

Summary
Innate lymphoid cells (ILCs) have emerged as important players regulating the balance between protective immunity and immunopathology at mucosal surfaces. However, mechanisms that regulate ILCs effector functions during mucosal pathogenic challenge are poorly defined. Using mice infected with the natural mouse enteric pathogen Citrobacter rodentium, we demonstrate that lymphotoxin (LT) is essential for IL-22 production by intestinal ILCs. Blocking of LTβR signaling dramatically reduced intestinal IL-22 production after C. rodentium infection. Conversely, stimulating LTβR signaling induced IL-22 protection pathway in LT-deficient mice. Furthermore, exogenous IL-22 expression rescued LTβR deficient mice. IL-22 producing ILCs were predominantly located in lymphoid follicles in the colon, and interacted closely with dendritic cells. Finally, we find that an LT-driven positive feedback loop controls IL-22 production by RORγt+ ILCs via LTβR signaling in dendritic cells. Altogether, we show that LTβR signaling in gut lymphoid follicles regulates IL-22 production by ILCs in response to mucosal pathogen challenge.
Introduction
Innate lymphoid cells (ILCs) represent a heterogeneous population of innate lymphocytes in the mucosa, that participate in regulation of mucosal immune homeostasis (Colonna, 2009; Sawa et al., 2010; Spits and Di Santo, 2011). Although ILCs do not express antigen specific receptors like adaptive lymphocytes, they have the capacity to produce several cytokines upon stimulation that function to regulate the balance between protective immunity and destructive inflammation in the gut. However, the pathways that mediate crosstalk between ILCs and intestinal epithelial cells in response to mucosal bacterial infection remain poorly understood. Citrobacter rodentium (C. rodentium) is a natural mouse extracellular enteric pathogen that mimics human enteropathogenic Escherichia coli and enterohaemorrhagic Escherichia coli infections (Mundy et al., 2005). C. rodentium uses attaching and effacing lesion formation as a major mechanism to target and infect the intestinal epithelial layer, and therefore represents an excellent model to define the role of ILCs in intestinal immune homeostasis.
Lymphoid tissue inducer cells (LTi) belong to ILCs, and are critical for development of secondary lymphoid tissues during fetal development (Eberl et al., 2004; Randall et al., 2008; Roozendaal and Mebius, 2011; Spits and Di Santo, 2011). However, their role in regulation of mucosal immune responses in the gut is poorly defined. Recently, a CD4+ population of LTi cells within the intraepithelial lymphocyte compartment has been implicated in controlling mucosal bacterial infection (Sonnenberg et al., 2011b). Another ILC subset that expressed the NK cell marker NKp46 and is located in small and large intestine lamina propria has also been suggested to control C. rodentium infection (Cella et al., 2009; Satoh-Takayama et al., 2008). Both the intraepithelial LTi cells and the lamina propria NKp46+ cells, as well as other ILCs in the gut, express the transcriptional factor nuclear hormone receptor retinoic acid receptor-related orphan receptor gamma t (RORγt) for their development (Colonna, 2009; Eberl et al., 2004; Spits and Di Santo, 2011).
Recent studies have suggested that development of LTi cells and mucosal NKp46+ cells is distinct from classical NK cells (Colonna, 2009; Spits and Di Santo, 2011). The developmental relationship between NKp46+ and LTi cells remains an active area of research. Several studies suggested that RORγt+NKp46+ cells can originate from RORγt+ LTi cells (Cupedo et al., 2009; Vonarbourg et al., 2010). However, another report indicated distinct developmental programs of these populations, originally generated from a common fetal liver progenitor (Sawa et al., 2010). In contrast to LTi cells, NKp46+ cells require commensal microflora for their development (Sanos et al., 2009; Satoh-Takayama et al., 2008). However, this requirement has not been confirmed by other studies (Sawa et al., 2010; Sawa et al., 2011). Additionally, inactivation of the Ncr1 gene (encoding NKp46) did not impact susceptibility to C. rodentium infection (Satoh-Takayama et al., 2009). Hence, the underlying function of mucosal NKp46+ cells in innate immune response is currently unclear.
The major functions of ILCs in mucosal immunity against C. rodentium infection as well as epithelial tissue repair are potentially mediated by the production of the cytokine, IL-22 (Cella et al., 2009; Luci et al., 2009; Sanos et al., 2009; Satoh-Takayama et al., 2008). IL-22 is a recently discovered cytokine of the extended IL-10 family that plays multiple roles in the regulation of mucosal immunity (Ouyang, 2010; Ouyang et al., 2011; Sonnenberg et al., 2011a). IL-22 signals through the IL-22R that is selectively expressed by intestinal epithelial cells. IL-22 can induce secretion of antimicrobial proteins from these epithelial cells, including RegIIIγ and RegIIIβ, to kill C. rodentium (Zheng et al., 2008). Accordingly, IL-22-deficient mice show increased morbidity and mortality after C. rodentium infection (Zheng et al., 2008). IL-23 has been shown to promote IL-22 production (Sonnenberg et al., 2011b; Zheng et al., 2008). Furthermore, IL-23-deficient mice display reduced IL-22 levels and succumb to C. rodentium infection (Zheng et al., 2008). However, other potential regulators of IL-22 production by ILCs remain poorly understood.
Lymphotoxin (LT) is a member of the TNF core family and plays a critical role in regulation of mucosal immune responses (Fu and Chaplin, 1999; Tumanov et al., 2007; Ware, 2005). The biologically active form of surface LT presents as a heterotrimeric complex (LTβ2LTα1) that interacts specifically with LTβR. While LT is expressed mostly by lymphocytes, including T, B, NK, and LTi cells, LTβR is expressed on stromal, epithelial, DC, and myeloid cells, but not on lymphocytes (Fu and Chaplin, 1999; Tumanov et al., 2007; Ware, 2005). LTβR signaling in DCs regulates DC homeostasis (Kabashima et al., 2005; Summers Deluca and Gommerman, 2011). Evidence of cross-talk between LT- expressing T cells and LTβR- bearing DC promotes T cell responses to a model antigen in mice (Summers-DeLuca et al., 2007; Summers Deluca and Gommerman, 2011). However, the role of LT in cross-talk between lymphocytes and DCs in host defense and autoimmune diseases remains to be fully elucidated. LTβR signaling plays a critical role in host defense against C. rodentium infection (Spahn et al., 2004), as all LTβR-deficient mice succumb early to C. rodentium infection (Spahn et al., 2004; Wang et al., 2010). In the present study, we explore the relationship between IL-22 and the LTβR pathway and define that LTβR signaling controls the IL-22 protective pathway in the gut via coordination of RORγt+ ILCs with DCs in lymphoid follicles.
