

Abstract
Aims/hypothesis
Recent studies have demonstrated that cannabinoid-1 (CB1) receptor blockade ameliorated inflammation, endothelial and/or cardiac dysfunction, and cell death in models of nephropathy, atherosclerosis and cardiomyopathy. However the role of CB1 receptor signalling in diabetic retinopathy remains unexplored. Using genetic deletion or pharmacological inhibition of the CB1 receptor with SR141716 (rimonabant) in a rodent model of diabetic retinopathy or in human primary retinal endothelial cells (HREC) exposed to high glucose, we explored the role of CB1 receptors in the pathogenesis of diabetic retinopathy.
Methods
Diabetes was induced using streptozotocin in C57BL/6J Cb1 (also known as Cnr1)+/+ and Cb1−/− mice aged 8 to 12 weeks. Samples from mice retina or HREC were used to determine: (1) apoptosis; (2) activity of nuclear factor kappa B, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), poly (ADP-ribose) polymerase and caspase-3; (3) content of 3-nitrotyrosine and reactive oxygen species; and (4) activation of p38/Jun N-terminal kinase/mitogen-activated protein kinase (MAPK).
Results
Deletion of CB1 receptor or treatment of diabetic mice with CB1 receptor antagonist SR141716 prevented retinal cell death. Treatment of diabetic mice or HREC cells exposed to high glucose with SR141716 attenuated the oxidative and nitrative stress, and reduced levels of nuclear factor κB, ICAM-1 and VCAM-1. In addition, SR141716 attenuated the diabetes- or high glucose-induced pro-apoptotic activation of MAPK and retinal vascular cell death.
Conclusions/interpretation
Activation of CB1 receptors may play an important role in the pathogenesis of diabetic retinopathy by facilitating MAPK activation, oxidative stress and inflammatory signalling. Conversely, CB1 receptor inhibition may be beneficial in the treatment of this devastating complication of diabetes.
Keywords: Apoptosis, Cannabinoid, CB1, ICAM-1, NFκB, Rimonabant, SR141716, VCAM-1
Introduction
Vascular inflammation and endothelial cell death are characteristic features of diabetic retinopathy [1, 2]. The early stages of the inflammatory reaction are characterised by leucocyte adhesion to the vessel wall, leading to altered vessel reactivity and subsequent activation of transcription factors including nuclear factor κB (NFκB), which ultimately results in capillary endothelial cell apoptosis and vascular cell loss in the diabetic retina [3, 4]. A critical role of increased oxidative and nitrative stress in mediating vascular inflammation and cell death is supported by previous studies [4, 5] and a review [6]. Based on the evidence that NFκB has a well-conserved cysteine residue, NFκB activity is tightly linked with its redox regulation [7]. Prior studies have shown that oxidative stress can induce production of inflammatory cytokines and adhesion molecules via activation of NFκB [8, 9]. Therefore, devising treatments that target oxidative stress and inflammation could be of great clinical significance for diabetic retinopathy.
The recently discovered endocannabinoid system, which consists of the endocannabinoids, their metabolising enzymes and the main cannabinoid 1 (CB1) and cannabinoid 2 (and perhaps other yet not determined) receptors, has been implicated as an important factor in regulation of energy balance, food intake, metabolism and inflammation in health and disease [10, 11]. While cannabinoid 2 receptors are predominantly localised on immune cells, CB1 receptor is mostly found in the central nervous system and the retina [12]; however, both receptors are also present in cardiovascular and virtually all other tissues, albeit at much lower levels [10]. It has been observed that: (1) the CB1 receptors [12] and endocannabinoids [13, 14] are present within the retina; (2) endocannabinoid anandamide level is elevated in the retina of patients with diabetic retinopathy [14]; and (3) CB1 receptor activation in coronary endothelial [15] and inflammatory [16, 17] cells by endocannabinoids or synthetic CB1 ligands mediates mitogen-activated protein kinase (MAPK) activation, reactive oxygen species (ROS) generation and inflammatory response [15–18], as well as promoting atherosclerosis [19]. These observations, coupled with the multiple beneficial effects of the CB1 receptor antagonist rimonabant (SR141716) on inflammatory markers as observed in obese and/or type 2 diabetic patients, and in various preclinical disease models [10, 11], and with the recently reported attenuation of albuminuria by CB1 receptor blockade in an experimental model of diabetic nephropathy [20], raise the possibility of a direct effect of CB1 receptor signalling in pro-inflammatory and pro-apoptotic response in retinal endothelial cells. Besides regulating photoreception and neurotransmission in the retina, the endocannabinoid system affects intraocular pressure and ocular blood vessels [21, 22], and plant-derived cannabinoids such as cannabidiol and tetrahydrocannabinol exert neuroprotective effects against retinal neurotoxicity [23], presumably by their antioxidant properties, independently of conventional cannabinoid receptors.
To assess the potential role of the CB1 receptor in the pathogenesis of retinal vascular injury in diabetes, we evaluated the effects of the selective CB1 receptor inhibitor, SR141716/rimonabant or of genetic deletion of CB1 receptors in a mouse model of diabetic retinopathy and in human primary retinal endothelial cells (HREC) exposed to high glucose. Our study demonstrates that pharmacological inhibition or genetic deletion of CB1 attenuates retinal oxidative stress, release of pro-inflammatory mediators and activation of p38/Jun N-terminal kinase (JNK) MAPK in streptozocin-induced diabetic mice, as well as in HREC exposed to high glucose.
