

Abstract
High-dose intravenous immunoglobulin (IVIG) therapy has been effective in treating various autoimmune and systemic inflammatory diseases. Here, we assessed the efficacy and safety of IVIG therapy with polyethylene glycol-treated human IgG (drug code GB-0998) for patients with corticosteroid-refractory polymyositis (PM) and dermatomyositis (DM) by means of a randomized, double-blind, placebo-controlled study. We randomly assigned 26 subjects (16 PM and 10 DM) to receive either GB-0998 or placebo. Intragroup comparison in the GB-0998 group showed statistically significant improvements due to GB-0998 administration in the primary endpoint (manual muscle test score) and secondary endpoints (serum creatine kinase level and activities of daily living score). However, significant improvements were also found in the placebo group, and comparison of the GB-0998 group with the placebo group did not show any significant difference between the groups. We discuss possible reasons for the absence of a clear intergroup difference in efficacy. Nineteen adverse drug reactions were observed in 11 of 26 subjects (42.3%), of which 2 events (decreased muscle strength and increased serum creatine kinase) were assessed as serious; however, they are previously known events. These results indicate that GB-0998 can be safely used with the same precautions as other current IVIG therapy.
Keywords: Intravenous immunoglobulin (IVIG); Manual muscle test (MMT); Polymyositis (PM); Dermatomyositis (DM); Randomized, double-blind, placebo-controlled study
Introduction
Polymyositis (PM) and dermatomyositis (DM) are diffuse inflammatory myopathies of unknown etiology, causing decreased strength in the limb proximal group muscles, cervical muscles, and pharyngeal muscles, and have been designated as targets for disease treatment research by the Japanese Ministry of Health, Labor, and Welfare. Oral corticosteroids are the first-line treatment for patients with PM and DM, and they produce good response in approximately 80% of patients [1]. For patients showing either no response or incomplete response, other therapies such as methylprednisolone pulse therapy and/or concomitant immunosuppressive drug therapy are empirically used, but are not always effective [2]. In addition, long-term and/or high-dose administration of corticosteroids and/or immunosuppressive drugs may cause severe side-effects, such as infection, and therefore new therapies are required.
Here, we present the results of a double-blind, placebo-controlled, crossover study designed to evaluate the efficacy and safety of polyethylene glycol-treated human IgG (GB-0998) in patients with corticosteroid-resistant PM or DM.
Patients and methods
Study design
This randomized, double-blind, placebo-controlled, crossover trial was performed in 47 medical institutions in Japan. The study protocol was approved by the Institutional Review Board of each participating institution, and the trial was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice. Patients gave written informed consent prior to registration for this study. This trial is registered with ClinicalTrials.gov, number NCT00335985.
The trial began with a 6-week run-in period, during which corticosteroid resistance was confirmed. Patients who had given informed consent, and had been confirmed to be resistant to corticosteroid treatment during the run-in period, were randomly allocated to receive GB-0998 (Benesis Corporation, Osaka, Japan), a polyethylene glycol-treated human normal immunoglobulin, followed by an indistinguishable placebo (GB-0998 group), or the same two treatments in the reverse order (placebo group), as shown in Fig. 1. GB-0998 at a dose of 400 mg (8 mL)/kg/day or the placebo was administered by intravenous infusion once daily for 5 consecutive days, in a double-blind manner. After an assessment duration of 8 weeks from the start of administration (the first period), subjects receiving the active drug were switched to receive the placebo in the second period, whereas those receiving the placebo in the first period were switched to receive the active drug in the second period. The assessment duration in the second period was 8 weeks, and there was an additional 4-week observation period (follow-up period) after the second period. Because of the seriousness of the disease, the second period was included in the study design to enable all subjects in the study to receive GB-0998. For ethical reasons, patients who did not show disease alleviation within 4 weeks after the start of the first period, rendering continuation of the study difficult, were given an early transition to the second period; i.e., these subjects were transferred to the second period without continuing in the first period for the full 8 weeks. For this reason, the primary evaluation was of efficacy in the first period.
Fig. 1.
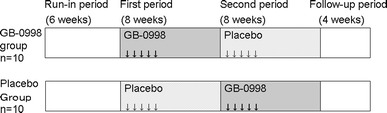
Outline of study design. GB-0998 (active drug) was administered to the GB-0998 group in the first period and to the placebo group in the second period. Differences between before and after the 8-week first period in the GB-0998 group were assessed in the primary analysis
Increases in corticosteroid and immunosuppressant doses, including methylprednisolone pulse therapy, were not allowed during the period from acquisition of informed consent until completion or termination of the study. Decreases in the dose were allowed when side-effects were elicited by corticosteroid or immunosuppressant during the period prior to the start of test drug administration, and were also allowed depending on the patients’ medical condition following the administration of the test drug.
