

Abstract
Rationale
Senescence of pulmonary artery-smooth muscle cells (PA-SMCs) caused by telomere shortening or oxidative stress may contribute to pulmonary hypertension (PH) associated with chronic lung diseases.
Objective
To investigate whether cell senescence contributes to pulmonary vessel remodeling and PH in chronic obstructive pulmonary disease (COPD).
Methods and results
In 124 patients with COPD, investigated by right heart catheterization, we found a negative correlation between leukocyte telomere length and PH severity. In-depth investigations of lung vessels and derived cultured PA-SMCs showed greater severity of remodeling and increases in senescent p16- and p21-positive PA-SMCs and proliferating Ki67-stained cells in 14 patients with COPD compared to 13 age- and sex-matched control smokers. Cultured PA-SMCs from COPD patients displayed accelerated senescence, with fewer cell-population doublings, an increased percentage of beta-galactosidase-positive cells, shorter telomeres, and higher p16 protein levels at an early cell passage, compared to PA-SMCs from controls. Both in situ and in vitro PA-SMC senescence criteria correlated closely with the degree of pulmonary vessel wall hypertrophy. Because senescent PA-SMCs stained for p16 and p21 were virtually confined to the media near the Ki67-positive cells, which predominated in the neointima and hypertrophied media, we evaluated whether senescent cells affected normal PA-SMC functions. We found that senescent PA-SMCs stimulated the growth and migration of normal target PA-SMCs through the production and release of paracrine soluble and insoluble factors.
Conclusion
PA-SMC senescence is an important contributor to the process of pulmonary vascular remodeling that underlies PH in chronic lung disease.
Keywords: Aged; Cell Aging; physiology; Cells, Cultured; Female; Humans; Male; Middle Aged; Muscle, Smooth, Vascular; pathology; physiopathology; Myocytes, Smooth Muscle; pathology; physiology; Pulmonary Artery; pathology; physiopathology; Pulmonary Disease, Chronic Obstructive; etiology; pathology; physiopathology
Keywords: pulmonary hypertension, senescence, smooth muscle cells, remodeling
INTRODUCTION
Pulmonary hypertension (PH) may occur as a complication of various chronic lung diseases. Among these diseases, chronic obstructive pulmonary disease (COPD) is becoming increasingly prevalent and is expected to become the third leading cause of death worldwide by 2020.1 COPD is characterized by slowly progressive airflow obstruction, resulting in dyspnea and exercise limitation. COPD is also one of the most common causes of pulmonary hypertension (PH) and cor pulmonale.2, 3 Extensive pulmonary vessel remodeling with prominent intimal thickening, medial hypertrophy, and muscularization of the small arterioles are cardinal pathological features of PH in COPD.4 These structural changes are considered the main cause of the increase in pulmonary vascular resistance (PVR), but their pathogenesis remains uncertain.
COPD is an age-related disease associated with telomere shortening.5 One consequence of reduced telomere length is early replicative senescence of somatic cells, characterized by growth arrest, loss of specialized cellular functions, and genomic instability.6, 7 Premature cell senescence may also occur through non-telomeric signals, in response to various types of stress such as oxidative stress.7, 8 Senescent cells survive in vivo but acquire many changes in the expression of genes encoding various cytokines, proteases, and growth factors.9, 10 These changes in gene expression may act not only to reinforce the senescence-related growth arrest, but also in a paracrine manner to promote degenerative or hyperproliferative changes in neighboring cells.9, 10 There is now widespread agreement that senescent cells can be deleterious and contribute to age-related diseases. Consistent with this view, senescent cells increase with age in mammalian tissues and are found at sites affected with age-related diseases such as osteoarthritis and atherosclerosis.11 Our previous report of marked telomere shortening in patients with COPD is consistent with the increased number of senescent cells found in lungs of patients with COPD compared to control smokers.5, 12 However, the role for cell senescence in the lung alterations characteristic of COPD has not yet been examined.
Here, we reasoned that senescent cells in COPD may contribute to the process of pulmonary vascular remodeling, and therefore to the pathogenesis of PH. First, to evaluate the hypothesis that telomere shortening was associated with PH in patients with COPD, we measured telomere length in circulating leukocytes from 124 patients with COPD investigated by right heart catheterization. Then, we assessed pulmonary vascular cell senescence by studying lung specimens and derived cultured pulmonary artery smooth muscle cells (PA-SMCs) from 14 patients with COPD and 13 age- and sex-matched control smokers. Finally, we investigated the propensity of senescent cells to release soluble and insoluble factors and to alter the migration and proliferation of normal target PA-SMCs, thereby contributing to the process of pulmonary vascular remodeling.
METHODS
Study population
We evaluated two groups of patients. The first group consisted of 124 patients with COPD who underwent right heart catheterization and telomere length measurement. The data from 91 of these patients in whom inflammatory biomarkers were assayed have been published previously13 (Table 1). The second group consisted of 27 patients treated with lung resection surgery for localized lung tumors and recruited prospectively at the Hotel-Dieu Teaching Hospital (Paris, France); of these 27 patients, 14 had COPD and 13 were defined as controls (Table 2). In this second group, lung tissue samples and derived cell cultures were studied. Inclusion criteria for COPD were an at least 10-pack-year history of tobacco smoking and a forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio <70%. Inclusion criteria for the control smokers were a smoking history greater than 10 pack-years; an FEV1/FVC ratio greater than 70%; and the absence of chronic cardiovascular, hepatic, and renal disease. None of these patients had received chemotherapy. This study was approved by the institutional review board of the Henri Mondor Teaching Hospital (Créteil, France). All patients and controls signed an informed consent document before study inclusion.
Table 1.
