinoma. J Pain. 2004;5(9):505–510. doi: 10.1016/j.jpain.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Davar G, Hans G, Fareed MU, Sinnott C, Strichartz G. Behavioral signs of acute pain produced by application of endothelin-1 to rat sciatic nerve. Neuroreport. 1998;9(10):2279–2283. doi: 10.1097/00001756-199807130-00025. [DOI] [PubMed] [Google Scholar]
- 8.Diener KM. Bisphosphonates for controlling pain from metastatic bone disease. Am J Health Syst Pharm. 1996;53(16):1917–1927. doi: 10.1093/ajhp/53.16.1917. [DOI] [PubMed] [Google Scholar]
- 9.Fujita T, Matsui M, Takaku K, Uetake H, Ichikawa W, Taketo MM, Sugihara K. Size- and invasion-dependent increase in cyclooxygenase 2 levels in human colorectal carcinomas. Cancer Res. 1998;58(21):4823–4826. [PubMed] [Google Scholar]
- 10.Greene FL American Cancer Society. American Joint Committee on Cancer. |. Title |, Vol. Volume |. City |: Publisher |, Year |. [Google Scholar]
- 11.Hammerlid E, Bjordal K, Ahlner-Elmqvist M, Boysen M, Evensen JF, Biorklund A, Jannert M, Kaasa S, Sullivan M, Westin T. A prospective study of quality of life in head and neck cancer patients. Part I: at diagnosis. Laryngoscope. 2001;111 (4 Pt l):669–680. doi: 10.1097/00005537-200104000-00021. [DOI] [PubMed] [Google Scholar]
- 12.Hodder SC, Edwards MJ, Brickley MR, Shepherd JP. Multiattribute utility assessment of outcomes of treatment for head and neck cancer. Br J Cancer. 1997;75(6):898–902. doi: 10.1038/bjc.1997.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honore P, Luger NM, Sabino MA, Schwei MJ, Rogers SD, Mach D, O’Keefe PF, Ramnaraine ML, Clohisy DR, Mantyh PW. Osteoprotegerin blocks bone cancer-induced skeletal destruction, skeletal pain and pain-related neurochemical reorganization of the spinal cord. Nat Med. 2000;6(5):521–528. doi: 10.1038/74999. [DOI] [PubMed] [Google Scholar]
- 14.Jarvis MF, Wessale JL, Zhu CZ, Lynch JJ, Dayton BD, Calzadilla SV, Padley RJ, Opgenorth TJ, Kowaluk EA. ABT-627, an endothelin ET(A) receptor-selective antagonist, attenuates tactile allodynia in a diabetic rat model of neuropathic pain. Eur J Pharmacol. 2000;388(1):29–35. doi: 10.1016/s0014-2999(99)00865-1. [DOI] [PubMed] [Google Scholar]
- 15.Jeronimo C, Henrique R, Campos PF, Oliveira J, Caballero OL, Lopes C, Sidransky D. Endothelin B receptor gene hypermethylation in prostate adenocarcinoma. J Clin Pathol. 2003;56(1):52–55. doi: 10.1136/jcp.56.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khodorova A, Fareed MU, Gokin A, Strichartz GR, Davar G. Local injection of a selective endothelin-B receptor agonist inhibits endothelin-1-induced pain-like behavior and excitation of nociceptors in a naloxone-sensitive manner. J Neurosci. 2002;22(17):7788–7796. doi: 10.1523/JNEUROSCI.22-17-07788.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khodorova A, Navarro B, Jouaville LS, Murphy JE, Rice FL, Mazurkiewicz JE, Long-Woodward D, Stoffel M, Strichartz GR, Yukhananov R, Davar G. Endothelin-B receptor activation triggers an endogenous analgesic cascade at sites of peripheral injury. Nat Med. 2003;9(8):1055–1061. doi: 10.1038/nm885. [DOI] [PubMed] [Google Scholar]
- 18.Knight LJ, Burrage J, Bujac SR, Haggerty C, Graham A, Gibson NJ, Ellison G, Growcott JW, Brooks AN, Hughes AM, Xinarianos G, Nikolaidis G, Field JK, Liloglou T. Epigenetic silencing of the endothelin-B receptor gene in non-small cell lung cancer. Int J Oncol. 2009;34(2):465–471. [PubMed] [Google Scholar]
- 19.Kolokythas A, Connelly ST, Schmidt BL. Validation of the university of California san francisco oral cancer pain questionnaire. J Pain. 2007;8(12):950–953. doi: 10.1016/j.jpain.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam D, Schmidt BL. Orofacial pain onset predicts transition to head and neck cancer. Pain. 2011 doi: 10.1016/j.pain.2011.02.009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo KW, Tsang YS, Kwong J, To KF, Teo PM, Huang DP. Promoter hypermethylation of the EDNRB gene in nasopharyngeal carcinoma. Int J Cancer. 2002;98(5):651–655. doi: 10.1002/ijc.10271. [DOI] [PubMed] [Google Scholar]
