

Abstract
Air trapped in glacial ice offers a means of reconstructing variations in the concentrations of atmospheric gases over time scales ranging from anthropogenic (last 200 yr) to glacial/interglacial (hundreds of thousands of years). In this paper, we review the glaciological processes by which air is trapped in the ice and discuss processes that fractionate gases in ice cores relative to the contemporaneous atmosphere. We then summarize concentration–time records for CO2 and CH4 over the last 200 yr. Finally, we summarize concentration–time records for CO2 and CH4 during the last two glacial–interglacial cycles, and their relation to records of global climate change.
Ice crystals near the surface of a glacier are compressed by the continual addition of snow at the surface. As the ice crystals travel down into the glacier, they grow and reorient themselves into a closer packing. Density rises, open porosity decreases, and by some depth between 40 and 120 m, the crystals are sintered together into an impermeable mass (“glacial ice”) in which about 10% of the volume is composed of isolated bubbles. The gas trapped in these bubbles is close in composition to contemporaneous air and allows us to reconstruct changes in atmospheric chemistry on three important time scales: the last 200 years (relating to anthropogenic change), the Holocene (last 10,000 years), and the last several hundred thousand years (relating to glacial–interglacial cycles). In this paper we discuss the gas trapping process and summarize data on the changes in the greenhouse gas concentrations of air over the three time scales of interest.
Physics of Gases in Glaciers
The density of ice at the surface of an ice sheet is typically 0.3–0.35 g cm−3; the corresponding porosity is 62–67%. Settling and packing cause the density to rise rapidly to about 0.55 g cm−3 by a depth of 10–30 m. Below, recrystallization and other processes drive a somewhat slower increase in density, which continues until individual crystals are fused together into an impermeable mass of glacial ice (1). At the “bubble closeoff depth,” about 10–15% of the volume is air, and the density is about 0.81–0.84 g cm−3 (2, 3). The “firn” is the zone of porous snow and ice above the closeoff depth, and the depth interval in which bubbles close is termed the “firn–ice transition.” Below the transition, densification continues by the compression of bubbles due to hydrostatic pressure.
When the snow accumulation rate at the surface of an ice sheet is greater than about 4 cm yr−1 (expressed as the ice-equivalent thickness of annual layers), discrete seasonal layers of snow are preserved that have characteristic physical and chemical properties. Wintertime layers initially have higher densities than summertime layers. This density contrast is maintained during the densification process (Fig. 1). The firn-ice transition occurs at the same density for wintertime and summertime layers, but wintertime layers attain the closeoff density at a shallower depth. Consequently, there is an interval of about 10 m in which wintertime layers are more extensively sealed than summertime layers. In this interval, permeable and impermeable layers alternate in the ice sheet (3).
Figure 1.

(From Martinerie et al., ref. 3). (Upper) Closed porosity vs. density in the firn at Summit, Greenland. Note the monotonic relationship between the two properties. When density = 0.83, open porosity = 0. The decrease in closed porosity at higher densities is due to compression of bubbles. (Lower) Closed porosity vs. depth for maximum-density layers and minimum-density layers. Note how maximum-density layers are completely closed by 75-m depth, and minimum-density layers by 83-m depth. In the intervening depth interval, parts of the firn remain permeable but gases cannot migrate vertically.
There are three regimes of gas transport in the firn (4, 5). The uppermost layer, which appears to extend down to about 10-m depth at some sites where firn air has been sampled and analyzed, is affected by convective mixing driven by surface wind stress. Underlying the convective zone is the “stagnant air column,” in which transport is by molecular and atomic diffusion only. Diffusivities of gases are typically about 1 m2 day−1 at 10-m depth. Below, they decrease with increasing density (5), due to a combination of lower porosity and higher tortuosity (the latter factor accounts for the extra distances gas atoms and molecules must travel as they wind their way through the ice crystals to move from one depth to another).
The diffusivity of the firn is such that air at the base of the stagnant column today has a “CO2 age” ranging from about 6 yr for the GISP2 core (central Greenland) to about 40 years at Vostok (East Antarctica). In point of fact, however, air in firn at a given depth is not of a single age. The composition of firn air is convoluted by a number of processes. Air in the convective zone responds instantaneously to changes in atmospheric chemistry. These changes then propagate down into the stagnant column by molecular or atomic diffusion. Schwander et al. (6) calculated that the age of maximum abundance at the base of the firn is about 0.65·zt2/D, where zt is the height of the stagnant column and D is the free air diffusivity of the gas at the ambient temperature and pressure of the site. A small fraction of the gas is younger, and there is a long tail to older ages.