Patients
Patients with corticosteroid-resistant PM/DM (Japanese people, resident in Japan) who were aged 16–75 years and fulfilled the diagnostic criteria of Bohan and Peter [3] for “definite” PM/DM were considered eligible to participate in this study. Among them, those that met one of the following criteria with respect to their history of corticosteroid therapy were enrolled for this study after obtaining their informed consent:
Had received high-dose corticosteroid therapy (≥50 mg/day or ≥1 mg/kg/day for 1 month or more), starting between 6 weeks and 1 year prior to acquisition of informed consent, and continued until the day of acquisition of informed consent.
In receipt of ≥50 mg/day or ≥1 mg/kg/day of corticosteroids on the day of acquisition of informed consent.
Had received two or more methylprednisolone pulse therapies within 6 weeks before the acquisition of informed consent, and receiving ≥30 mg/day or ≥0.6 mg/kg/day of corticosteroid on the day of acquisition of informed consent.
Patients with manual muscle test (MMT) scores of ≤80 points (normal score 90 points), assessed after having obtained their informed consent, and with twice the standard upper limit value of serum creatine kinase (CK) or more were tentatively enrolled, and followed for 6 weeks (run-in period). The patients who did not show any improvement in MMT scores and serum CK levels during these 6 weeks were formally registered, and received the investigational drug.
Patients excluded from participation in this study were those with malignant tumors; acute interstitial pneumonia including acute exacerbation of chronic phase; severe muscular atrophy for a long period; severe infectious disease; severe hepatic disorder or severe renal disorder; those who had a previous history of shock or hypersensitivity to the test drug; those who had a history of cerebral infarction or ischemic heart disease, or had symptom of these diseases; those who had a history of IgA deficiency diagnosed; pregnant, lactating, and possibly pregnant patients; patients who wanted to become pregnant; and those who had been administered immunoglobulin within 6 weeks of obtaining consent.
Efficacy assessment
Since muscle weakness is the most common symptom of PM/DM, changes in MMT scores were used as the primary endpoint, because the therapeutic effect could be assessed by observing the degree to which muscular strength was restored. MMT was scored on the following 18 muscles: the right and left of each of the following: deltoid, biceps brachii, brachioradialis, triceps brachii, iliopsoas, gluteus maximus, quadriceps femoris, and hamstring muscle; and the flexor and extensor muscles of the neck area. Each muscle was scored 0–5, with a healthy state thus being a score of 90 [4, 5]. In order to achieve consistency of evaluation, in principle, a single assessor evaluated the MMT for all patients throughout the study.
The secondary efficacy endpoints were serum CK level and the activities of daily living (ADL) score. Serum CK measurements and other clinical laboratory tests were carried out at a central laboratory, at Mitsubishi Chemical Medience Corporation (Tokyo, Japan). The upper limits of the normal serum CK range were taken as 270 and 150 IU/L for males and females, respectively. The ADL score was based on the following 15 actions, with each action being scored 0–3, and the normal total score being 45: raising arms, taking off outer clothes, turning on water tap, combing hair, fastening buttons, getting out of bed, lifting up feet, standing up from chair, climbing stairs, walking on flat surface, turning over in bed, holding up head, sitting down on bed, having conversation, and swallowing [4, 5].
In addition to the above endpoints, we considered the time (number of days) to discharge from hospital during the first period. Furthermore, two additional parameters evaluated were dysphagia, which was graded as none, mild, or severe by each attending physician, and the occurrence or nonoccurrence of early transition to the second period.
The safety endpoint was the occurrence or nonoccurrence of adverse events.
Target number of subjects
The number of patients meeting the criteria for inclusion in this study is very small. It was therefore considered that, with a study duration of 3 years, the number of subjects who could be enrolled would be limited to 20. Therefore, from the viewpoint of the feasibility of carrying out the study, the target number of subjects was set at 10 per group (a total of 20 subjects).
Statistical analysis
As described above, the number of target patients for this study was very small, and it was not practically possible to set the target number of subjects sufficiently high to permit a clear demonstration of the superiority of GB-0998 over placebo. Therefore, the primary analysis consisted of an intragroup comparison in the GB-0998 group, using the paired t test, of changes in MMT score, serum CK level, and ADL score between before initiation of study drug administration and after administration for 8 weeks in the first period.
An intergroup comparison between the GB-0998 and placebo groups was also carried out as a subsidiary analysis. For this analysis, descriptive statistics were calculated separately for the two groups, with respect to the changes in MMT score, serum CK level, and ADL score between before initiation of administration during the first period and after administration for 8 weeks. The serum CK levels were widely scattered, depending upon the severity of disease in each subject, and logarithmic transformation was therefore carried out for efficacy evaluation in relation to change in serum CK level. Then, using the mean changes in these parameters, the point-estimation value and 95% confidence interval were calculated for the difference between the GB-0998 and placebo groups.