Results
LTβR signaling controls innate IL-22 pathway in the gut
IL-22 is essential for control of early C. rodentium infection (Zheng et al., 2008). Since LTβR-deficient mice also succumb early to C. rodentium infection, we speculated that LTβR signaling might regulate IL-22 production. To test this hypothesis, we measured IL-22 levels in the colon of WT and LTβR-deficient mice early after C. rodentium infection. Compared to WT mice, expression of IL-22 was dramatically reduced in colon of Ltbr-/- mice (Figure 1A). mRNA expression of IL-22 dependent antimicrobial proteins RegIIIγ and RegIIIβ was also reduced in colon of Ltbr-/- mice at day 5 post-infection (Figure 1A). In contrast to infected mice, basal IL-22 levels were comparable in the colon of naïve WT and Ltbr-/- mice (Figure 1A). These results suggest the critical role of LTβR signaling to control IL-22 production in the gut in response to mucosal bacterial infection.
Figure 1. LTβR signaling controls innate IL-22 pathway in the gut.

A. IL-22 levels in the colon of naïve and infected WT and Ltbr-/- mice. LTbr-/- and WT mice (n=5/group/experiment) were orally inoculated with C. rodentium. mRNA expression of IL-22 and IL-22- dependent RegIIIγ and RegIIIβ antimicrobial proteins in the colon were measured by real-time PCR on day 5 post infection. Naïve WT mice were used as control. B. WT mice were treated with 150 μg of LTβR-Ig, or control human Ig (hIg) at day 0 and 3 post infection. Expression of IL-22 and RegIIIγ and RegIIIβ antimicrobial proteins were measured in colon by real-time PCR at day 5 post infection. C. Rag1-/- mice were treated intraperitoneally with 150 μg of LTβR-Ig at day 0 and 3 post infection, and expression of IL-22, and RegIIIγ and RegIIIβ antimicrobial proteins was measured by real-time PCR at day 5 post infection. A-C. Data represent means ± s.e.m. n=5, *p<0.05, **p<0.01. Data represent one of three independent experiments with similar results. Real-time PCR data were normalized to hprt expression.
LTβR-deficient mice display multiple immune abnormalities, including lack of lymph nodes and gut-associated lymphoid tissues, defects in DC and NK cell development and IgA production, and defects in central tolerance (Zhu et al., 2010). Therefore, in order to exclude developmental defects in Ltbr-/- mice that might affect IL-22 production in the gut, we treated WT mice early after infection with soluble LTβR-Ig fusion protein that transiently blocks LTβR signaling. WT mice treated with LTβR-Ig at days 1 and 3 after infection showed severe pathology and around 50% of LTβR-Ig treated mice died after acute infection, consistent with previous reports (Wang et al., 2010). Remarkably, expression of IL-22 and IL-22-dependent antimicrobial proteins was severely reduced in the colon of LTβR-Ig treated mice at day 5 post-infection, compared to control mice (Figure 1B). These results suggest that LTβR signaling at the time of infection is required for IL-22 induction in the gut.
To further define whether LTβR signaling regulates innate mechanisms of IL-22 production, we treated Rag1-deficient mice with LTβR-Ig. This treatment reduced expression of both IL-22 and antimicrobial proteins in colon (Figure 1C), indicating the critical role of LTβR to control IL-22 production by innate lymphoid cells. LTβR is known to regulate expression of several chemokines and adhesion molecules responsible for migration of lymphoid cells to the gut (Fu and Chaplin, 1999; Ware, 2005). Therefore, impaired migration or development of IL-22 producing cells could result in reduced IL-22 levels in the gut. However, analysis of gut lymphoid cells revealed similar numbers of innate cell populations within the colon of WT and Ltbr-/- mice (Figure S1). These results suggest that the defect in IL-22 production in LTβR-deficient mice is not due to the absence of innate cells capable to produce IL-22 in the gut. In fact, lamina propria lymphocytes from Ltbr-/- mice were capable to produce IL-22 after IL-23 stimulation in vitro (Figure S1, C and D), suggesting no intrinsic defect in production of IL-22 by these cells. Thus, our data demonstrate that direct LTβR signaling is essential for control of innate IL-22 pathway in the gut after mucosal bacterial infection.
IL-22 is an essential protection pathway downstream of LTβR signaling
Surface LT and LIGHT (TNFSF14) are two known ligands for LTβR (Fu and Chaplin, 1999; Ware, 2005). To define whether LIGHT or LT controls IL-22 production in the gut after infection, we orally infected WT, LTβ- and LIGHT- deficient mice with C. rodentium. Clearly, IL-22 expression was reduced in the colon of Ltb-/- (Figure S2A), but not in Tnfsf14-/- mice (Figure S2D), suggesting the essential role of surface LT to control IL-22 production after infection. Expression of LT on adaptive immune cells was dispensable for IL-22 production, because mice that lack surface LT specifically on T and B cells (T,B-Ltb-/-) were able to express similar mRNA levels of IL-22 and antimicrobial proteins RegIIIγ and RegIIIβ compared to WT mice after infection (Figure S2A-C). Consistently, Tnfsf14-/- mice and T,B-Ltb-/- mice survived C. rodentium infection (Wang et al., 2010). To define whether LTβR signaling is sufficient to induce IL-22 production in the gut, we treated LT-deficient mice with agonistic anti-LTβR antibody. We found that anti-LTβR treatment greatly induced expression of IL-22 in the colon early after infection (Figure 2A), increased production of antimicrobial proteins (Figure 2B) as well as reduced bacterial dissemination into peripheral organs (Figure 2C). These results suggest that LTβR signaling is sufficient to promote IL-22 pathway following mucosal bacterial infection in vivo. However, it was not clear whether IL-22 is the essential protection pathway downstream of LTβR signaling because LTβR could regulate other protection mechanisms. To address this question, we tested whether Ltbr-/- mice could be rescued from lethal C. rodentium infection by using hydrodynamic IL-22 injection. Remarkably, a single IL-22 injection 6 hours after infection was able to rescue Ltbr-/- mice from an otherwise lethal C. rodentium infection. More than 60% of Ltbr-/- mice treated with IL-22 survived the infection (Figure 2D, and E). Ltbr-/- mice treated with control vector showed severe colon inflammation, loss of goblet cells, ulcerations of epithelial layer, developed severe diarrhea and all died by day 12 post infection (Figure 2D-G). In contrast, histological analysis of colon sections at day 9 post-infection of IL-22 treated Ltbr-/-mice revealed reduced colon inflammation and reduced histopathology score (Figure 2F, and G). These data suggest that IL-22 is an essential pathway downstream of LTβR signaling to protect mice against mucosal bacterial pathogen.
Figure 2. IL-22 is an essential protection pathway downstream of LTβR signaling.