Methods
Animals and treatment
The animal procedures adhered to the National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee of the National Institute on Alcohol Abuse and Alcoholism (NIAAA)/NIH. Diabetes was induced in 8- to 12-week-old male C57/BL6J or in CB1 receptor knockout (Cb1 [also known as Cnr1]−/−) and wild-type (Cb1+/+) male mice (23–25 g; Jackson Laboratories, Bar Harbor, ME, USA) by multiple intra-peritoneal injection of streptozotocin as previously described [24]. After 1 week, blood glucose levels were measured using a glucometer (Ascensia Counter; Bayer Healthcare, NY, USA) by mandibular puncture blood sampling. Mice with blood sugar values >14 mmol/l (approximately 250 mg/dl) were used for the study. Diabetes was allowed to develop further for 1 additional week before animals were treated for 11 weeks with the selective CB1 receptor antagonist N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole carboxamide (SR141716A/rimonabant; 10 mg/kg daily, i.p.; NIDA Drug Supply Program, Research Triangle Park, NC, USA).
Cell culture
All human cell line experiments were approved by the NIH Office of Human Subjects Research. HREC were obtained from Cell Applications (San Diego, CA, USA) and grown in HREC growth medium (Cell Applications) in culture dishes coated with 0.2% (wt/wt) gelatin (Sigma, St Louis, MO, USA). HREC were used for the experiments in the passages 3 to 6. Cells were maintained in normal glucose (5 mmol/l) or high glucose (30 mmol/l) for 48 h. For osmotic controls, cells were maintained in either L-glucose (30 mmol/l) or mannitol (30 mmol/l) for the same duration.
Determination of retinal cell death in flat-mounted retina
TUNEL assay was performed using immunoperoxidase staining (ApopTag-Peroxidase; Roche Applied Science, Indiannapolis, IN, USA) in whole-mount retina as described previously [25, 26]. After permeabilisation, TUNEL-horseradish peroxidise staining with 3-amino-9-ethylcarbazole was performed following the manufacturer’s instructions. The total number of cells positive for TUNEL-horseradish peroxidise was counted in each retina using light microscopy. TUNEL was also performed in 10 μm eye sections frozen to optimal cutting temperature, using a kit (ApopTAG in situ cell death detection kit; TUNEL-FITC) as described previously [27].
Determination of apoptosis in HREC by flow cytometry
After various treatments, apoptosis/necrosis was determined with flow cytometry as described previously [15, 24].
Determination of glial activation and immunolocalisation studies
Retinal sections were fixed using 2% (wt/wt) paraformaldehyde in PBS and allowed to react overnight with polyclonal anti-glial fibrillary acidic protein (GFAP) antibody for glial activation (Affinity BioReagents, Rockford, IL, USA), monoclonal anti-NFκB p65 (BD Bioscience Pharmingen, San Diego, CA, USA) and monoclonal anti-vascular cell adhesion molecule 1 (VCAM-1) (R&D Systems, Minneapolis, MN, USA), followed by Texas Red- or Oregon Green-conjugated goat anti-mouse antibodies (Invitrogen, Carlsbad, CA, USA). Data (three fields per retina, n=4 in each group) were analysed using AxioObserver.Z1 Microscope (Carl Zeiss, Thornwood, NY, USA) and Axio-software to quantify the density of immunostaining.
Determination of cell surface intercellular adhesion molecule 1 and VCAM-1 levels
Measurements of cell surface intercellular adhesion molecule 1 (ICAM-1) and VCAM-1 levels in HRECs were done by in situ ELISA as described previously [28].
Western blot analysis
Total lysates from retinas or cells were extracted with RIPA buffer as described before [15, 26]. Blots were probed with polyclonal anti-phospho-p38, p38, phospho-JNK/JNK (Cell Signaling, Danvers, MA, USA), monoclonal anti-NFκB p65 (BD Bioscience), polyclonal anti-VCAM-1 (R&D Systems) and monoclonal anti-3-nitrotyrosine (Cayman Chemicals, Ann Arbor, MI, USA). Membranes were reprobed with β-actin (Millipore, Billerica, MA, USA) to confirm equal loading. The primary antibody was detected using appropriate horse-radish peroxidase-conjugated antibodies (GE Healthcare, Piscataway, NJ, USA) and enhanced chemiluminescence. The films were scanned, and band intensity was quantified using densitometry software (BioRad, Hercules, CA, USA) and expressed as relative optical density.
Determination of 3-nitrotyrosine content
Quantification of 3-nitrotyrosine levels in retinal extracts was performed using slot blot analysis as described [25, 26]. 3-Nitrotyrosine content in HREC extracts was determined using an ELISA kit (Hycult Biotechnology, Uden, the Netherlands) as described previously [29].
Determination of poly (ADP-ribose) polymerase and caspase 3/7 activities
Poly (ADP-ribose) polymerase (PARP) and caspase 3/7 activities in the HREC extracts were performed using kits (Trevigen, Gaithersburg, MD, USA, and Promega, Madison, WI, USA, respectively) as described previously by our group [29].
Determination of ROS generation in retina and HREC
2′,7′-Dichlorofluorescein (DCF) is the oxidation product of 2′,7′-dichlorodihydro-fluorescein diacetate (DHDCF) (Invitrogen), a marker of cellular oxidation by hydrogen peroxide, peroxynitrite and hydroxy radicals. For HREC, DCF was detected using flow cytometery techniques as described before [15]. Briefly, cells were incubated for 15 min with 5 μmol/l DHDCF at 37°C then measured at excitation of 488 nm with standard settings using a flow cytometer (FACS Calibur; Becton Dickinson, San Jose, CA, USA). For retina lysate, DCF was measured as described previously [30]. Briefly, equal volumes of retinal lysates were incubated with DHDCF (10 μmol/l) for 60 min at 37°C then measured at excitation 450 nm using a plate reader (BioTek, Winooski, VT, USA).
Statistical analysis
The results were expressed as mean ± SEM. Differences among experimental groups were evaluated by ANOVA and the significance of differences between groups was assessed by Tukey’s post-hoc test. The analysis was performed using a statistical software package (GraphPad-Prism 5; GraphPad, La Jolla, CA, USA). Significance was defined as p<0.05.