Pulmonary hemodynamic, physiological, and biological variables in 124 patients with COPD and correlations with telomere length in circulating leukocytes.
| Mean±SEM | Correlation with telomere length | ||
|---|---|---|---|
| r | P value | ||
| Females/males | 29/95 | - | - |
| Telomere length (T/S ratio) | 0.56±0.01 | - | - |
| Age, y | 64.1±0.7 | − 0.20 | 0.02 |
| Pack-years | 50.7±2.4 | 0.05 | 0.52 |
| FEV1, % | 41.5±1.6 | 0.04 | 0.65 |
| FEV1, L | 1.1±0.0 | 0.09 | 0.29 |
| FVC, L | 2.7±0.1 | 0.06 | 0.45 |
| FEV1/FVC, % | 47.4±1.2 | 0.02 | 0.82 |
| Pap, mmHg | 24.6±0.6 | − 0.20 | 0.04 |
| Pcwp, mmHg | 10.4±0.4 | 0.04 | 0.64 |
| Rap, mmHg | 5.7±0.3 | 0.02 | 0.82 |
| Cardiac index, L/min/m2 | 2.7±0.1 | 0.02 | 0.82 |
| PVR, Wood Units | 3,1±0.1 | − 0.29 | 0.01 |
| IL-6, pg/mL | 3.8±0.4 | − 0.20 | 0.04 |
| IL-8, pg/mL | 13.4±0.9 | 0.05 | 0.59 |
| IL-1β, pg/mL | 0.81±0.1 | 0.02 | 0.85 |
| TGF-β, pg/mL | 28,4±1,4 | 0.03 | 0.77 |
| TNF-α, pg/mL | 1.7±0.1 | − 0.03 | 0.73 |
| MCP-1, pg/mL | 530.7±23.0 | − 0.18 | 0.06 |
Table 2.
Comparison of clinical characteristics and pathological variables between patients with COPD and control smokers
| Patients (n = 14) | Controls (n = 13) | P value | |
|---|---|---|---|
| Females/males | 3/11 | 4/9 | |
| Age, y | 64.2 ± 1.8 | 59.1 ± 3.3 | 0.15 |
| Pack-years | 49.4 ± 6.1 | 37.1 ± 4.0 | 0.10 |
| FEV1, % | 74.7 ± 4.1 | 91.9 ± 5.5 | 0.003 |
| FEV1, L | 2.05 ± 0.15 | 2.69 ± 0.25 | 0.027 |
| FVC, L | 3.2 ± 0.2 | 3.8 ± 0.4 | 0.21 |
| FEV1/FVC | 63.6 ± 1.7 | 77.0 ± 1.4 | 0.0001 |
| Systolic Pap, mmHg | 33.2 ± 2.9 | 25.0 ± 1.6 | 0.04 |
| Wall thickness area, Ø 0–99 μm, 10−4 mm2 | 41.0 ± 3.3 | 27.4 ± 1.7 | 0.001 |
| Wall thickness area, Ø 100–199 μm, 10−4 mm2 | 123.2 ± 5.2 | 87.9 ± 5.4 | 0.00001 |
| Wall thickness area, Ø 200–399 μm, 10−4 mm2 | 292.0 ± 13.0 | 242.9±23.2 | 0.05 |
| Wall thickness area, % | 72.0 ± 1.9 | 55.8 ± 1.9 | 0.0001 |
Data are means±SEM. P values are for the comparisons between patients and smokers.
Laboratory investigations
Pulmonary vascular remodeling was quantified based on histomorphometric analyses and cell senescence was assessed by in situ analysis of lung tissue sections.4 Senescent PA-SMCs within the pulmonary vascular wall of distal pulmonary vessels were identified by p16 and p21 immunostaining and proliferative cells by Ki67 immunostaining (see the online data supplement).
Cultured PA-SMCs collected from pulmonary arteries of patients with COPD and controls were subjected to repeated cell passages to determine their threshold for replicative senescence and the total number of cell population doublings (PDL). Two main criteria were used for subsequent analyses: the PDL and the percentage of beta-galactosidase (β-gal)-positive cells at passage 2 and at senescence. The amounts of p53, p16, and p21 proteins were determined at passage 2 and at senescence by western blotting. Telomere length was assessed using a real-time quantitative polymerase chain reaction (PCR)-based assay5 and expressed as the ratio of the telomere repeat copy number over the single-gene copy number (36B4 gene). Genomic DNA was extracted either from blood samples or from smooth muscle cells at passage 2 and at senescence. Soluble factors (IL-6, IL-8, MCP-1, TNF-α, IL-1β and TGF-β) were measured in plasma and cell media using an ELISA. Functional studies were performed to investigate the impact of paracrine soluble and insoluble factors released by senescent cells on the proliferation and migration of target PA-SMCs. (See the online data supplement.)
Statistical analysis
Data are expressed as mean±SEM. Patients with COPD and controls were compared using the unpaired t-test for quantitative variables and the chi-square test for categorical variables. Correlations between variables were evaluated using least-square linear regression techniques. The effects of senescence in cells from patients with COPD and controls were assessed using a paired t-test. P values less than 0.05 were considered significant. Data were analyzed using Stata statistical software (release 8.0; StataCorp, College Station, TX, USA).
RESULTS
Relationship between telomere length and pulmonary hemodynamics in patients with COPD investigated by right heart catheterization
The characteristics of the 124 patients with COPD are reported in Table 1. Telomere length was associated with age but not with the degree of airflow limitation or with Sap or creatinine levels. Telomere length correlated negatively with Pap (range, 11–47 mmHg) and PVR (range, 1–9.5 Wood Units) (Table 1 and Online Figure I). Telomere length also correlated negatively with circulating IL-6 levels, which correlated positively with Pap (r=0.31; P<0.01) and PVR (r=0.42; P<0.001). Patients with telomere lengths lower than or equal to the median value (0.6, T/S ratio) had higher Pap and PVR values than did patients with telomere lengths greater than 0.6 (Online Figure II).