In addition, the Kaplan–Meier method was used to compare the time (number of days) until improvement of the MMT score in the GB-0998 and placebo groups. On the basis of a report by Dalakas et al. [6], the time at which “improvement” was taken to have occurred was the first time point when the MMT score was increased by 5 or more (and subsequently maintained at the higher level) during the period from before initiation of administration during the first period until transition to the second period. The median value of the time was calculated for each group, and the log-rank test was applied.
For the serum CK level, the Kaplan–Meier method was used to compare the time (number of days) until normalization in the GB-0998 and placebo groups. The event taken to constitute “normalization” was the serum CK reaching a level below the upper limit of the normal range for the first time during the period from before initiation of administration during the first period until transition to the second period. The median values for the groups were calculated, and the generalized Wilcoxon test was applied.
With respect to parameters other than the primary and secondary endpoints, the Kaplan–Meier method was used to compare the time (number of days) until first discharge from hospital during the first period. The median value was calculated for each of the groups.
Several supplementary analyses were also carried out, as follows. For MMT score, the percentage of subjects showing improvement was calculated for each group. In addition, for each of the individual muscles evaluated, an intergroup comparison using the paired t test was carried out for the change during the first period. For ADL score, the paired t test was used for intergroup comparison of the change in each evaluated individual action during the first period. In addition, the change in dysphagia during the first period was calculated, and the Fisher direct probability test was used for intergroup comparison of the elimination or nonelimination of dysphagia. Numbers of subjects undergoing early transition to the second period were also compared in the two groups.
Efficacy was assessed using the last observation carried forward (LOCF) approach at 8 weeks after the start of administration in the first period, when premature discontinuation occurred in the first 8-week period, or when the second period was started before the scheduled end of the first period. Similarly, when premature discontinuation occurred in the second 8-week period, the efficacy after 8 weeks for the second period was assessed using the LOCF approach.
Results
Background of subjects
Of 63 subjects who gave informed consent to participate in the study, corticosteroid resistance was confirmed in 26 (12 in the GB-0998 group and 14 in the placebo group). Study drug administration and evaluation of safety and efficacy were carried out with these subjects (Fig. 2).
Fig. 2.
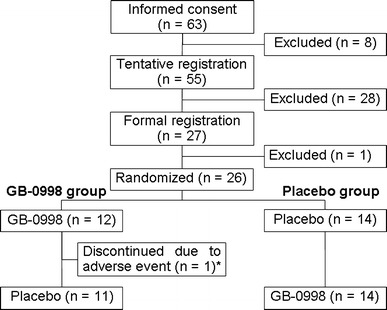
Flow diagram of patients participating in this trial. In this study, a total of 26 subjects received GB-0998. Asterisk Premature discontinuation occurred after the sixth week of the first period
Table 1 presents the backgrounds of these subjects, separately for each of the two groups. In the GB-0998 group there were 6 subjects with PM and 6 with DM, while in the placebo group, there were 10 with PM and 4 with DM.
Table 1.
Patients’ characteristics
| Characteristic | GB-0998 group (n = 12) | Placebo group (n = 14) | Total (n = 26) |
|---|---|---|---|
| Gender, n (%) | |||
| Male | 1 (8.3) | 5 (35.7) | 6 (23.1) |
| Female | 11 (91.7) | 9 (64.3) | 20 (76.9) |
| Age (years) | |||
| Mean | 50.6 | 48.1 | 49.3 |
| Standard deviation | 13.1 | 12.1 | 12.4 |
| Minimum value | 27 | 30 | 27 |
| Median | 49.0 | 50.5 | 49.0 |
| Maximum value | 69 | 68 | 69 |
| Diagnosis, cases (%) | |||
| Polymyositis | 6 (50.0) | 10 (71.4) | 16 (61.5) |
| Dermatomyositis | 6 (50.0) | 4 (28.6) | 10 (38.5) |
| Disease period, cases (%) | |||
| <6 months | 5 (41.7) | 3 (21.4) | 8 (30.8) |
| ≥6 months and <1 year | 0 (0.0) | 4 (28.6) | 4 (15.4) |
| ≥1 year | 7 (58.3) | 7 (50.0) | 14 (53.8) |
| Interstitial pulmonary disease, cases (%) | |||
| Absence | 6 (50.0) | 11 (78.6) | 17 (65.4) |
| Presence | 6 (50.0) | 3 (21.4) | 9 (34.6) |
| Daily dose of corticosteroid at formal registration (mg) | |||
| Mean | 34.79 | 40.71 | 37.98 |