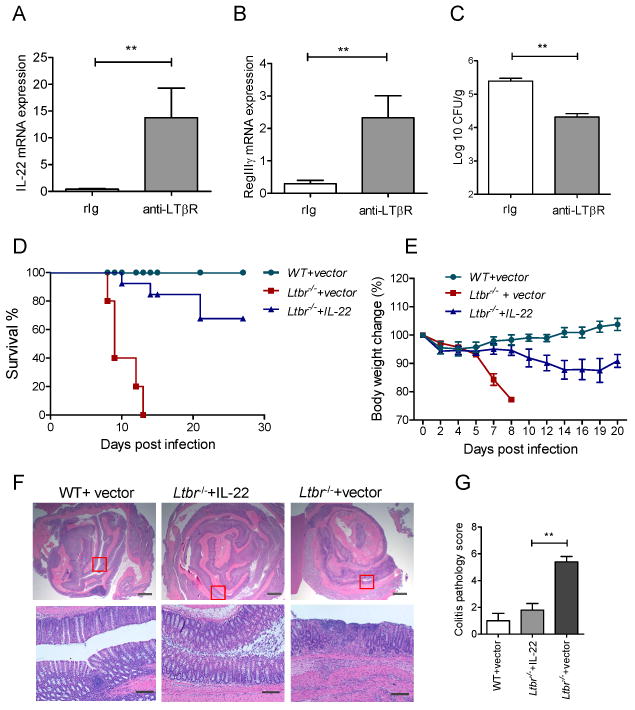
A-C. Stimulation of LTβR signaling promotes IL-22 protection pathway. Ltb-/- mice were injected intraperitoneally with 75μg of agonistic anti-LTβR antibody or control rat Ig (rIg) at day 0 and 3 post infection with C. rodentium. mRNA expression of IL-22 (A) and RegIIIγ (B) were measured in colon by real-time PCR at day 4 post infection. C. Bacterial titers in liver at day 4 post infection. n=4, **p<0.01, Data represent means ± s.e.m. D-G. Hydrodynamic injection of IL-22 expressing plasmid rescues Ltbr-/- mice from lethal C. rodentium infection. IL-22 expressing plasmid or control vector were intravenously injected to WT or Ltbr-/-mice 6h after C. rodentium infection. n=10-13/group. Survival (D) and body weight change (E) are shown. ***p<0.001 between IL-22 treated Ltbr-/- and control untreated Ltbr-/- mice by Mantel-Cox log-rank test. Data combined from two experiments with similar results. F. Representative hematoxylin and eosin staining of colons at day 9 post infection. Red boxes in top panel are shown at higher magnification at lower panel. Bars: 1mm for top panel, and 200μm for lower panel. G. Colitis histopathology score. n=5, *p<0.05. Data represent means ± s.e.m.
IL-22 is produced in lymphoid follicles by innate RORγt+ cells
Our data suggested that LTβR signaling controls the innate IL-22 production in response to mucosal bacterial infection. However, the innate cell populations that are regulated by LTβR signaling remain unclear. Analysis of IL-22 expression in total cell populations from colon revealed predominant expression of IL-22 in lamina propria lymphocytes (LPL), compared to intraepithelial lymphocytes (IEL) (Figure 3A). To further define the innate cell source of IL-22, we next measured expression of IL-22 in purified innate cell populations from lamina propria after infection. Because all ILCs require the transcriptional factor RORγt for their development (Eberl et al., 2004; Spits and Di Santo, 2011), we utilized RORγt-GFP+/- reporter mice that are heterozygous for insertion of a green fluorescent protein (GFP) reporter into the Rorc gene (Eberl and Littman, 2004) to determine IL-22 expression from the sorted cells. We found that LTi cells were the predominant IL-22 producing population in the lamina propria (Figure 3B, and Figure S3). Furthermore, we crossed Ltbr-/- mice to RORγt-GFP+/- reported mice to allow isolation of RORγt+ ILCs from LTβR-deficient mice by flow cytometry. Expression of IL-22 was greatly reduced in LTi cells from RORγt-GFP+/-/Ltbr-/- mice, compared to RORγt-GFP+/-/Ltbr+/+ control mice (Figure 3B), suggesting that, in the absence of LTβR signaling the LTi cells are unable to produce IL-22 after infection.
Figure 3. IL-22 is produced in lymphoid follicles by innate RORγt+ cells.
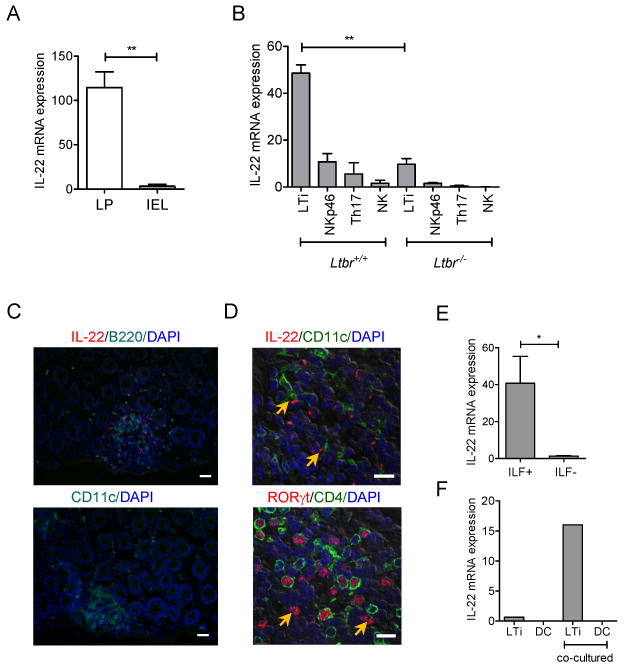
A. IL-22 is predominantly expressed in lamina propria. Colon lamina propria (LP) and intraepithelial lymphocyte (IEL) cell populations from WT mice were purified at day 5 post infection. IL-22 expression was measured by real-time PCR. Data normalized to hprt expression. Data represent means ± s.e.m. n=5 mice, **p<0.01. B. LTi cells are predominant IL-22 producing cells in the lamina propria. Colon LP innate cell populations from RORγt-GFP+/-/Ltbr+/+ and RORγt-GFP+/-/LTbr-/- mice at day 5 post infection were purified by flow cytometry. IL-22 expression was measured by real-time PCR. Sorted cell populations are indicated as: LTi: CD45+CD3-GFP+NK1.1-NKp46-, NKp46: CD45+CD3-GFP+NKp46+, NK: CD45+CD3-GFP-NK1.1+, Th17: CD45+CD3+GFP+NK1.1-NKp46-. Data combined from two experiments, means ± s.e.m, n=4, **p<0.01. IL-22 data were normalized to hprt expression. C. IL-22 expressing cells are predominantly located in lymphoid follicles. WT mice were orally infected with C. rodentium and colon sections at day 5 post infection were stained with indicated antibodies. Nuclei were stained with DAPI. Bars: 20μm. D. IL-22 producing cells interact with DC in lymphoid follicles. Colon sections of mice at day 5 post infection were stained with indicated antibodies and analyzed by confocal microscopy. Nuclei were stained with DAPI. Arrows indicate contact of DC (green) with IL-22 producing cells (red) on top panel, and RORγt+CD4- cells on bottom panel. Bars: 10μm. E. IL-22 is expressed predominantly in isolated lymphoid follicles. WT mice were orally infected with C. rodentium, and colon tissue collected at day 5 post infection. Isolated lymphoid follicles (ILF+) and surrounded tissue (ILF-) were microdissected under stereo microscope, and IL-22 expression measured by real-time PCR. IL-22 data were normalized to hprt expression. *p<0.05, n=3 mice. F. Co-culture of LTi cells with DC promotes IL-22 production by LTi cells. Lamina propria LTi cells from naïve RORγt-GFP+/- mice were co-cultured in vitro for 18h with lamina propria DCs from WT mice at day 5 post infection. After co-culture, LTi and DC cells were separated by flow cytometry and IL-22 measured by real-time PCR. Data normalized to b-actin. One of two independent experiments with similar results is shown.