Characteristics of patients with COPD and controls included in the study of lung tissue and derived cultured cells
Table 2 reports the clinical features of the surgical patients with and without COPD, as well as the histological findings from their lung specimens. The group with COPD did not differ significantly from the group of control smokers regarding age, sex ratio, smoking history, body mass index, or mean Sap. Systolic Pap was higher in the patients with COPD than in the controls and correlated positively with the wall thickness area (r=0.38, P<0.05). Pulmonary vascular remodeling (wall thickness area) was more severe in patients with COPD than in controls, whether remodeling was assessed based on selected vessel-size categories or on all vessel-size categories in a given individual.
In situ analysis of p16-, p21-, and Ki67-stained PA-SMCs, of p16- and p21- stained endothelial cells, and of collagen in distal pulmonary vessels from patients with COPD and controls
Senescent PA-SMCs were identified as p21- and p16-stained cells. Preliminary experiments were performed in proximal pulmonary arteries to verify that α-SMA-positive cells, when positive for β-galactosidase activity, were also positive for p16 and p21, indicating that they were senescent PA-SMCs. In distal pulmonary vessels, the number of senescent cells expressed as the percentage of α-SMA-positive PA-SMCs also positive for p16 and p21 was considerably higher in pulmonary vessels from patients with COPD than in those from controls (Figure 1A). Similar differences in Ki67-positive proliferating cells were observed (Figure 1A). Of note, senescent p16- or p21-positive PA-SMCs and Ki67-stained cells predominated in remodeled pulmonary arteries, as shown by the tight relationship between the percentage of cells stained for p16, p21, or Ki67 and the wall thickness area ratio (Figure 1A). In addition, the percentage of Ki67+ cells correlated positively with the percentage of p16-stained (r=0.81, P<0.001) and p21-stained (r=0.84, P<0.001) PA-SMCs. At sites of vascular hypertrophy, proliferating cells were found chiefly in the neointima or hypertrophied media, whereas senescent cells were virtually confined to the media, with only a few senescent cells in the neointima (Figure 2).
Figure 1.
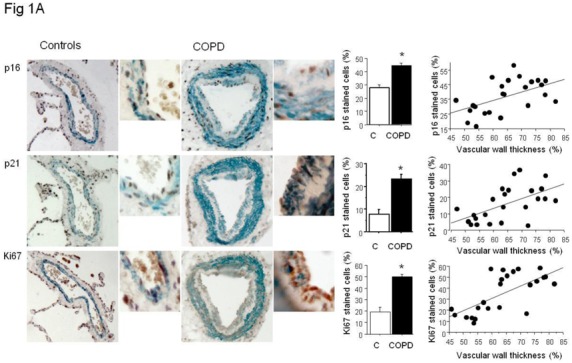
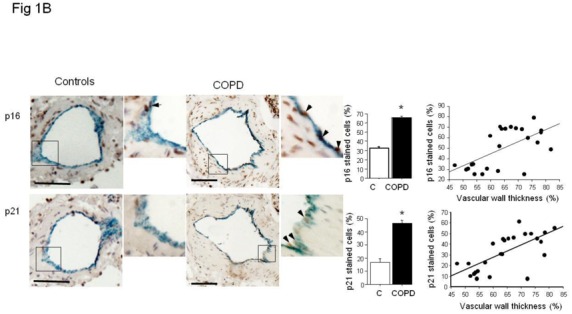
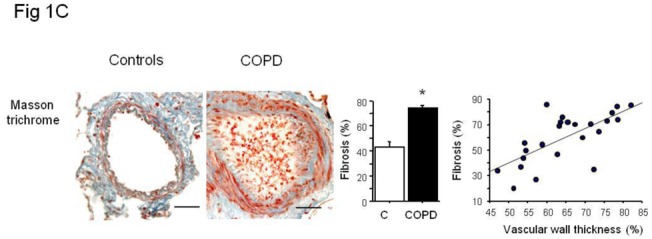
(A) Immunolocalization and quantification of α-SMA-positive smooth muscle cells (blue staining) stained for p16, p21, and Ki67 (brown staining) in sections of pulmonary vessels from patients with COPD and controls. Scale bar: 100 μm. Bar graphs represent the percentages of p16-, p21-, and Ki67-positive cells in pulmonary vessels from patients with COPD and controls. Values are means±SEM. *P<0.01 compared with values from controls. Correlations between the vascular wall area ratio and the percentage of p16-positive cells (r=0.54, P<0.01), p21 (r=0.56, P<0.01) and Ki67-positive cells (r=0.67, P<0.001 ) in patients with COPD and controls. (B) Similar representations for vWF positive endothelial cells stained for p16- and p21. Correlation between vascular wall thickness, and p16- (r=0.61, P<0.01), and p21-positive cells (r=0.68, P<0.001). (C) Similar representations for Masson trichrome staining where collagen appears in blue and cells in red. Correlation with collagen content (r=0.69, P<0.001).
Figure 2.
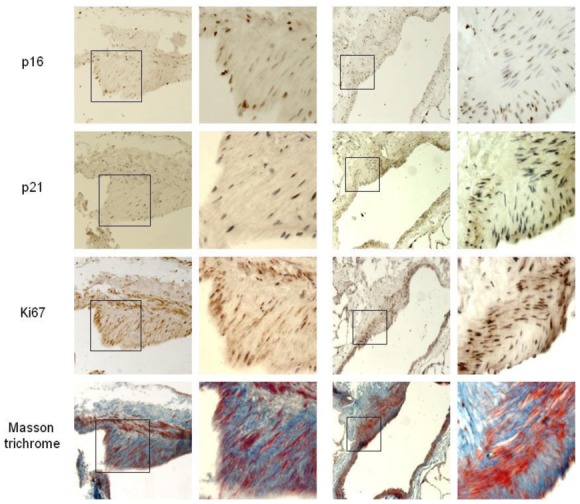
Immunolocalization of p16-, p21-, and Ki67-positive cells and collagen accumulation visualized by Masson trichrome staining in sections of remodeled pulmonary vessels from patients with COPD. Senescent PA-SMCs stained for p16 and p21 were virtually confined to the media, whereas Ki67-stained cells predominated in the neointima and hypertrophied media. Some p16 and p21 immunostaining was also associated with endothelial cells or PA-SMCs near the vessel lumen. In Masson trichrome-stained sections, the collagen is blue and the embedded cells are red.