The diffusivity of an element or compound decreases with increasing mass and increasing atomic or molecular diameter. Thus each element or compound diffuses at a different rate, and each isotope of a compound diffuses at a different rate. In consequence, the covariation between the composition of one gas and another (e.g., CO2 and CH4) in firn is different from their historical covariation in air. The isotopic composition of a gas (e.g., CO2) in firn air also varies with the concentration of that gas in a way that is different from the historical relationship. The concentrations of gases and isotopes that diffuse most rapidly will be closest to their current atmospheric concentrations. Because light isotopes diffuse more rapidly, the concentration of a gas in firn air will be more depleted in heavy isotopes than was the atmosphere at the time it had the same concentration as a firn air sample. Differential diffusivity is a first-order effect that must be taken into account when interpreting data on the concentration and isotopic composition of gases in firn air and ice cores (7).
Several additional factors influence the composition of air in the firn. Gravitational fractionation is one (8–10). The pressure of gases increases with depth below the surface of the firn according to the barometric equation:
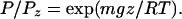 |
m = mass in units of g mol−1, g is the gravitational acceleration constant, z is depth, R is the ideal gas constant, and T is Kelvin temperature.
As Craig et al. (8) and Schwander (10) recognized, this equation applies not only to bulk air but to each individual constituent of air in that (dominant) depth interval of the firn where transport is essentially entirely by diffusion (the stagnant air column). The rate at which the enrichment-per-mass unit increases with depth, expressed in the δ notation, is (Δmg/RT −1)·1,000, or about 0.005‰/amu per meter at typical firn air temperatures. The relative enrichment with depth for different species is directly proportional to the mass difference. The firn air data for the GISP2 site, central Greenland, demonstrate the expected enrichment for the δ15N of N2 (Fig. 2). The enrichment or depletion is significant for nearly all species, corresponding to 3 ppmv of CO2 at the base of deep firn profiles, for example.
Figure 2.

δ15N (Left) and CH4 (Right) vs. depth in firn air from the GISP2 site at Summit, Greenland. The subsurface maximum in δ15N is due to thermal fractionation (15N is enriched at 5- and 10-m depth because the firn at these depths, which remains at the mean annual temperature, is colder than air at the surface during summertime, when sampling was done). The increase below 20-m depth is due to gravitational fractionation. CH4 decreases very slowly to the top of the bubble closeoff zone at 70-m depth. Below it decreases very rapidly because gases cannot migrate vertically and the age of the gas in the firn increases as rapidly as the age of the ice (about 4 yr/m).
Seasonal changes in the concentrations of gases in air cause seasonal variations in firn air chemistry. The magnitude of these variations relative to their secular trends depends on location and property. The effect is perhaps largest for O2, CO2, and δ13C of CO2 in Greenland. Seasonal variations are damped out with depth and become very small below 30–50 m.
Thermal fractionation also affects the isotopic and elemental composition of firn air. Severinghaus (11) and Severinghaus et al. (12) first recognized the importance of thermal fractionation in porous environmental media in their studies of the composition of air in sand dunes. Temperature gradients cause fractionation, with heavier gases or isotopes being enriched in colder regions. For 15N, the fractionation is about 0.025‰/°C. Thermal fractionation is large in firn because gases diffuse faster than heat. In consequence, steep seasonal temperature gradients occur in the upper ≈5 m of the firn and gases nearly equilibrate with these temperature gradients. This effect produces large seasonal variations in isotopic compositions and in the O2/N2 ratio in the top few meters of the firn. The seasonal anomalies decrease with depth, and for most species are insignificant below 30 m. O2 is an exception; the concentration of this gas in air is changing so slowly (on a percentage basis) that seasonal thermal gradients are significant down to 60-m depth.