Results
Metabolic variables
Induction of diabetes by multiple low doses of streptozotocin led to marked reduction in the bodyweight with concomitant increase in blood glucose levels in wild-type (Cb1+/+) and Cb1−/− mice respectively (Electronic supplementary material [ESM] Fig. 1). However, blood glucose levels were unaffected during the 12 week study period in Cb1−/− mice compared with wild-type mice (ESM Fig. 1). Similarly treatment of diabetic mice with SR 141716A for 11 weeks did not significantly alter body weight or blood glucose levels (ESM Fig. 2).
CB1 receptor plays a role in diabetes-induced retinal cell death
The CB1 receptor is highly abundant in the inner and outer plexiform layers of the retina [12]. However, the exact role of CB1 receptor in modulating retinal function in response to diabetes is not fully understood. Diabetes induced greater than sevenfold increases in retinal cell death as indicated by quantitative analysis of TUNEL-positive cells in flat-mounted retina (Fig. 1a, c). Deletion of Cb1 completely protected diabetic animals from retinal cell death, suggesting a potential role of CB1 receptor activation in mediating cell death. Co-localisation studies in diabetic retinal sections demonstrated that several TUNEL-positive cells were located within endothelial cells as indicated by isolectin B4 (Fig. 1b). We next evaluated the effect of the CB1 receptor antagonist SR 141716A in diabetic animals. As shown in Fig. 1d, treatment of diabetic animals with SR 141716A significantly reduced TUNEL-positive cells compared with diabetic animals treated with vehicle.
Fig. 1.

CB1 receptor plays a role in diabetes-induced retinal cell death. a Representative images (×200 magnification, scale bars 25 μm) and (c) statistical analysis showing that diabetes induced significant increases in retinal cell death as indicated by quantitative analysis of TUNEL-positive cells in flat-mounted retina. Deletion of Cb1 completely protected diabetic animals from retinal cell death (n=6). Arrows (a) indicate TUNEL-positive cells in retinal flat mounts. b Co-localisation studies in diabetic retinal sections demonstrated that several TUNEL-positive cells (green) are located within ganglion layer (GCL) and localised with endothelial cells as indicated by isolectin B4 (red); ×200 magnification; scale bars 25 μm. IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer. d Statistical analysis showing that treatment of diabetic animals (Diab) with SR 141716A (SR1) significantly reduced TUNEL-positive cells compared with diabetic animals treated with vehicle (Veh, n=6). *p<0.05 vs vehicle group; †p<0.05 vs wild-type diabetes
CB1 receptor inhibition attenuates diabetes-induced oxidative and nitrative stress in vivo
To explore the possible mechanism of the protective effects of CB1 receptor blockade, we next examined its effects on the well established phenomenon of diabetes-induced oxidative and nitrative stress. As shown in Fig. 2a, b, mice retinal lysate showed twofold increase in 3-nitrotyrosine formation measured by slot blot and twofold increase in ROS formation as indicated by DCF fluorescence in diabetic retina as compared with controls. Treatment of diabetic animals with SR 141716A significantly reduced 3-nitrotyrosine and ROS formation but did not alter control levels.
Fig. 2.
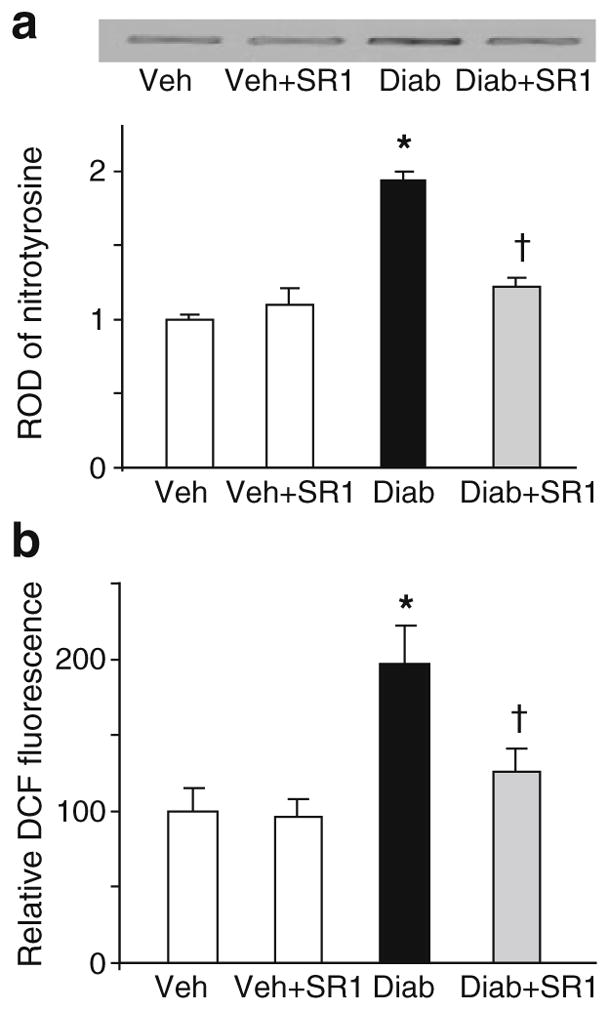
CB1 receptor inhibition attenuates diabetes-induced oxidative and nitrative stress in vivo. a Slot blot and statistical analysis of mouse retinal lysate showing twofold increase in 3-nitrotyrosine formation in diabetic mice as compared with controls (n=5–6). b Statistical analysis showing twofold increase in ROS formation as indicated by DCF fluorescence in diabetic mice as compared with controls (n=6). *p<0.05 vs vehicle group; †p<0.05 vs diabetes. Diab, diabetic animals; SR1, SR 141716A; Veh, vehicle-treated
CB1 receptor inhibition attenuates high glucose-induced oxidative stress in HREC
As shown in Fig. 3a–g, flow cytometry of DCF showed 2.4-fold increase in ROS. ELISA assay of nitrotyrosine showed a fivefold increase in 3-nitrotyrosine generation in HREC cells as compared with those maintained in normal glucose (Fig. 3h). Treatment of the cells with CB1 antagonist, SR 141716A (2 μmol/l) blocked high glucose-induced increase in oxidative and nitrative stress. Interestingly, treatment of HREC with SR 141716A alone or osmotic controls did not alter ROS or 3-nitrotyrosine formation.