The percentage of senescent endothelial cells expressed as the percentage of vWF stained cells also positive for p21 or p16 was higher in pulmonary vessels from patients with COPD than in those from controls and correlated positively with the wall thickness area ratio (Figure 1B). Similarly, more fibrosis was found in vessels from patients with COPD than controls (Figure 1C).
Replicative senescence of cultured PA-SMCs from patients with chronic obstructive lung disease and controls
Cultured PA-SMCs from pulmonary arteries of patients with COPD and controls were subjected to repeated cell passages to determine their threshold for replicative senescence and the total number of cell population doublings (PDL). As shown in Figure 3A, PA-SMCs from patients with COPD began senescing after passage 2–3, whereas those from controls began senescing after passage 5–6. Consequently, the PDL was twice as high in controls as in patients with COPD (Figure 3B). The percentage of β-galactosidase-positive cells was higher in patients with COPD than in controls at passage 2 then increased with subsequent passages and reached similar values in COPD patients and controls at the stage of cellular senescence (Figure 3C and D). In the overall population of surgical patients with and without COPD, PDL was tightly and inversely related to the wall thickness area ratio (r= −0.61; P<0.001), indicating a close relationship between in vitro criteria for senescence and the severity of pulmonary vascular remodeling (Figure 3E). A less significant relationship was found between the percentage of β-galactosidase-positive cells and the wall thickness area ratio (r=0.38; P<0.05).
Figure 3.
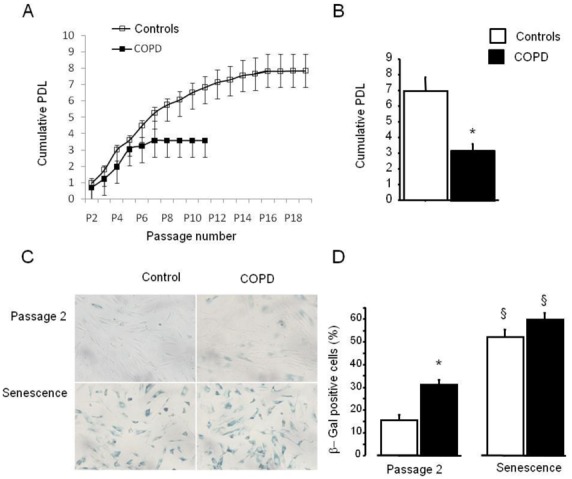
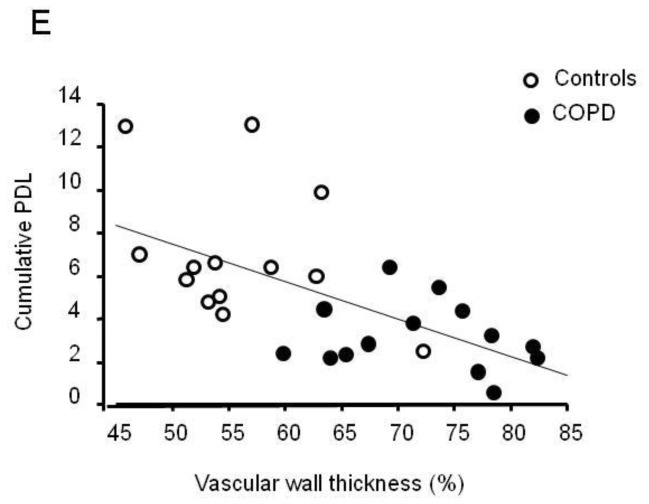
(A) Replicative senescence of PA-SMCs from patients with COPD and controls. Cells were subjected to repeated passages and counted at each passage, and the population doubling level (PDL) was calculated for patients with COPD and controls. (B) Data are means±SEM. *P<0.01, versus controls. (C) PA-SMCs were stained for senescence-associated β-galactosidase (β-Gal) activity at passage 2 and at senescence when cells began to exhibit proliferative arrest. (C): Representative photographs of cells stained for senescence-associated β-Gal activity at passage 2 and at senescence. (D) Percentage of β-Gal-positive cells. Data are means±SEM. *P<0.01 versus controls; §P<0.05, versus corresponding values at passage 2. (E) Correlations between the vascular wall area ratio and the PDL (r=−0.61; P<0.001).
Telomere length; telomerase activity; and levels of p53, p21, and p16 protein during replicative senescence of PA-SMCs from patients with COPD and controls
The expression of senescent regulatory proteins and telomere length in cultured PA-SMCs was assessed at passage 2 and at replicative senescence. With repeated PA-SMC passages, p53 and p21 increased, p16 decreased, and telomeres shortened until senescence was reached (Online Figure III). At passage 2, patients with COPD and controls differed regarding telomere length and p16 protein but not regarding p53 or p21. At senescence, telomere length was no longer significantly different between patients and controls, whereas the difference in p16 persisted. No telomerase activity was detected in PA-SMCs at any passage. Of note, PDL correlated strongly with the p16 level measured at passage 2 (r=−0.61; P<0.001) and less strongly with telomere length (r=0.37; P<0.05).
Factors secreted by PA-SMCs from patients with COPD and controls during replicative senescence
Because the cell senescent phenotype is not limited to an arrest of cell proliferation but includes widespread changes in protein expression and secretion, we measured the amounts of several cytokines and growth factors released by PA-SMCs from patients with COPD and controls at passage 2 and at senescence. As shown in Figure 4, soluble factors that increased from passage 2 to senescence included IL-6, IL-8, TNF-α, MCP-1, and TGF-β measured in the culture medium of PA-SMCs deprived of serum for 48 hours (IL1-β was not detectable in any of the samples). Among these factors, IL-6, IL-8, and TNF-α were found in higher concentrations in culture media of passage-2 cells from patients with COPD than from controls. At senescence, the differences were no longer significant, except for the difference in TNF-α. Of note, the amount of IL-6 released by passage-2 cells correlated with the percentage of β-galactosidase-positive cells (r= 0.40, P<0.05).