These processes combine to influence the composition of gas throughout the firn, and at its base where gases are trapped as bubbles in impermeable ice. Here, two modes of trapping are possible. First, seasonal layering may be absent and air may be trapped throughout the bubble closeoff zone. In this case, the composition of the bulk trapped gases in ice cores will be further convoluted because of the finite closeoff interval. At Vostok, for example, the bubble closeoff zone is about 8 m thick. A single layer of ice traps bubbles throughout the ≈300 yr it moves through this zone. This process accounts for the largest share of the dispersion of gas ages in a single sample of ice. At Summit, Greenland, on the other hand, high-density layers are completely sealed as they pass through the top of the bubble closeoff zone. Sealing forms vertically impermeable layers that prevent additional diffusive mixing and “locks in” the composition of gas present in the open, intervening, low-density layers. In such a case, individual ice samples can resolve time periods as short as a decade. The influence of lock-in at Summit can be seen from the firn air data at GISP2 (Fig. 2). δ15N ceases to rise below 70 m depth. CH4 and CO2 concentrations fall rapidly below this depth; CO2 and CH4 “ages” of firn air increase about as rapidly as ice ages below 70 m (4 yr/m). These firn air results are similar to those of Schwander et al. (4), who developed the method for firn air sampling and studied the composition of air in the firn at the nearby GRIP site.
The final process influencing the composition of gas in polar firn and ice cores is effusion. Craig et al. (8) suggested this process to account for the depletion of O2 and Ar relative to N2 in polar ice samples, as measured first by Raynaud and Delmas (13) and later by Craig et al. (8) and Sowers et al. (9). Craig et al. (8) pointed out that O2 and Ar, with diameters of about 3 Å, were smaller than N2 (diameter 3.3 Å). They suggested that cracks and imperfections in the ice of about 3 Å spacing would allow the effusive loss of O2 and Ar while selectively retaining nitrogen. It is not clear whether effusion takes place in situ during bubble closeoff or after ice cores are retrieved. Some indications for the former possibility are that deep firn air samples are enriched in O2 and Ar relative to N2, and that the O2 and Ar depletions of ice core samples do not increase after cores have been on the surface for a few days.
The processes affecting gases in ice cores need to be taken into account in reconstructions of the composition of the past atmosphere. First, measured concentrations of gases in ice cores and firn air need to be corrected for effects of gravitational fractionation and, where appropriate, thermal fractionation. Second, gas records are useful only when dated absolutely or on a time scale common to other records. Because bubbles close at depths of 40–120 m, gases are younger than the ice enclosing them. The gas age–ice age difference (Δage) is as great as 7 kyr in glacial ice from Vostok; it is as low as 30 yr in the rapidly accumulating Antarctic core DE 08. There are substantial uncertainties associated with Δage, limiting our ability to interpret some records. This is not a problem when reconstructing the anthropogenic transient from ice core studies, because one can align the recent part of ice core records with direct observations and assume that Δage is constant below the interval of overlap.
Once trapped in bubbles, air in ice cores is subject to two additional processes. First, bubbles are compressed under hydrostatic pressure (14). Second, gases eventually begin to dissolve in the ice as air hydrates (14, 15). Nucleation is kinetically limited, and at ambient temperatures and pressures occurs over order 104 yr (15). As a result, air hydrates form at depths of about 400–1,500 m; cold temperatures and slow accumulation favor formation at shallower depths.
Different gases form clathrates at different pressures (16). In the long zone over which air hydrates and bubbles coexist in ice cores, the composition of gases in bubbles must be different from the composition in bulk ice. When gas samples are extracted after crushing of ice for a sufficiently long period of time, as is done for CO2 analysis, individual compounds are apparently not fractionated despite the fact that overall extraction efficiency is <100%. The evidence for this statement is that coherent records of CO2 are obtained by analyzing different ice cores, despite the fact that the dissolution of gases occurs in samples of different ages.
Reconstructions of the Anthropogenic Transient from Ice Core and Firn Air Chemistry
CO2.
The most extensive study of the preindustrial CO2 concentration of air and its anthropogenic rise is that of Etheridge et al. (17). Their results are based largely on studies of the DE 08 ice core, from Law Dome, Antarctica (66° 43′ S, 113° 12′ E; elevation 1,250 m). The high accumulation rate, about 1.2 m/yr, and warm annual temperature (−19°C) at the site of this core (which causes the closeoff depth to be relatively shallow) allow time to be resolved exceptionally well. Etheridge et al. (17) estimate the gas age–ice age difference to be only 30 yr and the duration of the bubble closeoff process to be 8 yr. They supplement their results from DE-08 with the nearby DSS core and compare their CO2–time curve with that from Siple Dome, analyzed by Neftel et al. (18).