To better define the location of IL-22 producing cells in the gut, we stained colon sections with anti-IL-22 antibody at day 5 post infection. Surprisingly, most of IL-22 expressing cells were not randomly scattered in lamina propria, but were predominantly located in isolated lymphoid follicles (ILF) and colonic patches (Figure 3C-D). Consistently, we found predominant IL-22 expression in ILF-containing areas microdissected from colon tissue, compared to ILF-negative areas (Figure 3E). Although the DCs concentrated in B cell areas, they did not produce IL-22. However, we observed that they were in close contact with RORγt+ IL-22 producing cells (Figure 3C, and D). Furthermore, colon lamina propria DCs purified from infected mice were able to activate naïve LTi cells to produce IL-22 when co-cultured in vitro (Figure 3F), suggesting that cooperation of DC and LTi cells promotes efficient IL-22 production after infection. Thus, our results suggest that IL-22 is produced predominantly by innate RORγt+ cells that are located within gut-associated lymphoid tissues.
LT expression on RORγt+ cells is essential for control of IL-22 production and protection of mice against C. rodentium infection
Since our data suggested the critical role of LTβR signaling in controlling innate IL-22 pathway, we next tested whether LT expression on innate RORγt+ cells is essential for IL-22 production. In order to test this, we utilized mice with specific inactivation of surface LTβ2LTα1 complex on RORγt+ expressing cells (RORγt-Ltb-/- mice), by crossing LTβ floxed mice (Tumanov et al., 2002) with RORγt-Cre transgenic mice (Eberl et al., 2004). We found that IL-22 expression in colon tissue or in colon lamina propria cells was dramatically reduced in RORγt-Ltb-/- mice, compared to WT mice (Figure 4A). Reduced expression of IL-22 in colon correlated with reduced expression of IL-22 dependent antimicrobial proteins RegIIIγ and RegIIIβ (Figure 4B). Thus, our data suggest that surface LT expression on RORγt+ cells is essential to control the IL-22 production in the gut.
Figure 4. LT expression on RORγt+ cells is essential for control of IL-22 production and protection of mice against C. rodentium infection.
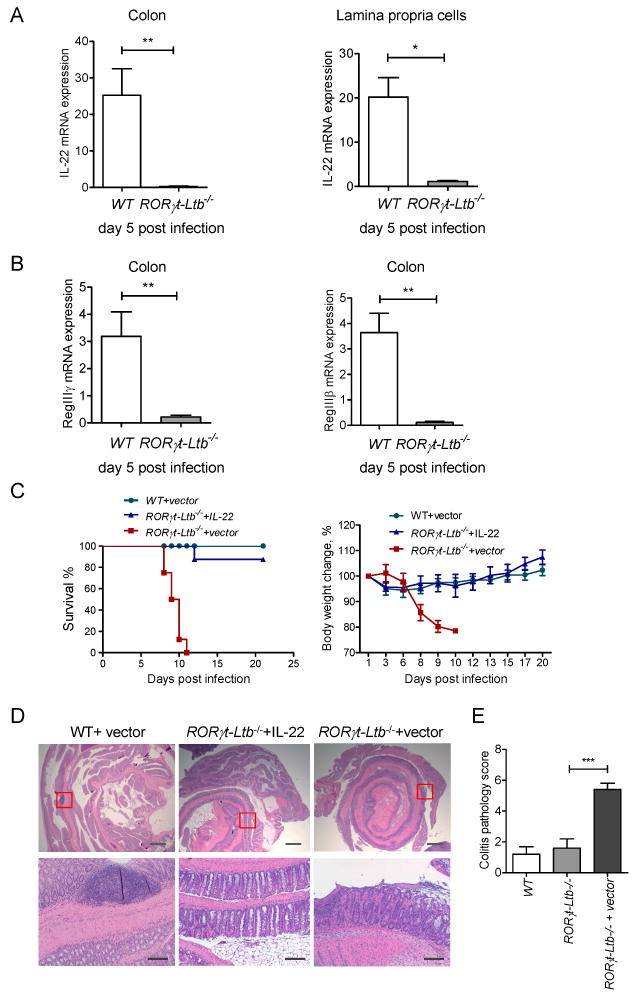
A-B. LT expression on RORγt+ cells controls IL-22 protection pathway. WT and RORγt-Ltb-/-mice were orally infected with C. rodentium. Expression of IL-22 in colon and purified lamina propria cells (A), and antimicrobial proteins RegIIIγ, and RegIIIβ (B) was measured by real-time PCR at day 5 post infection. Data represent means ± s.e.m. One out of three independent experiments with similar results is shown. n=5 mice, *p<0.05, **p<0.01. Expression data were normalized to hprt expression. C. IL-22 expression is sufficient to rescue RORγt-Ltb-/- mice from lethal C. rodentium infection. WT and RORγt-Ltb-/- mice were intravenously injected with IL-22 expressing plasmid or control vector at 6h after C. rodentium infection. n=10/group. Data combined from two experiments with similar results. Survival and body weight change are shown. D. Representative hematoxylin and eosin staining of colon at day 9 post infection. Red boxes in top panel are shown at higher magnification at lower panel. Bars: 1mm for top panel, and 200μm for lower panel. E. Colitis histopathology score for mice in panel A. n=5, ***p<0.001. Data represent means ± s.e.m. One of two experiments with similar results is shown.
To define whether the IL-22 pathway is an essential protection mechanism downstream of LT signaling we delivered exogenous IL-22 by hydrodynamic injection of IL-22 expressing plasmid to RORγt-Ltb-/- mice. Exogenous IL-22 rescued RORγt-Ltb-/- mice, as 90% of mice treated with IL-22 survived the infection and did not lose weight compared to mice treated with control vector (Figure 4C). RORγt-Ltb-/- mice rescued by IL-22 delivery showed reduced colonic inflammation and bacterial dissemination to peripheral organs, compared to RORγt-Ltb-/- mice treated with control vector (Figure 4D, and E). Thus, LT expression on innate RORγt+ cells is essential to control the IL-22 protective pathway.