Figure 4.
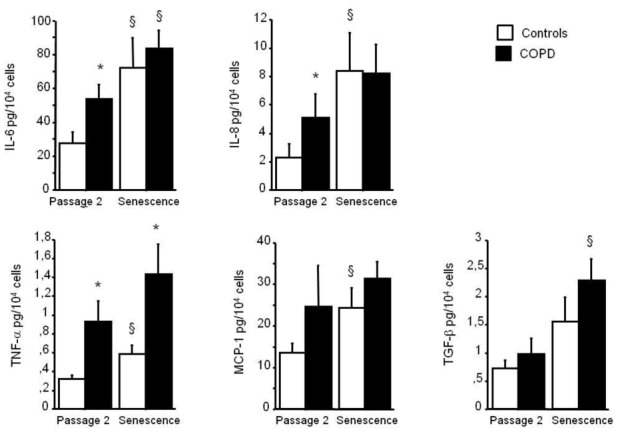
Levels of IL-6, IL-8, TNF-α, MCP-1, and TGF-β in PA-SMC media from the 14 patients with COPD and 13 controls, collected at passage 2 and at senescence. Each bar is the mean±SEM. * P<0.01 compared with values for PA-SMCs from controls. § P<0.05 compared with corresponding values for PA-SMCs at passage 2.
Contribution of secreted soluble and insoluble factors to PA-SMC proliferation and migration
To investigate whether secretion of soluble factors by senescent PA-SMCs affected the function of the target PA-SMCs, we evaluated the proliferation and migration of nonsenescent PA-SMCs treated with media from presenescent and senescent PA-SMCs. Growth stimulation of target PA-SMCs was more marked with medium of senescent PA-SMCs than with medium of presenescent cells (Figure 5A and C). Similarly, the medium of senescent PA-SMCs was more potent in stimulating PA-SMC migration than was the medium of presenescent cells (Figure 6A and B). Neutralizing antibodies to IL6 and MCP-1, but not to IL8 and TNF-α, markedly reduced PA-SMC proliferation induced by PA-SMC culture media (Figure 5D). The stimulatory effects of PA-SMC media from senescent and presenescent cells was no longer significantly different in the presence of anti-MCP-1- or IL6-antibodies (Figure 5D). In contrast, neutralizing antibodies to IL8, TNF-α, IL6, or MCP-1, did not affect PA-SMC migration in response to culture media from nonsenescent cells, whereas they inhibited PA-SMC migration induced by culture media from senescent cells, by about 25% (Figure 6D). Thus, neutralizing antibodies to IL-8, TNF-α, IL-6, and -MCP-1 abolished the differences in PA-SMC migration in response to culture media from senescent versus presenescent cells.
Figure 5.
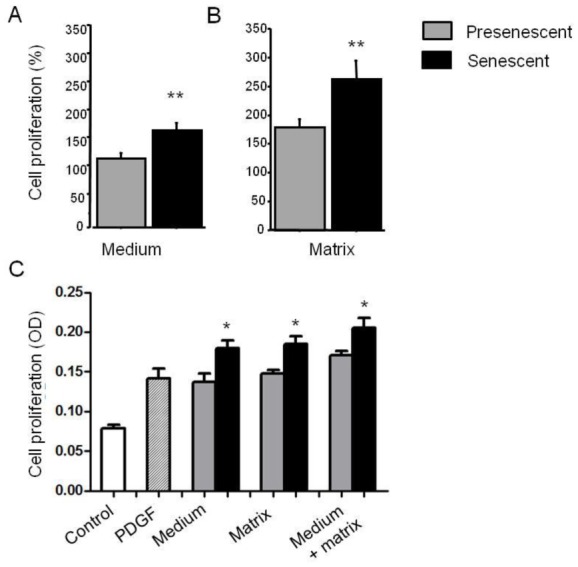
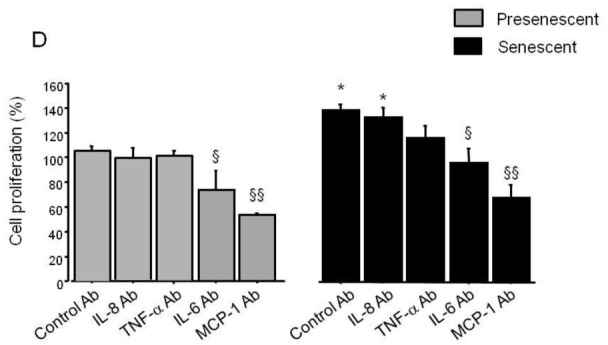
Growth stimulation of normal PA-SMCs by soluble (medium) or insoluble (matrix) factors secreted by presenescent (gray bars) or senescent (black bars) PA-SMCs. Presenescent cells were passage 3–4 cells from controls and senescent cells were passage 3–4 cells from patients with COPD. (A) Percentage of cells after exposure to the medium of presenescent or senescent PA-SMCs. (B) Percentage of cells plated onto matrices deposited by presenescent or senescent PA-SMCs. Values are mean±SEM of 12 values obtained from six independent experiments. (C) PA-SMC proliferation (OD: optical density, in arbitrary units) in a typical experiment where cells were stimulated by PDGF (20 ng/mL), medium, matrix, or medium combined with matrix from presenescent or senescent cells. (D) Percentage of cells after exposure to the medium of presenescent or senescent PA-SMCs in the presence of control or neutralizing antibodies to IL-8, TNF-α, IL-6, and MCP-1. *P<0.05 compared with values corresponding to stimulation by presenescent cells; § p<0.05 versus values with control antibodies.
Figure 6.