Results are summarized in Fig. 3. The CO2 concentration of air varies in the range 275 ± 5 ppm between 1350 and 1800. The anthropogenic rise begins between 1780 and 1870, the later age corresponding to the time when CO2 clearly begins rising above the highest level of the previous 500 yr. As inferred from the study of the DE 08 core, CO2 generally rises at a rate that tends to increase with time. The most dramatic exception is the interval of constant concentration inferred for the period between 1935 and 1945. The DE 08 record is in good agreement with the DSS and Siple data. The exception is the period between 1935 and 1945, when CO2 concentrations from the latter two cores fall below those of DE 08 by up to 5 ppmv.
Figure 3.

(Upper) CO2 vs. time before present, as inferred by Etheridge et al. (17) from ice core studies. (Lower) CH4 vs. time before present, as inferred by Etheridge et al. (19) from ice core studies.
Box-diffusion model deconvolutions, which assume CO2 concentrations to be in good agreement with the Siple results, invoke a net biosphere release of about 0.1–0.5 Gt C/yr during this period (20, 21). Etheridge et al. (17) suggest that one can balance the carbon cycle between 1935 and 1945 with a combination of lower respiration, increased growth due to CO2 fertilization and changes in temperature and precipitation, and suppressed CO2 input to the atmosphere as a result of exceptional El Nino events. Otherwise the new data of Etheridge et al. (17) are consistent with the Siple CO2 data and estimates of carbon fluxes derived therefrom. These invoke rising CO2 beginning around 1780, with the biosphere as the major source. Fossil fuel becomes the dominant source around 1910. The biosphere continues to be a source until 1940 in the deconvolution of Siegenthaler and Oeschger (20), when it becomes neutral, and 1970 in the deconvolution of Keeling et al. (21), when it becomes a sink.
CH4.
Two recent records (19, 22) supplement earlier work of Craig and Chou (23), Etheridge et al. (24), Khalil and Rasmussen (25), and Stauffer et al. (26) to provide a detailed curve of the anthropogenic increase in atmospheric CH4. The results of Blunier et al. (22) suggest a stable preindustrial value of about 720 ppbv over Antarctica between about 1000 A.D. and 1750 A.D. The Antarctic concentration rise after 1840, as measured in the DE 08 core by Etheridge et al. (24), is shown in Fig. 3.
The factors maintaining the preanthropogenic CH4 background, and those responsible for the anthropogenic increase, have been discussed in a number of papers (refs. 27 and 28 and citations therein). According to Chappellaz et al. (29, 30), preindustrial CH4 production was dominated by tropical wetlands, with grassland and temperate to boreal forests also making significant contributions. They argue that the natural source has diminished by 20% since preindustrial times. Anthropogenic sources are, of course, far greater. Chappellaz et al. (29) estimate that an anthropogenic decrease of about 20% in the atmospheric OH concentration has caused the atmospheric lifetime of CH4 to rise and thereby contributed to the anthropogenic increase.
Other Gases.
Studies of trapped gases in ice cores have revealed preanthropogenic levels of two other properties. The preanthropogenic N2O concentration is estimated to have been about 275–285 ppbv, compared with a contemporary value of about 311 ppbv (24, 31, 32). Because of its high solubility, the precise measurement of this gas in ice cores is very difficult. The δ13C of CO2 over Antarctica decreased from a preindustrial level of about −6.5‰ to approximately −8.0‰ today (33). The decrease roughly mirrors the CO2 increase.
Firn Air Studies.
Sampling and analysis of gas in the firn is an emerging approach for improving existing records of anthropogenic transients (4, 7). Firn air sampling allows one to collect 1,000 liters or more of isotopically unfractionated air as old as 90 yr. These samples can be used to measure concentrations of trace organic compounds, O2/N2 ratios, and the isotopic composition of greenhouse gases, all measurements that are far more difficult to make on ice core samples.
Glacial–Interglacial Changes in Atmospheric Chemistry Recorded in Ice Cores
Selected climate records are summarized in Figs. 4 and 5, covering the periods from 0 to 350 kyr and from 0 to 100 kyr before the present (B.P.), respectively. The δ18O of calcitic foraminifera from deep sea sediments is a proxy indicator for ice volume. The δD or δ18O of ice from ice cores is a proxy indicator of temperature in the area of the ice core. Recent inversions of borehole temperature data conclude that the glacial–interglacial temperature change in Greenland was about 20°C (39, 40). The dust content of ice cores is a proxy for dryness of the source areas. High dust contents are generally attributed to dry source areas from which dust is readily suspended into the atmosphere. CO2 and CH4 concentrations are measured in bubbles of polar ice as described above. June solar insolation at 65° N latitude is plotted at the bottom of the figure.