| Standard deviation | 14.56 | 14.69 | 14.65 |
| Minimum value | 10 | 7.5 | 7.5 |
| Median | 35.00 | 47.50 | 42.50 |
| Maximum value | 60 | 55 | 60 |
| Therapeutic history with immunosuppressant, cases (%) | |||
| Absence | 5 (41.7) | 7 (50.0) | 12 (46.2) |
| Presence | 7 (58.3) | 7 (50.0) | 14 (53.8) |
| Manual muscle test score at formal registration (points) | |||
| 0–50 | 2 | 1 | 3 |
| 51–60 | 4 | 3 | 7 |
| 61–70 | 4 | 8 | 12 |
| 71–80 | 2 | 2 | 4 |
| Mean ± SD | 61.8 ± 10.6 | 64.7 ± 9.0 | – |
| Minimum value | 42 | 43 | 42 |
| Maximum value | 77 | 77 | 77 |
| Activities of daily living score at formal registration (points) | |||
| 0–15 | 3 | 2 | 5 |
| 16–20 | 3 | 1 | 4 |
| 21–25 | 0 | 1 | 1 |
| 26–30 | 2 | 0 | 2 |
| 31–35 | 2 | 5 | 7 |
| 36–40 | 2 | 3 | 5 |
| 41–45 | 0 | 2 | 2 |
| Not done | 0 | 0 | 0 |
| Mean ± SD (points) | 23.8 ± 11.1 | 30.2 ± 10.3 | – |
| Minimum value | 4 | 11 | 4 |
| Maximum value | 40 | 45 | 45 |
Results of principal analyses: intragroup comparison during the first period
With respect to the principal analyses of the primary and secondary endpoints, Table 2 presents the changes in efficacy endpoints between before initiation of study drug administration, and after administration for 8 weeks during the first period.
Table 2.
Differences of assessment parameters between before administration and at 8 weeks after start of administration for the first phase and corresponding between-group differences
| No. of subjects | Mean | Standard deviation | Minimum value | Median | Maximum value | Corresponding t test | Difference of mean (GB-0998 group − placebo group) 95% confidence interval |
|
|---|---|---|---|---|---|---|---|---|
| MMT | ||||||||
| GB-0998 | 12 | 11.8 | 8.0 | −4 | 11.0 | 25 |
t = 5.0655 p = 0.0004 |
1.9 (−4.8 to 8.5) |
| Placebo | 14 | 9.9 | 8.3 | 0 | 8.0 | 22 |
t = 4.4334 p = 0.0007 |
|
| CK | ||||||||
| GB-0998 | 12 | −1.1633 | 1.4123 | −3.006 | −1.4389 | 1.229 |
t = −2.8533 p = 0.0157 |
0.1029 (−0.8382 to 1.0439) |
| Placebo | 14 | −1.2662 | 0.8900 | −2.797 | −1.4353 | −0.134 |
t = −5.3229 p = 0.0001 |
|
| ADL | ||||||||
| GB-0998 | 12 | 7.3 | 6.6 | −1 | 6.0 | 26 |
t = 3.8169 p = 0.0029 |
3.3 (−1.8 to 8.3) |
| Placebo | 14 | 4.0 | 5.8 | −4 | 3.0 | 18 |
t = 2.5610 p = 0.0237 |
|
The primary before–after analysis was performed using t tests to evaluate the differences in MMT scores, serum CK levels, and ADL scores between before the administration of the drug and at 8 weeks after the start of the administration in the first phase. As a subanalysis, descriptive statistics were calculated by group and the before–after differences (between before the drug administration and at 8 weeks after the start of the administration in the first phase) were compared for each assessment parameter. The between-group differences (GB-0998 group versus placebo group) and confidence intervals for means of differences between before and after administration were estimated based on analysis of variance. Patients whose treatment was discontinued or who were transferred to the second phase before 8 weeks after the start of administration in the first phase were assessed after 8 weeks in the first phase using the LOCF approach
MMT manual muscle test, CK creatine kinase, ADL activities of daily living
The MMT score was the primary endpoint, and the mean (±standard deviation, SD) of the change in this score in the GB-0998 group during the first period was 11.8 ± 8.0, this change being statistically significant according to the paired t test (p = 0.0004). In terms of classification by disease type, the mean scores in subjects with PM and DM were 12.2 ± 11.0 and 11.3 ± 4.5, respectively; thus, no marked difference was found between PM and DM subjects.
Serum CK level and the ADL score were the secondary endpoints. The mean (±SD) of the change in serum CK level was −1.1633 ± 1.4123 Ln (IU/L) (paired t test, p = 0.0157), and the mean (±SD) of the change in the ADL score was 7.3 ± 6.6 (paired t test, p = 0.0029). The changes in both these parameters were statistically significant.
Statistically significant increases during the first period were also found in the intragroup comparison in the placebo group. The mean change in MMT score was 9.9 ± 8.3 (paired t test, p = 0.0007), the mean change in serum CK level was −1.2662 ± 0.8900 Ln (IU/L) (paired t test, p = 0.0001), and the mean change in ADL score was 4.0 ± 5.8 (paired t test, p = 0.0237).