LTβR signaling on DCs is required for IL-22 production
Our results suggested that LT expression directly regulates IL-22 production from RORγt+ cells. Therefore, we considered two potential mechanisms to explain how LT regulates IL-22 production. We first considered that direct interactions among innate RORγt+ cells expressing both surface LT and LTβR leads to IL-22 production. We also considered an indirect mechanism, where RORγt+ cells produce IL-22 via interaction with accessory LTβR expressing cells. To test these possibilities, we used conditional gene targeting approach by inactivating ltbr in different cell types. We first ruled out the direct mechanism, because mice with specific inactivation of ltbr in RORγt+ cells produced similar levels of IL-22 and antimicrobial proteins after infection, compared to WT mice (Figure S4). Since our data showed a close interaction of RORγt+ cells with DCs in lymphoid follicles, we proposed that LT signaling by RORγt+ cells could trigger LTβR on DCs to provide additional signals for IL-22 production by RORγt+ cells as a positive feedback loop mechanism. In order to resolve this hypothesis, we inactivated the ltbr gene specifically in DCs by crossing LTβR floxed mice (Wang et al., 2010) with CD11c-Cre transgenic mice (Stranges et al., 2007), generating CD11c-Ltbr-/- mice. We found an efficient deletion of the ltbr gene and mRNA in bone marrow derived DCs from CD11c-Ltbr-/- mice (Figure S5A-B). Since CD11c is also expressed at lower levels in NK cells (Laouar et al., 2005; Stranges et al., 2007), we cannot exclude the possibility of ltbr gene deletion in a fraction of NK cells. However, CD11c-Cre expression unlikely affects the development or function of NK cells, since the number of NKp46+ and LTi cells was not affected in CD11c-Ltbr-/- mice (Figure S5A-B, and data not shown). CD11c-Ltbr-/- mice infected with C. rodentium showed significant reduction of body weight compared to WT mice, but were able to survive infection (Figure 5A). CD11c-Ltbr-/- mice showed focal colon bacterial lesions in colon (Figure 5B), and were not able to control bacterial dissemination to peripheral organs (Figure 5C-E). These data suggest that LTβR signaling in DCs is required for efficient immune response to mucosal pathogen. To define whether LTβR signaling on DCs is required for IL-22 production, we measured IL-22 production in colon of CD11c-Ltbr-/- mice early after infection. We found that both IL-22 mRNA and protein expression were greatly reduced in the colon of CD11c-Ltbr-/- mice compared to WT mice (Figure 5F, and G). Consistently, expression of IL-22- dependent antimicrobial protein RegIIIγ was reduced in colon of CD11c-Ltbr-/- mice (Figure 5H). These results suggest that LTβR signaling in DCs is required for effective IL-22 production in the gut post-infection.
Figure 5. LTβR signaling in DCs is required for IL-22 production.
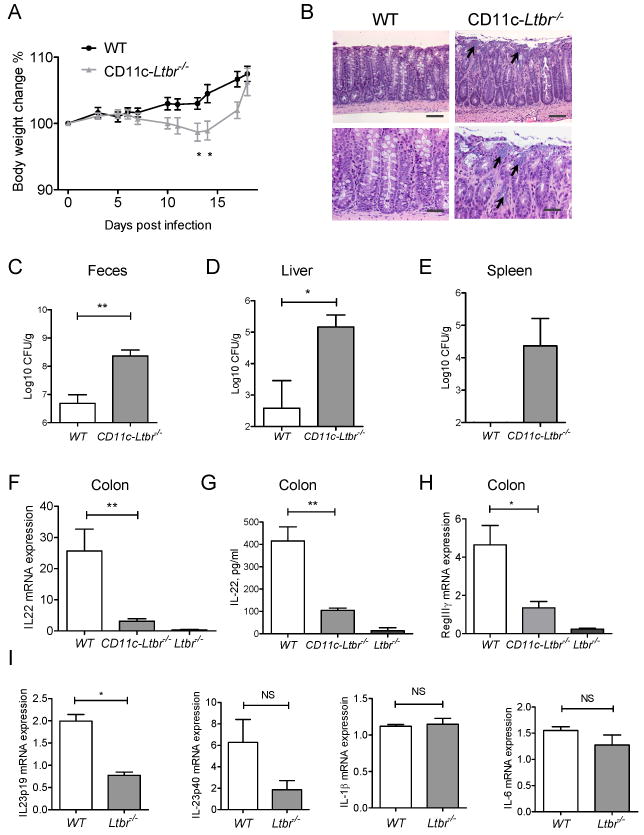
WT, CD11c-Ltbr-/-, and Ltbr-/- mice were orally infected with C. rodentium. Body weight change (A) and representative hematoxylin and eosin staining of colon at day 12 post infection (B). Arrows indicate bacterial lesions. Bars: 50μm (top panels), and 20 μm (lower panels). Bacterial titers were measured in feces (C) on day 14, and liver (D), and spleen (E) on day 10 post infection. F. IL-22 expression in colon at day 5 post infection. G. IL-22 levels in colon supernatants at day 5 post infection. H. Expression of RegIIIγ mRNA in colon at day 5 post infection. C-H. Data represent means ± s.e.m. n=5 mice, *p<0.05, **p<0.01. Represents one of three independent experiments with similar results. Real-time PCR data were normalized to hprt expression. I. LTβR signaling in DCs regulates IL-23 production. CD11c+CD11b+ DCs from colon lamina propria of WT and LTbr-/- mice were purified by flow cytometry at day 5 post infection. Expression of IL23p19, IL23p40, IL-1b, IL-6 cytokines was measured by real-time PCR. Data combined from two experiments, means ± s.e.m, n=4, *p<0.05, NS-not significant. RT-PCR data normalized to hprt expression.
To further define the mechanism of IL-22 production by LTi cells, we measured expression of cytokines in colon lamina propria DCs isolated from WT and Ltbr-/- mice at day 5 post infection. We found that expression of IL-23, a potent inducer of IL-22 (Sonnenberg et al., 2011b; Zheng et al., 2008), was reduced in DCs purified from colon of Ltbr-/- mice, compared to WT mice (Figure 5I). In contrast to IL-23 expression, DCs from WT and Ltbr-/- mice expressed similar levels of IL-1β and IL-6 (Figure 5I). These results suggest that LTβR signaling activates IL-22 pathway indirectly via IL-23 production by DCs.
Discussion
Innate lymphoid cells (ILCs) represent a mechanism to regulate the immune response to pathogens at mucosal barriers. However, the mechanism that controls their effector functions is not well defined. In this study, we identified LTβR signaling as an essential regulator of IL-22 protection pathway in the gut. We demonstrate that LT controls IL-22 production by ILCs in response to enteric bacterial pathogen C. rodentium. LT from innate RORγt+ cells but not from adaptive T or B cells was essential for IL-22 production. IL-22-producing RORγt+ cells localized predominantly in gut B cell follicles and interacted closely with DCs. We demonstrate that LT controls IL-22 production by RORγt+ ILCs indirectly via LTβR signaling in DCs. Therefore, LT represents a regulator of IL-22 protection pathway by ILCs to control mucosal bacterial infection.