Figure 4.

Selected ice core records for the last 350 kyr. Data from refs. 34–37.
Figure 5.

Selected ice core records for the last 100 kyr. Data from refs. 30, 34, 35, and 38.
On a broad scale, the atmospheric CO2 concentration is clearly linked to climate change. Lorius et al. (41) argued that the glacial–interglacial CO2 change (with a small contribution from CH4) was responsible for a 2°C temperature change when all feedbacks are considered. Obviously this number is highly uncertain but it is very likely that increasing CO2 concentrations in air contributed to global warming during glacial terminations 1 and 2. The link between CO2 and higher-frequency climate changes is weak. For example, there is no minimum in CO2 corresponding to the cold period (Glacial Stage 5d) at about 110 kyr B.P. Similarly, CO2 does not rise during the interstadial events of the past 35 kyr (ref. 42; Fig. 5).
Many factors have been invoked to explain the glacial-interglacial change in the CO2 concentration of air. The cause of this change was vigorously debated for a decade beginning in 1982. Recently attention to this subject has diminished, more because the protagonists are exhausted than because the issue is resolved. It is widely accepted that the pCO2 of the atmosphere is regulated by the pCO2 of surface seawater, because about 99% of the CO2 in the ocean/atmosphere system resides in the oceans. In his classic paper initiating the debate, Broecker (43) noted that lower glacial temperatures would cause the CO2 concentration of air to fall, whereas higher salinities would cause CO2 to rise. A glacial ocean temperature decrease of 1.5°C would cause CO2 to fall by 20 ppmv. This change would be roughly offset by a salinity increase of 1‰. Recent controversial results showing that equatorial temperatures may have increased by up to 5°C suggest that the glacial–interglacial change in sea surface temperatures may have been much larger than Broecker initially estimated. If this is correct, the temperature change may have contributed 20–30 ppm of the glacial–interglacial change in atmospheric pCO2 after correcting for the compensating effect of salinity (the temperature dependence of pCO2 on sea surface temperature is about 13 ppmv/°C; ref. 43).
Clearly other factors must contribute to the glacial-interglacial difference in atmospheric pCO2. These factors must act either by decreasing the total CO2 concentration of surface seawater or by increasing the surface alkalinity. Changes in ocean circulation, the nutrient/carbon ratio of organic matter, and varying rates of nutrient utilization in Antarctic waters have been invoked to account for the changing surface water total CO2 concentrations. Removal of CaCO3 during sea level rise, changing ratios of CaCO3 to organic carbon in biogenic debris falling out of the surface ocean, and changes in the calcium carbonate compensation depth in the deep ocean have been invoked to change the alkalinity of surface waters.
There have been large changes in the CH4 concentration of air during the last ≈200 kyr (Figs. 4 and 5). CH4 changes are very closely linked with changes in climate (29, 44). CH4 differs from CO2 in two important ways. First, CO2 changes are about five times more important than those of CH4 in driving glacial–interglacial temperature changes (41). Second, CH4 changes, which are believed to be caused mainly by changes in emission rates, are somewhat better understood and serve much better as a climate proxy. Chappellaz et al. (27) generally interpreted changes in atmospheric CH4 as reflecting increases in the extent of low-latitude wetlands due to increased precipitation and perhaps temperatures.
Recent records of atmospheric CH4 variations over the last 200 kyr (Figs. 4 and 5) reveal several very interesting features. As noted above, there is a large glacial–interglacial change, with Antarctic CH4 concentrations varying between glacial levels of 350 ppbv and interglacial levels of 700 ppbv. There are changes of somewhat smaller magnitude associated with climate changes linked to the 20-kyr climate cycles associated with precession. Examples are CH4 maxima associated with warm periods at 80 kyr and 100 kyr B.P. Third, there are large CH4 variations associated with interstadial events of the last 40 kyr. These events, recorded most dramatically in Greenland ice cores (e.g., ref. 30; Fig. 5), reflect rapid warming over Greenland, slow cooling, and rapid cooling back to baseline glacial values. During the last 40 kyr at least, interstadial warmings are accompanied by increases of about 150 ppbv in the CH4 concentration of air. Chappellaz et al. (30) invoked wetter and warmer conditions in the tropics to explain the CH4 increases. Support for this idea comes from the fact that the longer interstadial events, at least, are recorded in Antarctica, illustrating their global nature (45), and are sometimes coincident with deposition of cave deposits in Botswanaland, which Holmgren et al. (46) linked to increased tropical precipitation. Finally, the CH4 concentration of the atmosphere was surprisingly variable during the Holocene. Methane was at a maximum early in the Holocene, fell to a broad minimum about 5 kyr B.P., and has risen during the last 3 kyr (22). Only the abrupt minimum at about 8.2 kyr B.P. has been directly linked to a climate event, namely an abrupt cooling that occurred at that time. The causes of the mid-Holocene minimum and subsequent slow rise are topics of current investigation.