- 22.Mantyh PW, Clohisy DR, Koltzenburg M, Hunt SP. Molecular mechanisms of cancer pain. Nature Rev Cancer. 2002;2(3):201–209. doi: 10.1038/nrc747. [DOI] [PubMed] [Google Scholar]
- 23.Mercadante S. Malignant bone pain: pathophysiology and treatment. Pain. 1997;69(1–2):1–18. doi: 10.1016/s0304-3959(96)03267-8. [DOI] [PubMed] [Google Scholar]
- 24.Mercadante S, Dardanoni G, Salvaggio L, Armata MG, Agnello A. Monitoring of opioid therapy in advanced cancer pain patients. J Pain Symptom Manage. 1997;13(4):204–212. doi: 10.1016/s0885-3924(96)00302-8. [DOI] [PubMed] [Google Scholar]
- 25.Morton RP. Life-satisfaction in patients with head and neck cancer. Clin Otolaryngol. 1995;20(6):499–503. doi: 10.1111/j.1365-2273.1995.tb01588.x. [DOI] [PubMed] [Google Scholar]
- 26.Nelson JB, Carducci MA. The role of endothelin-1 and endothelin receptor antagonists in prostate cancer. BJU Int. 2000;85 (Suppl 2):45–48. doi: 10.1046/j.1464-410x.2000.00063.x. [DOI] [PubMed] [Google Scholar]
- 27.Parkin DM, Pisani P, Ferlay J. Global cancer statistics. CA Cancer J Clin. 1999;49(l):33–64. 31. doi: 10.3322/canjclin.49.1.33. [DOI] [PubMed] [Google Scholar]
- 28.Peters CM, Lindsay TH, Pomonis JD, Luger NM, Ghilardi JR, Sevcik MA, Mantyh PW. Endothelin and the tumorigenic component of bone cancer pain. Neuroscience. 2004;126(4):1043–1052. doi: 10.1016/j.neuroscience.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 29.Pickering V, Jay Gupta R, Quang P, Jordan RC, Schmidt BL. Effect of peripheral endothelin-1 concentration on carcinoma-induced pain in mice. Eur J Pain. 2008;12(3):293–300. doi: 10.1016/j.ejpain.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pickering V, Jordan RC, Schmidt BL. Elevated salivary endothelin levels in oral cancer patients--a pilot study. Oral Oncol. 2007;43(1):37–41. doi: 10.1016/j.oraloncology.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 31.Piovezan AP, D’Orleans-Juste P, Souza GE, Rae GA. Endothelin-1-induced ET(A) receptor-mediated nociception, hyperalgesia and oedema in the mouse hind-paw: modulation by simultaneous ET(B) receptor activation. Br J Pharmacol. 2000;129(5):961–968. doi: 10.1038/sj.bjp.0703154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quang PN, Schmidt BL. Endothelin-A receptor antagonism attenuates carcinoma-induced pain through opioids in mice. J Pain. 2010;11(7):663–671. doi: 10.1016/j.jpain.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quang PN, Schmidt BL. Peripheral endothelin B receptor agonist-induced antinociception involves endogenous opioids in mice. Pain. 2010;149(2):254–262. doi: 10.1016/j.pain.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosano L, Salani D, Di Castro V, Spinella F, Natali PG, Bagnato A. Endothelin-1 promotes proteolytic activity of ovarian carcinoma. Clin Sci (Lond) 2002;103 (Suppl 48):306S–309S. doi: 10.1042/CS103S306S. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt BL, Pickering V, Liu S, Quang P, Dolan J, Connelly ST, Jordan RC. Peripheral endothelin A receptor antagonism attenuates carcinoma-induced pain. Eur J Pain. 2007;11(4):406–414. doi: 10.1016/j.ejpain.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 36.Silverman SJ. Epidemiology. D.C: Decker Inc; 1998. [Google Scholar]
- 37.Wacnik PW, Eikmeier LJ, Ruggles TR, Ramnaraine ML, Walcheck BK, Beitz AJ, Wilcox GL. Functional interactions between tumor and peripheral nerve: morphology, algogen identification, and behavioral characterization of a new murine model of cancer pain. J Neurosci. 2001;21 (23):9355–9366. doi: 10.1523/JNEUROSCI.21-23-09355.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yohn JJ, Smith C, Stevens T, Morelli JG, Shurnas LR, Walchak SJ, Hoffman TA, Kelley KK, Escobedo-Morse A, Yanagisawa M, et al. Autoregulation of endothelin-1 secretion by cultured human keratinocytes via the endothelin B receptor. Biochim Biophys Acta. 1994;1224(3):454–458. doi: 10.1016/0167-4889(94)90282-8. [DOI] [PubMed] [Google Scholar]
- 39.Zhao BJ, Sun DG, Zhang M, Tan SN, Ma X. Identification of aberrant promoter methylation of EDNRB gene in esophageal squamous cell carcinoma. Dis Esophagus. 2009;22(1):55–61. doi: 10.1111/j.1442-2050.2008.00848.x. [DOI] [PubMed] [Google Scholar]