Previous studies identified IL-22 as a critical cytokine to control C. rodentium infection (Zheng et al., 2008). This IL-22 protective effect is mediated by production of antimicrobial proteins by epithelial cells that specifically express IL-22R. Additionally, IL-22R signaling is required for survival and regeneration of intestinal epithelial cells after injury as well as for mucus production (Sugimoto et al., 2008; Zenewicz et al., 2008). Innate immune cells are critical for production of this IL-22 that provides host protection after enteric C. rodentium infection (Zheng et al., 2008). IL-23 was shown as a critical regulator to promote IL-22 production by ILC in vitro and in vivo (Sonnenberg et al., 2011b; Zheng et al., 2008). However, other pathways that influence IL-22 production remained largely unknown. Our data show that the LTβR pathway is an essential regulator of IL-22 production by innate immune cells in response to enteric bacterial infection. Since LTβR deficient mice display multiple defects in components of both innate and adaptive immune system, it is important to define whether IL-22 pathway is an essential LTβR-dependent mechanism that might impart protection against C. rodentium. By using both agonistic anti-LTβR stimulation in LT deficient mice and reconstitution of IL-22 expression in LTβR-deficient mice we demonstrate that IL-22 is a critical protection pathway downstream of LTβR signaling.
Our next experiments aimed to define which LT-expressing cell populations are essential for IL-22 production after infection. Our results suggest that LT expression by innate but not adaptive immune cells is critical for IL-22 production and host protection, because mice with specific inactivation of surface LT on T and B cells did not show defects in IL-22 production. Recent studies identified a heterogeneous population of ILCs in the gut capable of producing IL-22 upon stimulation (Colonna, 2009; Sonnenberg et al., 2011a; Spits and Di Santo, 2011). In particular, mucosal RORγt+NKp46+ cells were suggested as a major source of IL-22 production in the small intestine lamina propria (Luci et al., 2009; Sanos et al., 2009; Satoh-Takayama et al., 2008). In contrast to RORγt+NKp46+ cells, LTi cells did not produce IL-22 in the small intestine lamina propria from naïve mice (Luci et al., 2009; Sanos et al., 2009), although other studies reported IL-22 expression also by LTi cells (Marchesi et al., 2009; Sawa et al., 2010). It remains possible that cytokine production by LTi cells might change under ongoing bacterial infection conditions. A recent study reported that LTi cells in the intraepithelial lymphocyte compartment were, in fact, the major IL-22 producing cells after C. rodentium infection (Sonnenberg et al., 2011b). Yet another study suggested that DCs could also contribute significantly to IL-22 levels soon after C. rodentium infection (Zheng et al., 2008). In contrast to this earlier study, we found that DCs were not major producers of IL-22 after infection. However, we observed that DCs were in close contact with IL-22 producing RORγt+ cells in B cell follicles. Our results using RORγt-GFP+/- reporter mice showed that LTi cells were the predominant producers of IL-22 in the lamina propria early after infection. Furthermore, expression of IL-22 was greatly reduced in LTi cells from Ltbr-/- mice, suggesting that in the absence of LTβR signaling the LTi cells are unable to produce IL-22 after infection. However, we found no intrinsic defect in IL-22 production by lamina propria ILCs from Ltbr-/- mice since these cells were able to produce IL-22 after in vitro stimulation with IL-23, consistent with a recent report (Satoh-Takayama et al., 2011). Our results are also consistent with the recent study (Sonnenberg et al., 2011b) and extend our current understanding to suggest that LTi subset of ILCs actively participate in host defense by regulating IL-22 protection pathway in the gut.
ILCs produce various TNF family cytokines and chemokine receptors, including TNF, LTα, LTβ, LIGHT, TRANCE, CCR7, CCR6, and CXCR5 (Luci et al., 2009; Marchesi et al., 2009; Vonarbourg et al., 2010; Wang et al., 2010). Our study revealed that a genetic inactivation of a single cytokine, surface LT on RORγt+ ILCs, resulted in a dramatic defect in the production of IL-22 by these cells, leading to a 100% mortality rate to C. rodentium infection in mice. These results demonstrate that LT expression on RORγt+ ILCs is critical for control of IL-22 protection pathway in the gut.
In contrast to LT, expression of LTβR on ILCs was dispensable for IL-22-mediated protection, suggesting an indirect LT-dependent mechanism of IL-22 production by RORγt+ ILCs. Remarkably, IL-22 producing cells were not randomly scattered in the gut, but predominantly located in organized secondary lymphoid tissues in the colon, and closely interacted with DCs. By using a conditional gene targeting approach, we demonstrate that LTβR signaling in DC is important for IL-22 production and bacterial clearance. We also cannot exclude the potential contribution of LTβR signaling by other cells, in addition to DC, because IL-22 levels were only partially reduced in colon of CD11c-Ltbr-/- mice compared to Ltbr-/- mice. Ltbr-/- mice have multiple defects in development of gut-associated lymphoid tissues which may additionally affect IL-22 production independently of LTβR signaling in DCs.
Based on our results, we propose a model of regulation of IL-22 production in the gut to control mucosal bacterial infection as shown in Figure 6. LT expression by RORγt+ ILCs is necessary for IL-22 production following invasion of mucosal bacterial pathogen. LT signaling by RORγt+ ILC promotes the development of lymphoid follicles in the gut. Lymphoid follicles provide the necessary microenvironment for close interaction between RORγt+ ILCs and DCs. Interplay between RORγt+ ILCs and DCs then promotes IL-23 secretion by DCs that, in turn, activates IL-22 production by RORγt+ cells as a positive feedback loop. Since both LT (LTβ2LTα1) and LTβR exist only as membrane bound molecules, DCs and RORγt+ ILCs interaction likely require direct cell contact. IL-22 activates IL-22R on epithelial cells which leads to production of antimicrobial proteins RegIIIγ and RegIIIβ to eliminate the mucosal bacterial pathogen. Thus, our study opens avenues to explore the role of ILCs and DC cooperation in host defense and in pathogenesis of autoimmune diseases.
Figure 6. Proposed model to control IL-22 pathway against mucosal bacterial pathogen.

LT expression by RORγt+ ILCs is necessary for IL-22 production following invasion of mucosal bacterial pathogen. LT signaling by RORγt+ ILC promotes the development of lymphoid follicles in the gut. Lymphoid follicles provide necessary microenvironment for interaction between innate RORγt+ cells and DC. Interplay between RORγt+ ILCs and DCs promotes IL-23 production by DCs that, in turn, activates IL-22 synthesis by RORγt+ cells as a positive feedback loop. IL-22 stimulates IL-22R on epithelial cells which triggers production of antimicrobial proteins RegIIIγ and RegIIIβ to eliminate mucosal bacterial pathogen.
It was recently proposed by several studies that host-protective functions of RORγt+ ILCs evolutionary preceded the acquisition of lymphoid tissue-inducing functions (Lane et al., 2009; Sonnenberg et al., 2011b). However, our study suggests that gut-associated lymphoid tissues provide an environment where RORγt+ ILCs coordinate closely with cells of adaptive immune system. It is also possible that LT signaling by RORγt+ ILCs also participates in the maintenance of gut-associated lymphoid tissues. Therefore, the mechanism of interplay and coordination of ILCs with components of adaptive immune system will be an exciting area of future research.