Acknowledgments
This research was supported by grants from the New England Regional Center of the National Institute of Global Environmental Change, Department of Energy (Subagreement 901214-HAR), and the Office of Polar Programs, National Science Foundation.
ABBREVIATION
- kyr
thousands of years
References
- 1.Herron M M, Langway C C., Jr J Glaciol. 1980;25:373–385. [Google Scholar]
- 2.Raynaud D, Lorius C. Proceedings of the Grenoble Symposium, August/September 1975. Vol. 118. United Kingdom: International Association of Hydrological Sciences; 1977. pp. 326–335. [Google Scholar]
- 3.Martinerie P, Raynaud D, Etheridge D M, Barnola J-M, Mazaudier D. Earth Planet Sci Lett. 1992;112:1–13. [Google Scholar]
- 4.Schwander J, Barnola J-M, Andrie C, Leuenberger M, Ludin A, Raynaud D, Stauffer B. J Geophys Res. 1993;98:2831–2838. [Google Scholar]
- 5.Sowers T, Bender M, Raynaud D, Korotkevich Y S. J Geophys Res. 1992;97:15683–15697. [Google Scholar]
- 6.Schwander J, Stauffer B, Sigg A. Annu Glaciol. 1988;10:141–145. [Google Scholar]
- 7.Battle M, Bender M, Sowers T, Tans P, Butler J, Elkins J, Ellis J, Conway T, Zhang N, Lang P, Clarke A. Nature (London) 1996;383:231–235. [Google Scholar]
- 8.Craig H, Horibe Y, Sowers T. Science. 1988;242:1675–1678. doi: 10.1126/science.242.4886.1675. [DOI] [PubMed] [Google Scholar]
- 9.Sowers T, Bender M, Raynaud D. J Geophys Res. 1989;94:5137–5150. [Google Scholar]
- 10.Schwander J. In: The Environmental Record in Glaciers and Ice Sheets. Oeschger H, Langway C, editors. New York: Wiley; 1989. [Google Scholar]
- 11.Severinghaus, J. P. (1995) Ph.D. dissertation (Columbia University, New York).
- 12.Severinghaus J P, Bender M L, Keeling R F, Broecker W A. Geochim Cosmochim Acta. 1996;60:1005–1018. [Google Scholar]
- 13.Raynaud D, Delmas R. Proceedings of the Grenoble Symposium, August/September 1975. Vol. 118. United Kingdom: International Association of Hydrological Sciences; 1977. pp. 377–381. [Google Scholar]
- 14.Gow A J, Williamson T. J Geophys Res. 1975;80:5101–5108. [Google Scholar]
- 15.Price P B. Science. 1975;267:1802–1804. doi: 10.1126/science.267.5205.1802. [DOI] [PubMed] [Google Scholar]
- 16.Miller S L. Science. 1969;165:489–490. doi: 10.1126/science.165.3892.489. [DOI] [PubMed] [Google Scholar]
- 17.Etheridge D M, Steele L P, Langenfelds R L, Francey R J, Barnola J-M, Morgan V I. J Geophys Res. 1995;101:4115–4128. [Google Scholar]
- 18.Neftel A, Moor E, Oeschger H, Stauffer B. Nature (London) 1985;315:45–47. [Google Scholar]
- 19.Etheridge D M, Pearman G I, Fraser P J. Tellus. 1992;44B:282–294. [Google Scholar]
- 20.Siegenthaler U, Oeschger H. Tellus. 1987;39B:140–154. [Google Scholar]
- 21.Keeling, C. D., Bacastow, R. B., Carter, A. F., Piper, S. C., Whorf, T. P., Heimann, M., Mook, W. G. & Roeloffzen, H. (1989) in Aspects of Climate Variability in the Pacific and Western Americas, Geophysical Monograph, ed. Peterson, J. H. (Amer. Geophysical Union, Washington, DC), Vol. 55, pp. 165–236.