Fig. 3.

CB1 receptor inhibition attenuates high glucose-induced oxidative stress in HREC. a Representative histograms of DCFDA fluorescence with D-glucose (5 mmol/l), (b) SR 141716A (SR1), (c) L-glucose (30 mmol/l), (d) mannitol (30 mmol/l), (e) high glucose (HG; D-glucose 30 mmol/l) and (f) high glucose + SR1. Mean fluorescence intensity–height at ~525 nm (FL1-H) (a) 13.5, (b) 11.5, (c) 13.9, (d) 16.0, (e) 31.6 and (f) 20.9. g Summary data of DCFDA fluorescence from the indicated representative treatment condition (n=4–6). High glucose (30 mmol/l D-glucose) induced an approximately 2.4-fold increase in DCF fluorescence in HREC cells. h Statistical analysis showing approximately fivefold increase in 3-nitrotyrosine (NT) formation in HREC cells maintained in high glucose as compared with those maintained in normal glucose. Treatment of the cells with CB1 antagonist SR 141716A (2 μmol/l) almost completely prevented high glucose-induced increase in oxidative and nitrative stress. Treatment of HREC with SR1 alone or osmotic controls did not alter ROS or 3-nitrotyrosine formation. *p<0.05 vs vehicle group; †p<0.05 vs diabetes
CB1 receptor inhibition attenuated diabetes-induced glial activation and NFκB production
As shown in Fig. 4a, diabetes enhanced glial activation as indicated by increases in the intensity of GFAP immunoreactivity in the filaments of Müller cells that extend from the nerve fibre and inner plexiform layers into the outer nuclear layer of retina as compared with controls. In addition, diabetes enhanced NFκB activation (twofold), which was mainly localised in the vascular layers of the retina, compared with controls (Fig. 4b, c). Additional studies with isolectin B4, a marker of endothelial cells confirmed that NFκB was colocalised with endothelial cells (data not shown). Treatment of diabetic animals with SR 141716A blocked these effects but did not alter control levels.
Fig. 4.

CB1 receptor inhibition attenuated diabetes-induced glial activation and NFκB production. a Representative images showing a substantial increase in the intensity of GFAP immunoreactivity in the filaments of Müller cells in diabetes (Diab) vs vehicle (Veh). This increase extended from the nerve fibre layer and inner plexiform layer (IPL) into the outer nuclear layer (ONL) of retina as compared with controls. b Representative images of NFκB in mouse retinal sections showing an approximately 2.5-fold increase in diabetic mice as compared with the controls. NFκB was mainly localised within retinal capillaries (n=6). Magnification (a, b) ×200; scale bars 25 μm. GCL, ganglion cell layer; INL, inner nuclear layer; SR1, SR 141716A. c Statistical analysis of above findings (b); *p<0.05 vs vehicle group; †p<0.05 vs diabetes. ROD, relative optic density
CB1 receptor inhibition attenuated diabetes-induced adhesion molecule production in vivo
We next examined levels of the adhesion molecules ICAM-1 and VCAM-1 in rat retinas. As shown in Fig. 5a, c, diabetes enhanced VCAM-1 production (~twofold), which was mainly localised in retinal capillaries, compared with controls. This effect was paralleled by significant increases (1.9-fold) in ICAM-1 production in diabetic animals (Fig. 5b, d). Treatment of diabetic animals with SR 141716A blocked these effects but did not alter control levels.
Fig. 5.

CB1 receptor inhibition attenuated diabetes-induced adhesion molecule production in vivo. a Representative images of VCAM-1 in mice retinal sections showing an approximately twofold increase in diabetic (Diab) mice as compared with vehicle (Veh)-treated controls. VCAM-1 was mainly localised within retinal capillaries; n=5, ×200 magnification, scale bar 25 μm. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer. c Statistical analysis of above (a) findings, expressed as relative optical density (ROD). b Western blot analysis showing increase in adhesion molecule ICAM-1 levels in diabetic animals as compared with the controls (n=5–6). Treatment of diabetic animals with SR 141716A (SR1) blocked these effects, but did not alter control levels. d Statistical analysis of above findings (b) showing 1.9-fold increase in diabetic animals.*p<0.05 vs vehicle group; †p<0.05 vs diabetes
CB1 receptor inhibition attenuated high glucose-induced ICAM-1 and VCAM-1 production in HREC
As shown in Fig. 6, treatment of HRECs with high glucose significantly increased levels of ICAM-1 and VCAM-1 by 2.5- to 3.5-fold, respectively, compared with normal glucose. When the cells were incubated with SR 141716A, high glucose-induced adhesion molecules levels were significantly reduced, but not completely blocked by SR 141716A. Treatment of HREC with SR 141716A alone or osmotic controls did not alter levels of adhesion molecules.
Fig. 6.