Results of subsidiary analyses: intergroup analyses
Changes during the first period
The results for intergroup comparison of MMT score, serum CK level, and ADL score during the first period are presented in Table 2.
For the primary endpoint, the intergroup difference in mean MMT scores during the first period was 1.9, with the 95% confidence interval being −4.8 to 8.5. As for the secondary endpoints, the difference between the mean logarithmic serum CK levels was 0.1029 Ln (IU/L), with the 95% confidence interval being −0.8382 to 1.0439, and the mean difference between the ADL scores was 3.3, with the 95% confidence interval being −1.8 to 8.3. The GB-0998 group showed numerically greater changes in the MMT and ADL scores than did the placebo group, but the differences were not statistically significant. In addition, the placebo group showed a greater change of serum CK level than did the GB-0998 group, but this difference was not statistically significant.
Time to improvement in MMT score
The time (number of days) until the improvement in the MMT score reached 5 or more was compared between the two groups.
The Kaplan–Meier survival curves for the time until improvement are shown in Fig. 3. The median times until improvement were 16.5 days in the GB-0998 group, and 29.0 days in the placebo group, but the difference between the two groups was not statistically significant (log-rank test, p = 0.1154).
Fig. 3.
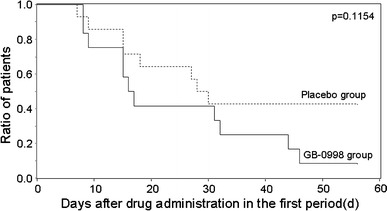
Kaplan–Meier curve of patients for whom the MMT scores increased by 5 points or more in the first period. Event occurrence was defined as the time point when MMT scores initially increased by 5 points or more, and subsequently remained at the higher level, during the period from before drug administration until transfer to the second period. When the score was increased by less than 5 points upon transfer to the second period, the test was discontinued on day 56
Time until normalization of serum CK level
The Kaplan–Meier survival curves for time (number of days) until the first normalization of serum CK level after initiation of study drug administration during the first period are shown in Fig. 4. The median time until normalization of the serum CK level was significantly less in the GB-0998 group, at 22.0 days, than in the placebo group, in which it was 57.5 days (generalized Wilcoxon test, p = 0.0301).
Fig. 4.
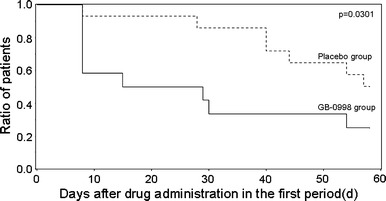
Kaplan–Meier curve of patients in whom serum CK level was normalized in the first period. Event occurrence was defined as the first decline of the serum CK level to below the upper limit of the normal range during the first phase. In one subject whose serum CK level was not normalized on day 57, the test was discontinued on day 58
Time until discharge from hospital
The time until the first discharge from hospital after initiation of administration during the first period was analyzed using the Kaplan–Meier method, as shown in Fig. 5. The median time (number of days) until discharge was 11.5 days in the GB-0998 group, and 41.5 days in the placebo group. In the GB-0998 group, all 12 subjects were discharged from hospital during the first period, whereas only 8 of 14 subjects in the placebo group were discharged during the first period.
Fig. 5.
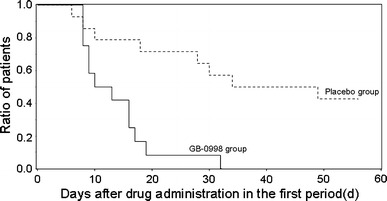
Kaplan–Meier curve of patients discharged from hospital in the first period. Event occurrence was defined as the first discharge from hospital after test drug administration in the first period
Results of supplementary analyses and investigations
Percentage of subjects showing improvements in MMT score
The time-courses of percentage of subjects showing improvement in MMT score are shown in Fig. 6a. Even after week 8 of the first period, no statistically significant intergroup difference in the percentage of subjects showing improvement was found (Fisher direct probability test, p = 0.0809). However, in the GB-0998 group the percentage showing improvement at week 8 was 91.7% (11 of 12 subjects); further, the percentage showing improvement increased up to week 6, and was then maintained even after the week-8 time-point. In the placebo group, on the other hand, the percentage showing improvement at week 8 was 57.1% (8 of 14 subjects), this percentage having shown no increase from week 4. In addition, at week 8 of the second period, the percentage of the GB-0998 group (administered placebo during the second period) that showed improvement was 81.8% (9 of 11 subjects), and the percentage of the placebo group (administered GB-0998 during the second period) that showed improvement was 84.6% (11 of 13 subjects). In the GB-0998 group, the percentage that showed improvement at week 8 of the second period was approximately the same as that at week 8 of the first period. However, in the placebo group the percentage that showed improvement increased during the second period, when they were receiving GB-0998, and became approximately the same as the percentage in the GB-0998 group during the first period (when they were receiving GB-0998) (Fig. 6b).