- 22.Blunier T, Chappellaz J, Schwander J, Stauffer B, Raynaud D. Nature (London) 1995;374:46–49. [Google Scholar]
- 23.Craig H, Chou C C. Geophys Res Lett. 1982;9:1221–1224. [Google Scholar]
- 24.Etheridge D M, Pearman G I, de Silva F. Annu Glaciol. 1988;10:28–33. [Google Scholar]
- 25.Khalil M A K, Rasmussen R A. Atmos Environ. 1987;21:2445–2452. [Google Scholar]
- 26.Stauffer B, Fischer G, Neftel A, Oeschger H. Science. 1985;229:1386–1388. doi: 10.1126/science.229.4720.1386. [DOI] [PubMed] [Google Scholar]
- 27.Aselmann I, Crutzen P J. J Atmos Chem. 1989;8:307–358. [Google Scholar]
- 28.Fung I, John J, Lerner J, Matthews E, Prather M, Steele L P, Fraser P J. J Geophys Res. 1991;96:13033–13065. [Google Scholar]
- 29.Chappellaz J, Fung I, Thompson A. Tellus. 1993;45B:228–241. [Google Scholar]
- 30.Chappellaz J, Blunler T, Raynaud D, Barnola J M, Schwander J, Stauffer B. Nature (London) 1993;366:443–445. [Google Scholar]
- 31.Khalil M A K, Rasmussen R. Annu Glaciol. 1988;10:73–80. [Google Scholar]
- 32.Machida T, Nakazawa T, Fujii Y, Aoki S, Watanabe O. Geophys Res Lett. 1995;22:2921–2924. [Google Scholar]
- 33.Friedli H, Lotscher H, Oeschger H, Siegenthaler U, Stauffer B. Nature (London) 1986;324:237–238. [Google Scholar]
- 34.Shackleton J H, Pisias N G. In: The Carbon Cycle and Atmospheric CO2: Natural Variations Archean to Present, Geophysical Monograph Series. Sundquist E T, Broecker W S, editors. Vol. 32. Washington, DC: Amer. Geophysical Union; 1985. pp. 303–317. [Google Scholar]
- 35.Jouzel J, Barkov N I, Barnola J M, Bender M L, Chappellaz J, Genthon C, Kotlyakov V M, Lipenkov V, Lorius C, Petit J R, Raynaud D, Raisbeck G, Ritz C, Sowers T, Stievenard M, Yiou F, Yiou P. Nature (London) 1993;364:407–412. [Google Scholar]
- 36.Barnola J M, Pimienta P, Raynaud D, Korotkevich Y S. Tellus. 1991;43B:83–90. [Google Scholar]
- 37.Petit J R, Mounier L, Jouzel J, Korotkevich Y, Kotlyakov V, Lorius C. Nature (London) 1990;343:56–58. [Google Scholar]
- 38.Grootes P M, Stuvier M, White J W C, Johnsen S, Jouzel J. Nature (London) 1993;366:552–555. [Google Scholar]
- 39.Cuffey K, Alley R, Grootes P, Bolzan J, Anandakrishnan S. J Glaciol. 1994;40:341–349. [Google Scholar]
- 40.Johnsen S, Dahl-Jensen D, Dansgaard W, Gundestrup N. Tellus. 1995;47B:624–629. [Google Scholar]
- 41.Lorius C, Jouzel J, Raynaud D, Hansen J, Le Treut H. Nature (London) 1990;347:139–145. [Google Scholar]
- 42.Sowers T, Bender M. Science. 1995;269:210–214. doi: 10.1126/science.269.5221.210. [DOI] [PubMed] [Google Scholar]
- 43.Broecker W. Geochim Cosmochim Acta. 1982;46:1689–1705. [Google Scholar]
- 44.Chappellaz J, Barnola J M, Raynaud D, Korotkevich Y S, Lorius C. Nature (London) 1990;345:127–131. [Google Scholar]
- 45.Bender M, Sowers T, Dickson M L, Orchardo J, Grootes P, Mayewski P A, Meese D A. Nature (London) 1994;372:663–666. [Google Scholar]
- 46.Holmgren K, Karlen W, Shaw P A. Quat Res. 1995;43:320–328. [Google Scholar]