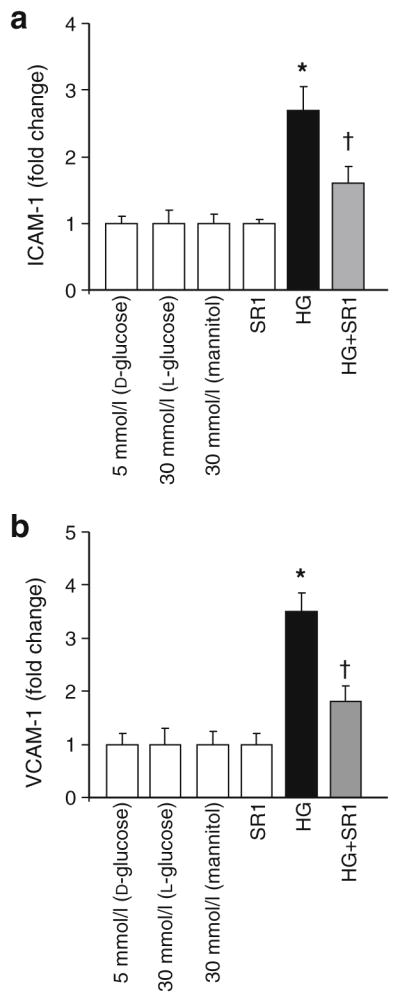
CB1 receptor inhibition attenuated high glucose-induced ICAM-1 and VCAM-1 production in HREC. a Statistical analysis of ICAM-1 and (b) VCAM-1 levels measured by ELISA and showing 2.5- and 3.5-fold increases, respectively in HREC cells maintained in high glucose (HG; 30 mmol/l D-glucose) as compared with those maintained in normal glucose. When the cells were incubated with SR 141716A (SR1; 2 μmol/l), high glucose-induced adhesion molecule production was significantly reduced. Treatment of HREC with SR 141716A alone or osmotic controls did not alter levels of adhesion molecules. *p<0.05 vs vehicle group; †p<0.05 vs diabetes
CB1 receptor inhibition attenuates diabetes-induced proapoptotic MAPK activation in vivo and in HREC
Activation of proapoptotic MAPK pathways, including p38 MAPK and JNK, is a known downstream target of oxidative stress and inflammatory mediators [24]. In comparison to controls, diabetes increased activation of p38 MAPK (2.4-fold), this increase being mitigated by SR 141716A treatment (Fig. 7a, b). Moreover, treatment of HRECs with high glucose significantly increased MAPK (p38 and JNK) activation as compared with cells maintained in normal glucose. This was attenuated upon treatment with SR 141716A (Fig. 7c, d).
Fig. 7.

CB1 receptor inhibition attenuates diabetes-induced proapoptotic MAPK activation in vivo and in HREC. a Western blot of mice retinal lysate showing an increase in the activation of p38MAPK in diabetic (Diab) as compared with the vehicle (Veh)-treated controls; the increase was mitigated by SR 141716A (SR1) treatment (n=4–6). b Statistical analysis of above results (a), indicating a 2.4-fold increase. c Western blot of HREC cell lysate showing significant increases in activation of p38MAPK and JNK in cells maintained in high glucose (HG; 30 mmol/l D-glucose) as compared with cells maintained in normal glucose. These increases were mitigated upon treatment with SR 141716A (2 μmol/l). d, e Statistical analysis of above (c) findings for MAPK and JNK respectively. *p<0.05 vs vehicle group; †p<0.05 vs diabetes
CB1 receptor inhibition mitigates high glucose-induced apoptosis in HREC
HREC cells maintained in high glucose for 48 h showed significant increases in the activity of cell death markers PARP and cleaved caspase-3, as compared with those maintained in normal glucose (Fig. 8a, b). In addition, HREC cells maintained in high glucose showed significant increase in apoptosis as indicated by Annexin V using flow cytometry; this increase was attenuated by SR 141716A (Fig. 8c, d).
Fig. 8.

CB1 receptor inhibition mitigates high glucose–induced cell death in HREC. a Western blot statistical analysis of HREC cell lysate showing significant increases in the activity of PARP and (b) caspase-3 in cells maintained in high glucose (HG) as compared with cells maintained in normal glucose. These increases were mitigated upon treatment with SR 141716A (SR1; 2 μmol/l). c Flow cytometric analysis of cell death with D-glucose (5 mmol/l), (d) SR 141716A (SR1), (e) L-glucose (30 mmol/l), (f) mannitol (30 mmol/l), (g) high glucose (HG; D-glucose 30 mmol/l) and (h) high glucose + SR1. Cells were maintained in different media as indicated and treated with CB1 antagonists for 1 h, followed by incubation with different media for 48 h in the continuous presence of CB1 antagonists. c–h Sytox green (y-axes), x-axes: annexin V-APC. i Summary of the results showing significant increase in apoptosis in HREC cells maintained in high glucose (30 mmol/l D-glucose) compared with those maintained in normal glucose. When the cells were incubated with SR 141716A (2 μmol/l), the high glucose-induced apoptosis was significantly reduced. Treatment of HREC with SR 141716A alone or osmotic controls did not alter cell death. n=4–6; *p<0.05 vs vehicle group; †p<0.05 vs diabetes. FL4-H, fluorescence intensity–height at ~675 nm
Discussion
The main findings of the current study are that: (1) deletion or pharmacological inhibition of the CB1 receptor with SR141716 prevents diabetes-induced retinal vascular cell death; and (2) CB1 inhibition ameliorates diabetes-induced retinal oxidative stress, cellular adhesion molecule production and inflammation. Although pharmacological inhibition of CB1 receptor has been associated with numerous cytoprotective and anti-inflammatory effects in models of ischaemia–reperfusion injury, cardiomyopathy, nephropathy and atherosclerosis [17–19, 29, 31–33], a potential role of the CB1 receptor in the pathogenesis of diabetic retinopathy has not been explored previously.
Diabetes-induced retinal oxidative and nitrative stress have been well documented in patients and animals, and have been positively correlated with vascular cell death [34–36]. The biochemical mechanisms involved in promoting oxidative stress are complex and include activation of several cellular pathways, as reviewed by Caldwell at al. [37]. Recent studies have demonstrated that activation of CB1 receptors with endocannabinoids or synthetic ligands can promote oxidative stress, inflammation, cell death and/or organ dysfunction in models of cardiomyopathy [17, 18, 38], atherosclerosis [19] and nephropathy [20, 29, 32], and likewise in macrophages [16], neutrophils [17], murine or human cardiomyocytes [18, 38], and human coronary artery endothelial cells [15]. In agreement with the above-mentioned studies, our results demonstrate that pharmacological inhibition of CB1 confers marked protection against diabetes or high glucose-induced oxidative/nitrative stress in retinas or in cultured HREC.