Fig. 6.
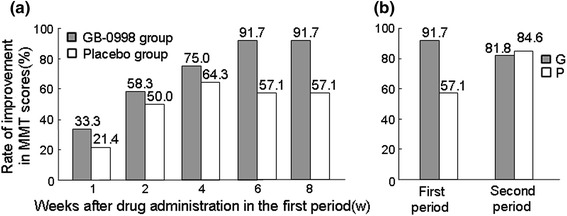
a Time-dependent changes in the rate of improved MMT scores in the first period. When MTT score increased by 5 points or more compared with that before administration of the drug, this was defined as an improvement. The time-dependent changes in the rate of improvement are shown by group. For cases discontinued or transferred to the second period before 8 weeks after the start of administration in the first period, the assessment in each assessment period was performed using the LOCF approach. Fisher’s exact test was used to compare the rates of improved MMT scores between the two groups at the end of the first 8-week period. b An MTT score that increased by 5 points or more compared with before drug administration in the first period was considered as an improvement, and improvement rates after 8 weeks in the first period and after 8 weeks in the second period were compared by group. For subjects discontinued or transferred to the second period before the end of the 8-week first period, the assessment after the first period was performed using the LOCF approach. For subjects discontinued before the end of the 8-week second period, the assessment after the second period was performed using the same method
We found that 42.9% (6 of 14 subjects) of the placebo group had no improvement in MMT score during the first period. These subjects did not show improved muscle strength at the initiation of the second period, despite having undergone corticosteroid therapy by that time, but four of them did show improvements in MMT score with GB-0998 administration during the second period.
MMT scores classified by evaluated muscle
The intergroup differences in changes of individual MMT scores in the 18 muscles evaluated in this study, during the first period, are shown in Fig. 7. The point-estimation values were greater in the GB-0998 group than in the placebo group for 14 of the 18 muscles, although the differences were small for all individual muscles.
Fig. 7.
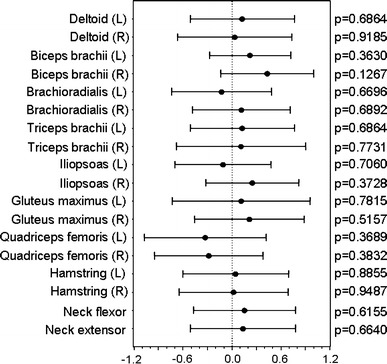
Forest plots of before–after differences in MMT scores for the 8-week first period. Black circles and lines, respectively, represent point estimates and 95% confidence intervals of between-group differences (GB-0998 group versus placebo group) concerning MMT scores for each assessed muscle
ADL scores classified by evaluated parameter
The intergroup differences in changes of ADL scores for the 15 actions evaluated in this study, during the first period, are shown in Fig. 8. The point-estimation values show that the changes were greater in the GB-0998 group than in the placebo group for 12 of the 15 actions, although the differences for individual actions were small. Among these actions, the difference (value 1.17) with respect to swallowing action at week 8 of the first period between an increase of 1.00 in the GB-0998 group and a decrease of −0.17 in the placebo group (the normal score is 3) was statistically significant (t test, p = 0.0222).
Fig. 8.
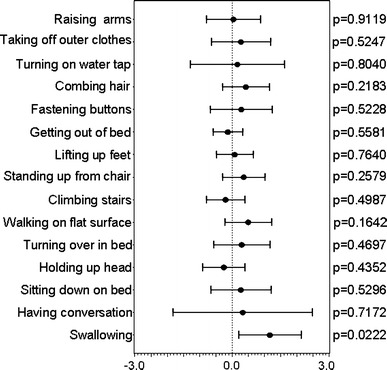
Forest plots of before–after differences in ADL scores for the 8-week first period. Black circles and lines, respectively, represent point estimates and 95% confidence intervals of between-group differences (GB-0998 group versus placebo group) in the ADL scores for each activity
Dysphagia
The number of subjects showing symptoms of dysphagia in the GB-0998 group before initiation of study drug administration in the first period was 7 of 12, whereas the number in the placebo group was 2 of 14. At week 8 of the first period, the symptoms had ceased in 5 of the 7 subjects in the GB-0998 group, whereas they had not ceased with either of the two subjects in the placebo group (Fisher direct probability test, p = 0.1667).
Transition to the second period
From week 4 of the first period, it was permissible to make an early transition to the second period if alleviation of the disease was not seen on the basis of the time-courses of MMT score and serum CK level, if continuation of the first period was judged to be inappropriate, or if it was considered that treatment intensification would have been necessary. The numbers of subjects experiencing such early period transitions were 1 of 12 in the GB-0998 group and 4 of 14 in the placebo group.