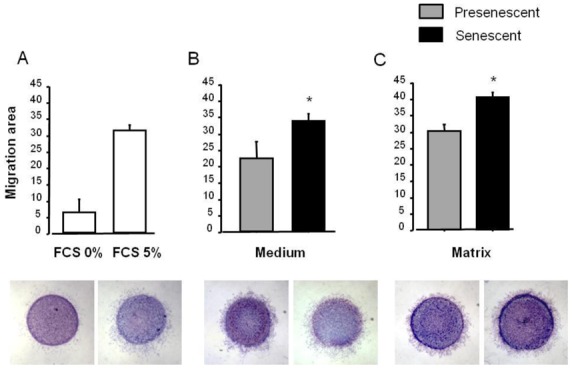
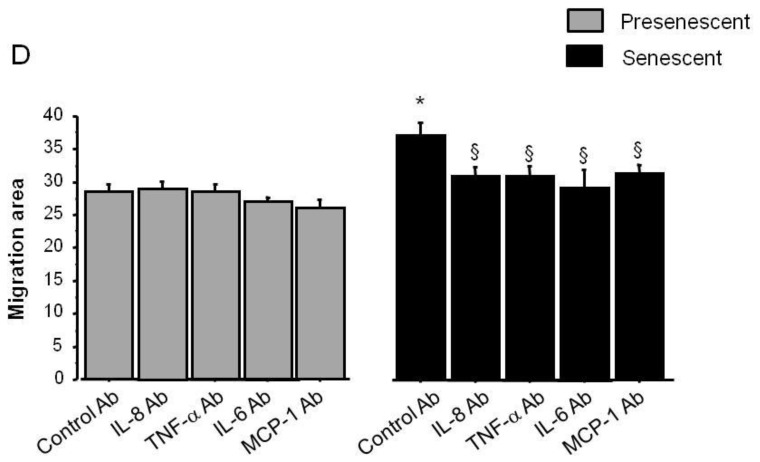
Stimulation of migration of normal PA-SMCs by soluble (medium) or insoluble (matrix) factors secreted by presenescent (gray bars) or senescent (black bars) PA-SMCs. Presenescent cells were passage 3–4 cells from controls and senescent cells were passage 3–4 cells from patients with COPD. (A) Migration area of cells measured 24 h after stimulation by 0% or 5% fetal calf serum. (B) Migration area of cells after exposure to the medium of presenescent or senescent PA-SMCs. (C) Migration area of cells onto matrices deposited by presenescent or senescent PA-SMCs. Values are mean±SEM of six values obtained from three independent experiments. Photomicrographs represent corresponding cells migrating from agarose droplets. (D) Migration area of cells after exposure to the medium of presenescent or senescent PA-SMCs in the presence of control or neutralizing antibodies to IL-8, TNF-α, IL-6, and MCP-1. *P<0.05, versus values corresponding to stimulation by presenescent cells; § P<0.05 versus values with control antibodies.
To determine the contribution of secreted matrices or insoluble factors, we allowed senescent or non-senescent PA-SMCs to deposit extracellular matrix onto culture dishes for 3 days, after which we removed the cells without altering the matrix and introduced new healthy PA-SMCs, which were assessed for growth or migration. We found that target PA-SMCs exhibited faster growth (Figure 5B and C) and greater migration (Figure 6C) in dishes coated with matrices secreted by senescent cells compared to non-senescent cells.
DISCUSSION
We show here that PA-SMC senescence is involved in the process of pulmonary vessel remodeling that underlies PH in patients with COPD. After finding a positive correlation between telomere shortening and PH severity in a large population of patients with COPD, we investigated the pulmonary vessels and derived cultured PA-SMCs from patients with or without COPD. We found that remodeled vessels were characterized by PA-SMC senescence and that cultured PA-SMCs from patients with COPD displayed accelerated senescence. Our finding that in situ and in vitro criteria for PA-SMC senescence correlated closely with the severity of pulmonary vascular remodeling, together with the presence of senescent cells near actively dividing cells at sites of vessel wall hypertrophy, strongly suggests a role for senescent cells in the remodeling process. Moreover, we found that accelerated PA-SMC senescence in COPD was associated with increased expression of soluble and insoluble factors that affected PA-SMC migration and proliferation. Taken together, these results support a role for PA-SMC senescence in the process of pulmonary vascular remodeling in COPD.
The role for telomere shortening as a pathogenic mechanism was recently highlighted in patients with familial idiopathic fibrosis who harbored a mutation in the telomerase gene14. Both COPD and pulmonary fibrosis are age-related diseases associated with telomere shortening.5, 14 Because short telomeres are associated with increased susceptibility to replicative cellular senescence, one current hypothesis is that cellular senescence represents one mechanism underlying the pathological alterations seen in these chronic lung diseases. Here, we focused on the process of pulmonary vascular remodeling that underlies PH in patients with COPD. In a population of patients with COPD investigated by right heart catheterization, we found that telomere shortening was associated with PH severity independently from the severity of airflow obstruction, age, and smoking history. [To evaluate whether cellular senescence was present in pulmonary vessels from patients with COPD and reflected a process related to pulmonary vascular remodeling, we compared pulmonary vessels and derived cultured PA-SMCs from patients with COPD and from sex- and age-matched control smokers. Pulmonary vessels from patients with COPD were characterized by increased wall hypertrophy compared to controls, in keeping with the higher Pap in the patients with COPD than in the controls. Immunohistochemical examination of pulmonary vessels revealed an increased percentage of pulmonary vascular cells stained for p21 and p16, including endothelial and smooth muscle cells, in patients with COPD compared to controls. These cells were identified as senescent cells by experiments performed in the proximal pulmonary arteries, in which β-galactosidase-positive cells were also positive for p16 and p21. We then evaluated whether PA-SMCs derived from pulmonary vessels also exhibited characteristic features of accelerated senescence when studied in vitro. Cultured PA-SMCs from patients with COPD exhibited premature senescence when compared to those of controls, with a marked decrease in cumulative PDL and a higher percentage of β-galactosidase-positive cells measured at an early passage].