Diabetic retinopathy has been perceived as an inflammatory disease, in the pathogenesis of which adhesion molecules may be involved [39, 40]. Previous studies have shown that oxidative stress can induce production of inflammatory cytokines and adhesion molecules via activation of the redox-regulated transcription factor NFκB [8, 9]. Upon activation, NFκB translocates to the nucleus, where it regulates the expression of a large number of genes including those encoding cellular adhesion molecules such as ICAM-1 and VCAM-1 [41]. In agreement with this, our results show significant increases in levels of NFκB, ICAM-1 and VCAM-1 in the retina of diabetic mice and in HREC cells maintained in high glucose. These results lend further support to previous reports showing enhanced NFκB p65 in diabetic rats [42] and enhanced levels of ICAM-1 and VCAM-1 in endothelial cells cultured in high glucose [3]. The notion that CB1 activation may promote inflammation and tissue injury is supported by several studies showing that genetic deletion or pharmacological inhibition of CB1 consistently exerts beneficial effects on the inflammation and oxidative/nitrative stress cell death cascade [17, 18, 31–33, 43, 44]. In agreement, chronic treatment of our diabetic animals with SR141716 almost completely blocked the increases in retinal activation of NFκB and production of ICAM-1 and VCAM-1, and similar findings were obtained in HREC maintained in high glucose.
There is increasing recognition that CB1 receptor activation may promote activation of stress signalling pathways including p38 and JNK MAPKs, leading to cell death [15, 18, 29, 45]. In agreement with this, we found marked increases (tenfold) in TUNEL-positive cells in retinal flat-mounts and sections of diabetic animals; these increases were largely attenuated by CB1 receptor deletion or treatment with SR141716, illustrating a causal role of CB1 receptors in mediating retinal cell death. This is also consistent with the elevated endocannabinoid anandamide (endogenous ligand for CB1 receptors) levels observed in retinas of patients with diabetic retinopathy [14]. Immunolocalisation studies using isolectin-B4, a marker for endothelial cells, showed colocalisation of several of TUNEL-positive cells within vascular endothelial cells, lending further support to previous reports showing apoptosis of retinal capillaries as early as 12 weeks of diabetes duration [46, 47]. Apoptosis of retinal capillary cells begins early in diabetes and is likely to contribute to the capillary obliteration that is an important feature of diabetic retinopathy. The increase in cell death was associated with increases in oxidative markers in vivo and in vitro. It is well known that oxidative and nitrative stress may also lead to activation of p38 and JNK–MAPKs, promoting cell death. Our results showed that SR141716 significantly reduced activation of p38 and JNK–MAPKs in diabetic mice and in retinal endothelial cells maintained in high glucose. In agreement with this, activation of p38 MAPK has been reported in diabetic retinas [5, 16, 25, 26, 48]. Previous studies have demonstrated a pro-apoptotic role of JNK activation in inducing vascular cell death in vivo and in vitro [15, 49, 50]. Cell death of retinal capillaries was further confirmed by significant increases in activity of cleaved caspase-3 and PARP in response to high glucose. Treatment with SR141716 only partially attenuated cell death, suggesting that activation of CB1 receptor is not the only player mediating high glucose-induced cell demise.
In summary, our results demonstrate that pharmacological blockade and/or genetic deletion of the CB1 receptors ameliorate diabetes-induced retinal oxidative stress and production of cellular adhesion molecules, and prevent cell death, strongly supporting an important role for activation of CB1 receptor in the pathogenesis of diabetic retinopathy.
Supplementary Material
Acknowledgments
This work was supported by intramural research grants to NIH-NIAAA to P. Pacher and grants from JDRF (2-2008-149) and Vision Discovery Institute to A. B. El-Remessy. B. Horváth was supported by an NKTH-OTKA-EU fellowship (MB08-A-80238). The authors are indebted to G. Kunos (Scientific Director of NIH-NIAAA) for providing key resources for the completion of this study.
Abbreviations
- CB1
Cannabinoid 1
- DCF
2′,7′-Dichlorofluorescein
- DHDCF
2′,7′-Dichlorodihydro-fluorescein diacetate
- GFAP
Glial fibrillary acidic protein
- HREC
Human primary retinal endothelial cells
- ICAM-1
Intercellular adhesion molecule 1
- JNK
Jun N-terminal kinase
- MAPK
Mitogen-activated protein kinase
- NFκB
Nuclear factor κB
- PARP
Poly(ADP-ribose) polymerase
- ROS
Reactive oxygen species
- VCAM-1
Vascular cell adhesion molecule 1
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00125-011-2061-4) contains supplementary material, which is available to authorised users.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Contributor Information
A. B. El-Remessy, Program in Clinical and Experimental Therapeutics, College of Pharmacy, University of Georgia, Augusta, GA, USA. Charlie Norwood VA Medical Center, Augusta, GA, USA
M. Rajesh, Section on Oxidative Stress Tissue Injury, Laboratory of Physiological Studies, National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism, 5625 Fishers Lane, MSC-9413, Bethesda, MD 20892-9413, USA
P. Mukhopadhyay, Section on Oxidative Stress Tissue Injury, Laboratory of Physiological Studies, National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism, 5625 Fishers Lane, MSC-9413, Bethesda, MD 20892-9413, USA
B. Horváth, Section on Oxidative Stress Tissue Injury, Laboratory of Physiological Studies, National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism, 5625 Fishers Lane, MSC-9413, Bethesda, MD 20892-9413, USA. Institute of Human Physiology and Clinical Experimental Research, Semmelweis University, Budapest, Hungary