Time-courses of MMT scores and serum CK levels of individual patients for 8 weeks from initial administration
The time-courses of MMT scores and serum CK levels in individual patients for 8 weeks after the initial administration are shown in Figs. 9, 10, respectively, for both the GB-0998 and placebo groups. The broken lines indicate the time-courses in patients after early transition to the second period.
Fig. 9.
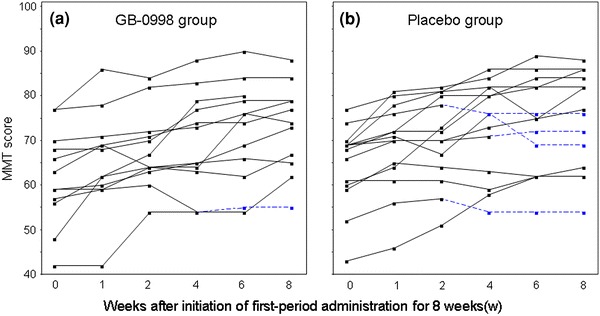
Time-courses of MMT scores of individual subjects for 8 weeks after initiation of first-period administration (a GB-0998 group, b placebo group). Broken lines indicate the time-courses after transfer to the second period in the cases of subjects who were transferred before the full 8 weeks in the first period
Fig. 10.
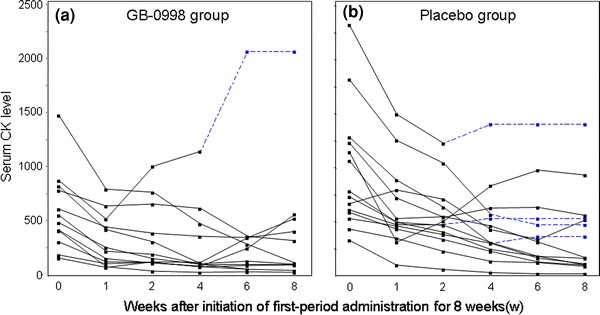
Time-courses of serum CK levels of individual subjects for 8 weeks after initiation of first-period administration (a GB-0998 group, b placebo group). Broken lines indicate the time-courses after transfer to the second period in the cases of subjects who were transferred before the full 8 weeks in the first period
Safety
Total numbers of adverse events occurring after initiation of GB-0998 administration were obtained for the 26 subjects receiving the study drug. One hundred one adverse events occurred in 23 of 26 subjects (88.5%) after initiation of the first period. This total does not include 91 cases of abnormal changes in clinical laboratory test results that occurred in 18 subjects. In terms of classification by severity, 89 adverse events in 15 subjects were mild, 11 events in 7 subjects were moderate, and 1 event in 1 subject was severe. The severe adverse event was cerebral infarction, but it was considered that there was no causal relationship between this and GB-0998 administration.
Nineteen adverse drug reactions occurred in 11 of 26 subjects (42.3%). These included gingivitis, hyperkalemia, disorders of glucose tolerance, diarrhea, dry skin, perspiration disorder, oral herpes, eructation, nausea, increased serum CK, decreased muscle strength, hot sensation, purpura, chest pain, headache, increased serum β-d-glucan, increased blood pressure, bronchitis, and fatigue. All these adverse drug reactions were mild to moderate in severity, and none occurred more than once.
Four serious adverse events occurred in 3 subjects. Of these, one case of cerebral infarction in a 36-year-old female occurred during the first period in the GB-0998 group, 50 days after initiation of first-period administration, and continued for 6 days until recovery. This event was thought to have been due to long-term corticosteroid administration, and a causal relationship to GB-0998 administration was considered unlikely. A second serious adverse event, Prinzmetal’s angina in a 56-year-old male, occurred during the second period, 55 days after initiation of second-period administration, and continued for 84 days until improvement. This patient was in the placebo group, and a considerable time had elapsed since completion of study drug administration, so a causal relationship with GB-0998 was considered unlikely. In addition, two adverse drug reactions in one 47-year-old female in the GB-0998 group occurred during the second period, 56 days after initiation of first-period administration. They were decreased muscle strength and increased serum CK level, which continued for 149 days until recovery. A causal relationship between these events and GB-0998 was judged to be probable.
These are all known adverse events.
Discussion
Since the first report of the use of immunoglobulin formulations to treat PM by Roifman et al. in 1987 [7], numerous clinical studies have confirmed the efficacy of intravenous immunoglobulin for treating PM, DM, and/or juvenile DM resistant to existing treatments. In addition, various textbooks [8] and guidelines [9, 10] present immunoglobulin formulations as treatment options for treatment-resistant PM and DM. However, the only published report of a placebo-controlled study relates to a double-blind, crossover study carried out by Dalakas et al. in 1993 [6]. Furthermore, the target disease for that study was treatment-resistant DM, and PM subjects were excluded.