In studies of pulmonary vessels and derived cultured PA-SMCs from patients with COPD and from sex- and age-matched control smokers, we found that in situ and in vitro criteria for cell senescence correlated with the severity of pulmonary vascular wall hypertrophy, suggesting a close relationship between cell senescence and the pulmonary vascular remodeling process. The large proportion of senescent cells within the walls of remodeled pulmonary vessels may seem paradoxical, since PH is primarily a proliferative disorder and cell senescence is associated with impaired regenerative capacity in a given tissue. We investigated PA-SMC proliferation in vessels from our patients and found that remodeled vessels from patients with COPD contained more proliferating Ki67-stained PA-SMCs and more accumulated extracellular matrix than those from controls. Thus, remodeled vessels from patients with COPD were paradoxically characterized by a combination of elevated senescent cell counts with an increased proportion of proliferating cells and increased extracellular matrix deposition. Of note, studies of remodeled vessels at sites of vascular hypertrophy revealed senescent cells to be virtually confined to the media, with only a few senescent cells in the neointima, whereas proliferating cells predominated in the neointima and hypertrophied media. Our results therefore support the concept that several PA-SMC subsets are present in the pulmonary vascular wall of remodeled vessels in COPD and that these subsets work in combination to participate in the remodeling process. Similar results have been reported in atherosclerotic lesions characterized by senescent cells and showing the presence in the neointima of actively dividing cells, possibly of monoclonal origin.11, 15 Similarly, endothelial cells with senescence-associated phenotypes are present in human atherosclerotic lesions.16
The mechanisms underlying premature PA-SMC senescence in COPD can only be speculated from the present study. We found increased p53 and p21 expression during replicative PA-SMC senescence, in parallel with a decrease in telomere length. Cells from patients with COPD studied at passage 2 had shorter telomeres than those from controls, in keeping with their increased susceptibility to replicative senescence. Although telomere loss is known to activate p53, with subsequent transcription of p21, we found similar levels of p53 and p21 in cells from patients with COPD and controls. In contrast, p16 expression was higher in cells from patients with COPD than in those from controls, suggesting a major role for p16 in driving premature senescence in COPD. Indeed, p16 activation by non-telomeric signals such as oxidative stress leads to premature senescence17 but may also occur during replicative senescence as a second barrier to cell proliferation.8 In accordance with this possibility, we found a strong inverse relationship between PDL and the amount of p16 measured in passage-2 cells, as well as a relationship with telomere length. Accelerated PA-SMC senescence in COPD may therefore be attributable to a combination of both telomere shortening and oxidative stress responsible for p16 activation. Other mechanisms may also interact with the senescence process, including reduced expression of HDAC2 and sirtuin, decreased proteasome activity, and decreased Akt signaling, which have been reported in patients with COPD.
It may be argued that cellular senescence was a consequence of pulmonary vascular remodeling rather than an active contributor, i.e., was due to the cell divisions involved in lesion development. This possibility is unlikely. Indeed, in patients with COPD, telomere shortening and accelerated cell senescence exist as a general process that is not restricted to the blood vessels.12 Cells stained for p16 were present not only in pulmonary vascular cells, but also in nondividing alveolar epithelial cells, in keeping with previous studies of patients with emphysema.12 Second, replicative senescence secondary to increased PA-SMC turnover would have been associated with prominent p21 expression. In contrast, we found a prominent difference in p16 expression, which appeared to be the main mechanism driving accelerated senescence of cultured PA-SMCs in patients with COPD. Moreover, the presence of senescent cells and actively dividing cells at different sites of the vascular lesions is not in favor of cell senescence occurring at exhaustion of their replicative potential. Finally, cells studied in culture were collected from proximal pulmonary arteries, which are not subjected to the same remodeling process as PA-SMCs from distal vessels. Thus, PA-SMC senescence in COPD does not seem to be a consequence of increased cell turnover at sites of vascular hypertrophy. Rather, PA-SMC senescence may be part of the pathogenic mechanisms associated with COPD that lead to pulmonary vessel remodeling and subsequent development of PH. Although we could not examine the relationship between telomere length in circulating leukocytes and in cultured PA-SMCs, such a positive relationship between telomere length in circulating leukocytes and in lung tissues have already been reported in patients with lung fibrosis 14.
The identification of the exact mechanisms by which PA-SMC senescence contributes to pulmonary vascular remodeling is challenging. Proliferation of neighboring cells may occur via a direct mitogenic effect or via indirect effects mediated by tissue damage or the recruitment of inflammatory cells.6, 18 In the present study, we found that PA-SMCs undergoing replicative senescence released excessive amounts of several cytokines and mediators. The amount of these secreted factors differed markedly between patients with COPD and controls at an early cell passage but not at senescence, indicating that this difference was mainly due to the higher proportion of senescent cells in patients with COPD. Several factors such as IL-6, IL-8, MCP-1, and IL-1β have been demonstrated to make a strong but indirect contribution to pulmonary vascular remodeling.19, 20 In particular, IL-6 is a major contributor to hypoxic PH and is closely linked to PH severity in patients with COPD13 or idiopathic PH.20 To better investigate the potential interplay between nonsenescent and senescent cells, we evaluated whether senescent cells affected the migration and proliferation of nonsenescent cells in a paracrine manner. We found that soluble and insoluble factors released by senescent cells stimulated the growth and migration of target PA-SMCs. A similar finding was obtained previously using senescent fibroblasts and cultured epithelial cells and was taken as evidence that senescent cells promoted cell proliferation and tumor growth.18 This possibility is consistent with our observation that actively dividing cells in the neointimal lesions of remodeled vessels were surrounded by senescent cells, suggesting cross-talk between the two cell subsets. In our study, cell proliferation in response to culture media from senescent cells were markedly reduced in the presence of neutralizing antibodies to IL6 and MCP-1, and cell migration was reduced in the presence of neutralizing antibodies to IL8, TNF-α, IL6 and MCP-1. The fact that the increased PA-SMC proliferation in response to culture media from senescent compared to presenescent cells was no longer observed in the presence of anti-MCP-1 and anti-IL6 antibodies suggests an important role for these cytokines in this process.19, 13, 20 The fact that anti-IL6 and anti-MCP-1 antibodies inhibited proliferation under both basal and stimulated conditions is also consistent with an autocrine effect of these mediators. Moreover, these results suggest that the global action of these soluble factors may be stimulation of growth and migration and not stimulation of senescence. Thus, senescent cells may create a microenvironment that facilitates the migration and growth of nonsenescent cells, thereby inducing neointima formation and vessel remodeling. Whether nonsenescent cells involved in neointima formation exhibit a normal or an abnormal phenotype remains to be elucidated.