In animal models of colitis, expression of IL-22 by both innate and adaptive immune cells regulates colon inflammation in both DSS-induced and T-cell induced colitis models (Sugimoto et al., 2008; Zenewicz et al., 2008). Other studies show that LT production by B cells promotes colon inflammation in a DSS-induced inflammation model (Jungbeck et al., 2008; Lochner et al., 2011), while RORγt+ ILCs reduce colon inflammation in this model (Sawa et al., 2011). In light of our results, it would be important to determine whether LT by innate RORγt+ cells can also participate in prevention of colon inflammation in DSS-induced colitis and other models of colon inflammation.
Elevated IL-22 levels have been reported in IBD patients (Ouyang, 2010). Although the role of IL-22 in IBD is complex, most studies suggest a protective role of IL-22 in IBD, by regulating epithelial barrier function, regeneration of epithelial cells, and production of antibacterial proteins (Sonnenberg et al., 2011a; Sugimoto et al., 2008; Zenewicz et al., 2008). However, a pathogenic role of IL-22 has been shown in other organs, for example, IL-22 promotes psoriasis (Ouyang, 2010). Therefore, careful design of IL-22 modulating agents is required for the development of potential therapeutic approaches. Our data suggest that LTβR signaling regulates IL-22 production by ILCs in the gut and that pharmacological manipulation of LTβR signaling could represent a therapeutic avenue in autoimmune diseases.
Experimental Procedures
Mice
C57BL/6 and Rag1-/- mice were purchased from Harland Teklad. Ltb-/-, Tnfsf14-/-, and Ltbr-/- mice were backcrossed onto C57BL/6 background 13, 11 or 10 generations, respectively, and maintained under specific pathogen-free conditions as described (Wang et al., 2010). Rorc-/- (Eberl and Littman, 2004) mice were purchased from The Jackson Lab. CD11c-Ltbr-/- and RORγt-Ltbr-/- mice were generated by crossing LTβR floxed mice (Wang et al., 2010) with CD11c-Cre (Stranges et al., 2007) and RORγt-Cre (Eberl et al., 2004), respectively. T,B-Ltb-/-mice were described previously (Tumanov et al., 2002; Tumanov et al., 2003). Animal care and use were in accordance with institutional and National Institutes of Health guidelines and all studies were approved by the Animal Care and Use Committee of the University of Chicago.
Citrobacter rodentium infection
To induce bacterial colitis in mice, mice were orally gavaged with 2×109 cfu C. rodentium strain DBS100 (ATCC 51459; American Type Culture Collection), as previously described (Zheng et al., 2008). Briefly, mice were fasted for 8 h before oral inoculation of C. rodentium culture in a total volume of 0.2ml per mouse. Bacteria were prepared by shaking at 37°C overnight in LB broth. Concentration was assessed by measuring absorbance at OD600. Bacterial culture was serially diluted and plated after each inoculation to confirm the colony-forming units (CFUs) administered. Body weight was measured a day before and then frequently during the course of disease.
LTβR-Ig and anti-LTβR antibody treatment
The LTβR-Ig used in this study has been previously described (Wang et al., 2010). Briefly, cDNA encoding the extracellular domain of murine LTβR was fused with the Fc portion of human IgG, transfected into BHK/VP16 cell, and the supernatant collected. The anti-LTβR agonistic antibody (3C8) was kindly provided by C. Ware (La Jolla Institute for Allergy and Immunology, La Jolla, CA).
Isolation of intraepithelial lymphocytes (IEL), lamina propria mononuclear (LPL) cells and epithelial cells from mouse colon
IELs, LPLs and colonic epithelial cells were isolated as described (Wang et al., 2010).
Tissue collection, histology, histological score and CFU counts
Colons were dissected from the mice and fixed in 10% neutral buffered formalin. Paraffin-embedded tissue sections were stained with H&E to evaluate tissue pathology. Histological scoring was performed using a modified scoring system described previously (Zheng et al., 2008). In brief, the presence of rare inflammatory cells in the lamina propria were counted as: 0; 1- confluence of inflammatory cells; 2- extending into the submucosa; 3- transmural extention of the inflammatory cell infiltrate. For epithelial damage, absence of mucosal damage was counted as 0, discrete focal lymphoepithelial lesions were counted as 1, mucosal erosion/ulceration was counted as 2, and a score of 3 was given for extensive mucosal damage and extension through deeper structures of the bowel wall. The two subscores were added and the combined histological score ranged from 0 (no changes) to 6 (extensive). For CFU count fecal samples were collected and weighted, then homogenized in sterile phosphate-buffered saline. Serially diluted homogenates were plated on MacConkey agar plates (Sigma). C. rodentium colonies were identified as pink colonies after overnight incubation at 37°C. Spleens and livers were aseptically removed and homogenized. Organs colonization was assessed as described for fecal specimens.
Hydrodynamic IL-22 delivery
Mice were restrained in conical restrainer with a heating element to dilate blood vessels. 10 μg of IL-22 expressing plasmid (pRK-mIL-22, Genentech) or control vector (pRK) were injected in tail vein 6h after C. rodentium infection in 1.6 ml of TransIt-EE Hydrodynamic Delivery Solution (MIR 5340, Mirus Inc) over a period of 3-5 sec.
ELISA
Terminal colon pieces 1 centimeter length were cut in small pieces and incubated in 0.3ml of RPMI 1640 medium containing 10% FCS, amphotericin, gentamicyn, penicillin and streptomycin for 48h in tissue plates, as previously described (Zheng et al., 2008). IL-22 in supernatants was measured by ELISA (R&D Systems) according to manufactures recommendations.
Flow cytometry and antibodies
Flow cytometry analysis was performed on FACSCanto, and FACSAria II (BD Biosciences) instruments and analyzed using FlowJo software (Tree Star Inc.). All antibodies were purchased from BD Biosciences or eBioscience. Intracellular for IL-22 and RORγt was performed as described previously (Sawa et al., 2010).
RNA isolation and real-time reverse transcriptase PCR
RNA from cells or tissues was isolated using RNeasy Micro or Mini Kit (Qiagen). cDNA synthesis and real-time RT-PCR was performed as described (Wang et al., 2010).
Immunohistochemistry
Colons was isolated from mice and extensively washed in PBS, then fixed for 1h in 4% (wt/vol) paraformaldehyde/PBS. After three washes in PBS, paraformaldehyde-fixed organs were incubated in a 30% (wt/vol) sucrose/PBS solution overnight. Fixed tissues were embedded in optimum cutting temperature compound (Sakura Finetek) and frozen at -70C. 6-8 mm cryostat sections were stained with IL-22 (Alexa 555-conjugated, clone 8E11, Genentech) and RORγt (hamster polyclonal antibody), as previously described (Eberl et al., 2004; Zheng et al., 2008). After being stained, slides were dried, mounted with ProLong Gold (Invitrogen) and examined with Nikon A1 confocal microscope (Nikon, Inc.), or Zeiss Axiovert 200M microscope (Carl Zeiss, Inc.). Images were processed with NIH ImageJ v.1.42 software.