V. Patel, Section on Oxidative Stress Tissue Injury, Laboratory of Physiological Studies, National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism, 5625 Fishers Lane, MSC-9413, Bethesda, MD 20892-9413, USA
M. M. H. Al-Gayyar, Program in Clinical and Experimental Therapeutics, College of Pharmacy, University of Georgia, Augusta, GA, USA. Charlie Norwood VA Medical Center, Augusta, GA, USA
B. A. Pillai, Program in Clinical and Experimental Therapeutics, College of Pharmacy, University of Georgia, Augusta, GA, USA. Charlie Norwood VA Medical Center, Augusta, GA, USA
P. Pacher, Email: pacher@mail.nih.gov, Section on Oxidative Stress Tissue Injury, Laboratory of Physiological Studies, National Institutes of Health/National Institute on Alcohol Abuse and Alcoholism, 5625 Fishers Lane, MSC-9413, Bethesda, MD 20892-9413, USA
References
- 1.Huysman E, Mathieu C. Diabetes and peripheral vascular disease. Acta Chir Belg. 2009;109:587–594. doi: 10.1080/00015458.2009.11680493. [DOI] [PubMed] [Google Scholar]
- 2.Talahalli R, Zarini S, Sheibani N, Murphy RC, Gubitosi-Klug RA. Increased synthesis of leukotrienes in the mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:1699–1708. doi: 10.1167/iovs.09-3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khalfaoui T, Lizard G, Ouertani-Meddeb A. Adhesion molecules (ICAM-1 and VCAM-1) and diabetic retinopathy in type 2 diabetes. J Mol Histol. 2008;39:243–249. doi: 10.1007/s10735-007-9159-5. [DOI] [PubMed] [Google Scholar]
- 4.Kowluru RA, Kanwar M, Kennedy A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp Diabetes Res. 2007;2007:21976. doi: 10.1155/2007/21976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Remessy AB, Al-Shabrawey M, Khalifa Y, Tsai N-t, Caldwell RB, Liou GI. Neuroprotective and blood–retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–244. doi: 10.2353/ajpath.2006.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brodsky M, Halpert G, Albeck M, Sredni B. The anti-inflammatory effects of the tellurium redox modulating compound, AS101, are associated with regulation of NFkappaB signaling pathway and nitric oxide induction in macrophages. J Inflamm (Lond) 2010;7:3. doi: 10.1186/1476-9255-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamei H, Iwabuchi K, Namba K, et al. Amelioration of experimental autoimmune uveoretinitis (EAU) with an inhibitor of nuclear factor-kappaB (NF-kappaB), pyrrolidine dithiocarbamate. J Leukoc Biol. 2006;79:1193–1201. doi: 10.1189/jlb.0805453. [DOI] [PubMed] [Google Scholar]
- 9.Wang T, Zhang X, Li JJ. The role of NF-kappaB in the regulation of cell stress responses. Int Immunopharmacol. 2002;2:1509–1520. doi: 10.1016/s1567-5769(02)00058-9. [DOI] [PubMed] [Google Scholar]
- 10.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Marzo V. The endocannabinoid system in obesity and type 2 diabetes. Diabetologia. 2008;51:1356–1367. doi: 10.1007/s00125-008-1048-2. [DOI] [PubMed] [Google Scholar]
- 12.Straiker A, Stella N, Piomelli D, Mackie K, Karten HJ, Maguire G. Cannabinoid CB1 receptors and ligands in vertebrate retina: localization and function of an endogenous signaling system. Proc Natl Acad Sci USA. 1999;96:14565–14570. doi: 10.1073/pnas.96.25.14565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bisogno T, Delton-Vandenbroucke I, Milone A, Lagarde M, Di Marzo V. Biosynthesis and inactivation of N-arachidonoylethanolamine (anandamide) and N-docosahexaenoylethanolamine in bovine retina. Arch Biochem Biophys. 1999;370:300–307. doi: 10.1006/abbi.1999.1410. [DOI] [PubMed] [Google Scholar]
- 14.Matias I, Wang JW, Moriello AS, Nieves A, Woodward DF, Di Marzo V. Changes in endocannabinoid and palmitoylethanolamide levels in eye tissues of patients with diabetic retinopathy and age-related macular degeneration. Prostaglandins Leukot Essent Fatty Acids. 2006;75:413–418. doi: 10.1016/j.plefa.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Rajesh M, Mukhopadhyay P, Hasko G, Liaudet L, Mackie K, Pacher P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br J Pharmacol. 2010;160:688–700. doi: 10.1111/j.1476-5381.2010.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han KH, Lim S, Ryu J, et al. CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc Res. 2009;84:378–386. doi: 10.1093/cvr/cvp240. [DOI] [PubMed] [Google Scholar]
- 17.Mukhopadhyay P, Horvath B, Rajesh M, et al. Fatty acid amide hydrolase is a key regulator of endocannabinoid-induced myocardial tissue injury. Free Radic Biol Med. 2011;50:179–195. doi: 10.1016/j.freeradbiomed.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukhopadhyay P, Rajesh M, Batkai S, et al. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc Res. 2010;85:773–784. doi: 10.1093/cvr/cvp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugamura K, Sugiyama S, Nozaki T, et al. Activated endocannabinoid system in coronary artery disease and antiinflammatory effects of cannabinoid 1 receptor blockade on macrophages. Circulation. 2009;119:28–36. doi: 10.1161/CIRCULATIONAHA.108.811992. [DOI] [PubMed] [Google Scholar]
- 20.Barutta F, Corbelli A, Mastrocola R, et al. Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes. 2010;59:1046–1054. doi: 10.2337/db09-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlicker E, Timm J, Gothert M. Cannabinoid receptor-mediated inhibition of dopamine release in the retina. Naunyn Schmiedebergs Arch Pharmacol. 1996;354:791–795. doi: 10.1007/BF00166907. [DOI] [PubMed] [Google Scholar]
- 22.Struik ML, Yazulla S, Kamermans M. Cannabinoid agonist WIN 55212-2 speeds up the cone response to light offset in goldfish retina. Vis Neurosci. 2006;23:285–293. doi: 10.1017/S0952523806232127. [DOI] [PubMed] [Google Scholar]