There are several important limitations to this study. First, cell senescence in COPD is probably a process applying to the whole body, and it remains unclear whether the observations described here are specific to the pulmonary circulation or apply also to other vascular beds. Second, because we could not study a third group with severe PH or with other forms of PH, we do not know whether our data are relevant to various types of PH or specific of diseases associated with telomere dysfunction. Indeed, telomere shortening in circulating leucocytes is found in patients with COPD and lung fibrosis but not in patients with idiopathic PH.21 Whether telomere shortening may represent a biomarker of disease severity in various types of PH also remains an open question. In patients with COPD, telomere shortening may constitute a biomarker of overall accelerated aging and, potentially, of its effects, including PH, cardiovascular disease, and cancer, which are the main causes of morbidity and mortality in patients with COPD.
Supplementary Material
Acknowledgments
Funding Sources
This study was supported by grants from the INSERM, Delegation a la Recherche Clinique de l’AP-HP, Fondation pour la Recherche Médicale (FRM), and the Carvsen foundation.
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- PA-SMCs
pulmonary artery smooth muscle cells
- COPD
chronic obstructive pulmonary disease
- PH
pulmonary hypertension
- Pap
mean pulmonary artery pressure
- Sap
mean systemic arterial pressure
- PVR
pulmonary vascular resistance
- FEV1
forced expiratory volume in 1 second
- FVC
forced vital capacity
- BMI
body mass index
- PDL
population doubling level
- β-gal
beta-galactosidase
- α-SMA
alpha-smooth muscle actin
- IL-6
interleukin-6
- IL-8
interleukin-8
- MCP-1
monocyte chemoattractant protein 1
- TNF-α
tumor necrosis factor-alpha
- IL-1β
interleukin-1-beta
- TGF-β
transforming growth factor-beta
- PDGF
platelet-derived growth factor
- FCS
fetal calf serum
- HDAC
histone deacetylase
- Akt
serine/threonine:kinase
- vWF
von Willebrand factor
Footnotes
Disclosures
None
References
- 1.Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23(6):932–946. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 2.Chaouat A, Bugnet AS, Kadaoui N, Schott R, Enache I, Ducolone A, Ehrhart M, Kessler R, Weitzenblum E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189–194. doi: 10.1164/rccm.200401-006OC. [DOI] [PubMed] [Google Scholar]
- 3.Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD: epidemiology, significance, and management: pulmonary vascular disease: the global perspective. Chest. 2010;137(6 Suppl):39S–51S. doi: 10.1378/chest.10-0087. [DOI] [PubMed] [Google Scholar]
- 4.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, Barbera JA. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 2002;19(4):632–638. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 5.Savale L, Chaouat A, Bastuji-Garin S, Marcos E, Boyer L, Maitre B, Sarni M, Housset B, Weitzenblum E, Matrat M, Le Corvoisier P, Rideau D, Boczkowski J, Dubois-Rande JL, Chouaid C, Adnot S. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;179(7):566–571. doi: 10.1164/rccm.200809-1398OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Mathon NF, Lloyd AC. Cell senescence and cancer. Nat Rev Cancer. 2001;1(3):203–213. doi: 10.1038/35106045. [DOI] [PubMed] [Google Scholar]
- 8.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22(16):4212–4222. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 10.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2009;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006;99(2):156–164. doi: 10.1161/01.RES.0000233315.38086.bc. [DOI] [PubMed] [Google Scholar]
- 12.Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174(8):886–893. doi: 10.1164/rccm.200509-1374OC. [DOI] [PubMed] [Google Scholar]
- 13.Chaouat A, Savale L, Chouaid C, Tu L, Sztrymf B, Canuet M, Maitre B, Housset B, Brandt C, Le Corvoisier P, Weitzenblum E, Eddahibi S, Adnot S. Role for Interleukin-6 in COPD-Related Pulmonary Hypertension. Chest. 2009 doi: 10.1378/chest.08-2420. [DOI] [PubMed] [Google Scholar]
- 14.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA, 3rd, Lansdorp PM, Loyd JE, Armanios MY. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murry CE, Gipaya CT, Bartosek T, Benditt EP, Schwartz SM. Monoclonality of smooth muscle cells in human atherosclerosis. Am J Pathol. 1997;151(3):697–705. [PMC free article] [PubMed] [Google Scholar]
- 16.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002;105(13):1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 17.Nyunoya T, Monick MM, Klingelhutz AL, Glaser H, Cagley JR, Brown CO, Matsumoto E, Aykin-Burns N, Spitz DR, Oshima J, Hunninghake GW. Cigarette smoke induces cellular senescence via Werner’s syndrome protein down-regulation. Am J Respir Crit Care Med. 2009;179(4):279–287. doi: 10.1164/rccm.200802-320OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98(21):12072–12077. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez O, Marcos E, Perros F, Fadel E, Tu L, Humbert M, Dartevelle P, Simonneau G, Adnot S, Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2007;176(10):1041–1047. doi: 10.1164/rccm.200610-1559OC. [DOI] [PubMed] [Google Scholar]
- 20.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, Trembath RC, Jennings S, Barker L, Nicklin P, Walker C, Budd DC, Pepke-Zaba J, Morrell NW. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122(9):920–927. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 21.Cronkhite JT, Xing C, Raghu G, Chin KM, Torres F, Rosenblatt RL, Garcia CK. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(7):729–737. doi: 10.1164/rccm.200804-550OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.