Statistical analysis
Comparisons of data were analyzed by two-tailed Student's t test using GraphPad Prism 5.0 program. Data from such experiments are presented as mean values ±S.E.M. P< 0.05 was considered significant. For survival curves statistics were done using the log rank (Mantel-Cox) test.
Supplementary Material
Highlights.
The Lymphotoxin pathway regulates IL-22 production by RORγt+ innate lymphoid cells
LTβR signaling in gut DCs triggers the innate immune response to mucosal pathogens
RORγt+ ILCs communicate with DCs in gut lymphoid follicles for IL-22 production
IL-22 represents an essential protection pathway downstream of LTβR signaling
Acknowledgments
This research was in part supported by US National Institutes of Health grants AI062026, CA115540 and DK58891 to Y.X.F; by Career Development Award from the Crohn's and Colitis Foundation (CCFA #2672) to A.V.T., by SFB633 from Deutsche Forschungs gemeinschaft and by MCB Program of the Russian Academy of Sciences. We are grateful to W. Ouyang (Genentech, CA) for providing IL-22 expressing plasmid and anti-IL-22 antibody, D. Littman and I. Ivanov (New York University, NY) for RORγt-Cre mice and hamster anti-RORγt antibody, C. Ware (La Jolla Institute for Allergy and Immunology, CA) for 3C8 LTβR antibody, and A. Chervonsky (University of Chicago, IL) for CD11c-Cre mice. We thank M. Miller for critical reading of the manuscript.
Footnotes
Authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, Colonna M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science. 2004;305:248–251. doi: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- Jungbeck M, Stopfer P, Bataille F, Nedospasov SA, Mannel DN, Hehlgans T. Blocking lymphotoxin beta receptor signalling exacerbates acute DSS-induced intestinal inflammation-Opposite functions for surface lymphotoxin expressed by T and B lymphocytes. Mol Immunol. 2008;45:34–41. doi: 10.1016/j.molimm.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005;22:439–450. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Lane PJ, McConnell FM, Withers D, Gaspal F, Saini M, Anderson G. Lymphoid tissue inducer cells: bridges between the ancient innate and the modern adaptive immune systems. Mucosal Immunol. 2009;2:472–477. doi: 10.1038/mi.2009.111. [DOI] [PubMed] [Google Scholar]
- Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, Eberl G. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J Exp Med. 2011;208:125–134. doi: 10.1084/jem.20100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol. 2009;10:75–82. doi: 10.1038/ni.1681. [DOI] [PubMed] [Google Scholar]
- Marchesi F, Martin AP, Thirunarayanan N, Devany E, Mayer L, Grisotto MG, Furtado GC, Lira SA. CXCL13 expression in the gut promotes accumulation of IL-22-producing lymphoid tissue-inducer cells, and formation of isolated lymphoid follicles. Mucosal Immunol. 2009;2:486–494. doi: 10.1038/mi.2009.113. [DOI] [PubMed] [Google Scholar]
- Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. 2005;7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- Ouyang W. Distinct roles of IL-22 in human psoriasis and inflammatory bowel disease. Cytokine Growth Factor Rev. 2010;21:435–441. doi: 10.1016/j.cytogfr.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]
- Randall TD, Carragher DM, Rangel-Moreno J. Development of secondary lymphoid organs. Annu Rev Immunol. 2008;26:627–650. doi: 10.1146/annurev.immunol.26.021607.090257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal R, Mebius RE. Stromal cell-immune cell interactions. Annu Rev Immunol. 2011;29:23–43. doi: 10.1146/annurev-immunol-031210-101357. [DOI] [PubMed] [Google Scholar]
- Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, Diefenbach A. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat Immunol. 2009;10:83–91. doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh-Takayama N, Dumoutier L, Lesjean-Pottier S, Ribeiro VS, Mandelboim O, Renauld JC, Vosshenrich CA, Di Santo JP. The natural cytotoxicity receptor NKp46 is dispensable for IL-22-mediated innate intestinal immune defense against Citrobacter rodentium. J Immunol. 2009;183:6579–6587. doi: 10.4049/jimmunol.0901935. [DOI] [PubMed] [Google Scholar]
- Satoh-Takayama N, Lesjean-Pottier S, Sawa S, Vosshenrich CA, Eberl G, Di Santo JP. Lymphotoxin-beta receptor-independent development of intestinal IL-22-producing NKp46+ innate lymphoid cells. Eur J Immunol. 2011;41:780–786. doi: 10.1002/eji.201040851. [DOI] [PubMed] [Google Scholar]
- Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–970. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, Cua D, Di Santo JP, Eberl G. RORgammat(+) innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011a;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) Lymphoid Tissue-Inducer Cells Promote Innate Immunity in the Gut. Immunity. 2011b;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn TW, Maaser C, Eckmann L, Heidemann J, Lugering A, Newberry R, Domschke W, Herbst H, Kucharzik T. The lymphotoxin-beta receptor is critical for control of murine Citrobacter rodentium-induced colitis. Gastroenterology. 2004;127:1463. doi: 10.1053/j.gastro.2004.08.022. [DOI] [PubMed] [Google Scholar]
- Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, Hartig H, Sundberg JP, Servick S, Kaufmann G, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–641. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers-DeLuca LE, McCarthy DD, Cosovic B, Ward LA, Lo CC, Scheu S, Pfeffer K, Gommerman JL. Expression of lymphotoxin-alphabeta on antigen-specific T cells is required for DC function. J Exp Med. 2007;204:1071–1081. doi: 10.1084/jem.20061968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers Deluca L, Gommerman JL. The lymphotoxin pathway as a novel regulator of dendritic cell function. Adv Exp Med Biol. 2011;691:363–374. doi: 10.1007/978-1-4419-6612-4_37. [DOI] [PubMed] [Google Scholar]
- Tumanov A, Kuprash D, Lagarkova M, Grivennikov S, Abe K, Shakhov A, Drutskaya L, Stewart C, Chervonsky A, Nedospasov S. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity. 2002;17:239–250. doi: 10.1016/s1074-7613(02)00397-7. [DOI] [PubMed] [Google Scholar]
- Tumanov AV, Christiansen PA, Fu YX. The role of lymphotoxin receptor signaling in diseases. Curr Mol Med. 2007;7:567–578. doi: 10.2174/156652407781695701. [DOI] [PubMed] [Google Scholar]
- Tumanov AV, Grivennikov SI, Shakhov AN, Rybtsov SA, Koroleva EP, Takeda J, Nedospasov SA, Kuprash DV. Dissecting the role of lymphotoxin in lymphoid organs by conditional targeting. ImmunolRev. 2003;195:106–116. doi: 10.1034/j.1600-065x.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu YX, Tumanov AV. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. 2010;32:403–413. doi: 10.1016/j.immuni.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- Zhu M, Brown NK, Fu YX. Direct and indirect roles of the LTbetaR pathway in central tolerance induction. Trends Immunol. 2010;31:325–331. doi: 10.1016/j.it.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.