- 23.El-Remessy AB, Khalil IE, Matragoon S, et al. Neuro-protective effect of (−)Delta9-tetrahydrocannabinol and cannabidiol in N-methyl-D-aspartate-induced retinal neurotoxicity: involvement of peroxynitrite. Am J Pathol. 2003;163:1997–2008. doi: 10.1016/s0002-9440(10)63558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajesh M, Mukhopadhyay P, Batkai S, et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J Am Coll Cardiol. 2010;56:2115–2125. doi: 10.1016/j.jacc.2010.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ali TK, Al-Gayyar MM, Matragoon S, et al. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia. 2011;54:657–668. doi: 10.1007/s00125-010-1935-1. [DOI] [PubMed] [Google Scholar]
- 26.Ali TK, Matragoon S, Pillai BA, Liou GI, El-Remessy AB. Peroxynitrite mediates retinal neurodegeneration by inhibiting nerve growth factor survival signaling in experimental and human diabetes. Diabetes. 2008;57:889–898. doi: 10.2337/db07-1669. [DOI] [PubMed] [Google Scholar]
- 27.Al-Gayyar MM, Abdelsaid MA, Matragoon S, Pillai BA, El-Remessy AB. Neurovascular protective effect of FeTPPs in N-methyl-D-aspartate model: similarities to diabetes. Am J Pathol. 2010;177:1187–1197. doi: 10.2353/ajpath.2010.091289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajesh M, Mukhopadhyay P, Batkai S, et al. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am J Physiol Heart Circ Physiol. 2007;293:H610–H619. doi: 10.1152/ajpheart.00236.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay P, Pan H, Rajesh M, et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol. 2010;160:657–668. doi: 10.1111/j.1476-5381.2010.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Shabrawey M, Bartoli M, El-Remessy AB, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muthian S, Rademacher DJ, Roelke CT, Gross GJ, Hillard CJ. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience. 2004;129:743–750. doi: 10.1016/j.neuroscience.2004.08.044. [DOI] [PubMed] [Google Scholar]
- 32.Janiak P, Poirier B, Bidouard JP, et al. Blockade of cannabinoid CB1 receptors improves renal function, metabolic profile, and increased survival of obese Zucker rats. Kidney Int. 2007;72:1345–1357. doi: 10.1038/sj.ki.5002540. [DOI] [PubMed] [Google Scholar]
- 33.Zhang M, Martin BR, Adler MW, et al. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J Neuroimmune Pharmacol. 2009;4:249–259. doi: 10.1007/s11481-009-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Remessy AB, Bartoli M, Platt DH, Fulton D, Caldwell RB. Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI 3-kinase tyrosine nitration. J Cell Sci. 2005;118:243–252. doi: 10.1242/jcs.01612. [DOI] [PubMed] [Google Scholar]
- 35.Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du Y, Sarthy VP, Kern TS. Interaction between NO and COX pathways in retinal cells exposed to elevated glucose and retina of diabetic rats. Am J Physiol Regul Integr Comp Physiol. 2004;287:R735–R741. doi: 10.1152/ajpregu.00080.2003. [DOI] [PubMed] [Google Scholar]
- 37.Caldwell RB, Bartoli M, Behzadian MA, et al. Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets. 2005;6:511–524. doi: 10.2174/1389450054021981. [DOI] [PubMed] [Google Scholar]
- 38.Mukhopadhyay P, Batkai S, Rajesh M, et al. Pharmacological inhibition of CB1 cannabinoid receptor protects against doxorubicin-induced cardiotoxicity. J Am Coll Cardiol. 2007;50:528–536. doi: 10.1016/j.jacc.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–653. [PMC free article] [PubMed] [Google Scholar]
- 40.Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 41.Singh R, Barden A, Mori T, Beilin L. Advanced glycation end-products: a review. Diabetologia. 2001;44:129–146. doi: 10.1007/s001250051591. [DOI] [PubMed] [Google Scholar]
- 42.Alves M, Calegari VC, Cunha DA, Saad MJ, Velloso LA, Rocha EM. Increased expression of advanced glycation end-products and their receptor, and activation of nuclear factor kappa-B in lacrimal glands of diabetic rats. Diabetologia. 2005;48:2675–2681. doi: 10.1007/s00125-005-0010-9. [DOI] [PubMed] [Google Scholar]
- 43.Pacher P, Hasko G. Endocannabinoids and cannabinoid receptors in ischaemia–reperfusion injury and preconditioning. Br J Pharmacol. 2008;153:252–262. doi: 10.1038/sj.bjp.0707582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schafer A, Pfrang J, Neumuller J, Fiedler S, Ertl G, Bauersachs J. The cannabinoid receptor-1 antagonist rimonabant inhibits platelet activation and reduces pro-inflammatory chemokines and leukocytes in Zucker rats. Br J Pharmacol. 2008;154:1047–1054. doi: 10.1038/bjp.2008.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dalton GD, Bass CE, van Horn CG, Howlett AC. Signal transduction via cannabinoid receptors. CNS Neurol Disord Drug Targets. 2009;8:422–431. doi: 10.2174/187152709789824615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohr S, Xi X, Tang J, Kern TS. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes. 2002;51:1172–1179. doi: 10.2337/diabetes.51.4.1172. [DOI] [PubMed] [Google Scholar]
- 47.Drel VR, Xu W, Zhang J, et al. Poly(ADP-ribose) polymerase inhibition counteracts cataract formation and early retinal changes in streptozotocin-diabetic rats. Invest Ophthalmol Vis Sci. 2009;50:1778–1790. doi: 10.1167/iovs.08-2191. [DOI] [PubMed] [Google Scholar]
- 48.Igarashi M, Wakasaki H, Takahara N, et al. Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J Clin Invest. 1999;103:185–195. doi: 10.1172/JCI3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alikhani M, Roy S, Graves DT. FOXO1 plays an essential role in apoptosis of retinal pericytes. Mol Vis. 2010;16:408–415. [PMC free article] [PubMed] [Google Scholar]
- 50.Poulaki V, Joussen AM, Mitsiades N, Mitsiades CS, Iliaki EF, Adamis AP. Insulin-like growth factor-I plays a pathogenetic role in diabetic retinopathy. Am J Pathol. 2004;165:457–469. doi: 10.1016/S0002-9440(10)63311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.