The present authors have previously carried out two clinical studies on the use of GB-0998 to treat corticosteroid-resistant multiple PM and DM. As in the present study, the efficacy endpoints in the previous studies were MMT score, ADL score, and serum CK level, and in those studies, GB-0998 was found to be effective against corticosteroid-resistant multiple PM and DM [4, 5].
However, both of the previous studies were uncontrolled, and therefore the present study was designed as a placebo-controlled, double-blind trial in order to obtain a more objective efficacy evaluation. In addition, the protocol for the present study was made stricter, in that the criteria for resistance to corticosteroid treatment were more clearly defined, including an increase of the prestudy run-in period for confirmation of resistance to 6 weeks from the 4 weeks used in the two previous studies.
With respect to the intragroup comparison in the GB-0998 group, which was the principal analysis, we found statistically significant improvements due to GB-0998 administration in the primary endpoint (MMT score) and the secondary endpoints (ADL score and serum CK level). Further, the GB-0998 group was statistically significantly superior to the placebo group with respect to the time to normalization of serum CK level and score of swallowing action. These results suggest that early normalization of serum CK level, which is an index of inflammation and increases during the active stage of PM or DM, can be achieved by administration of GB-0998. Normalization of serum CK is important for achieving improvement in muscle strength, which is the ultimate goal of treatment.
In addition, dysphagia, which is related to swallowing, occurs in an active and advanced stage of PM and DM, and is often refractory to corticosteroid treatment; this may result in aspiration pneumonia, which significantly affects the prognosis [11–13]. It is therefore noteworthy that GB-0998 also appeared to improve dysphagia, a life-threatening complication of PM and DM.
With respect to safety, no severe adverse drug reactions occurred, and all serious adverse drug reactions were previously known effects. It is therefore considered that GB-0998 can be administered safely, with the same precautions that are applied in current IVIG treatments. However, a case of cerebral infarction did occur in the present study, although a causal relationship with administration of the study drug was considered unlikely. In addition, asymptomatic myocardial infarction was found as an adverse event in a previous study by the authors [4]. Furthermore, there have been reports of cerebrovascular and cardiovascular disorders, thought to be due to circulatory disturbances associated with increased blood viscosity, when high doses of immunoglobulins were administered to patients at risk of cerebrovascular and cardiovascular events [14, 15]. In the treatment of patients who have, or have a history of, cardiovascular and/or cerebrovascular disorders, and patients who are at high risk of developing thromboembolism, careful consideration should be given before administration of GB-0998, and careful monitoring is needed after administration.
Thus, the safety and efficacy results in the present study indicate that GB-0998 has the potential to be a useful drug for treatment of PM and DM subjects in whom corticosteroid treatment has not been effective.
However, the intragroup comparison in the placebo group also revealed significant improvements, and intergroup comparison of the changes in efficacy endpoints between before initiation of study drug administration and after administration for 8 weeks during the first period did not show a significant difference between the groups. In order to clarify the reasons for this, we tried to identify features of the subjects’ backgrounds that might have led to significant improvement being achieved even with placebo administration. The results suggested that the inequality of changes in MMT scores during the prestudy run-in period may have been the factor that had the most effect on intergroup difference.
The number of subjects whose MMT scores increased by 1 or more during the prestudy run-in period was 7 of 26 in the two groups (26.9%). However, a breakdown of these subjects showed that they consisted of 1 of 12 subjects in the GB-0998 group, and 6 of 14 in the placebo group, so subjects who were already showing a tendency toward recovery thus tended to be more numerous in the placebo group. When these 7 subjects were excluded, an intergroup comparison of MMT scores during the first period among the other 19 subjects, who underwent either no change or deterioration during the prestudy run-in period, gave an increased intergroup difference, 3.8, with a 95% confidence interval of −4.4 to 11.9, although this is still not statistically significant. A detailed assessment of the above seven subjects indicated that they can be considered as subjects in whom corticosteroid treatment showed some, albeit minor, efficacy, and therefore it is conceivable there might have been a carry-over effect from the corticosteroid treatment in these subjects.
In addition to steroid carry-over effect, the possibility of steroid myopathy must be considered. The patients in the present study had been administered steroids at high doses and/or for extended periods, and steroid administration was continued during the study period. Therefore, development of steroid myopathy is a potential issue [8], and one patient did in fact develop this condition. However, because it is difficult to distinguish muscle weakness due to steroid myopathy and that due to the primary disease, PM or DM, no evaluation of steroid myopathy was carried out in this study. Therefore, the possibility that evaluation of the primary endpoint, MMT score, was biased by the effect of steroid myopathy cannot be ruled out.
Perhaps the greatest limitation of the present study is the small number of patients included, and it is considered that the resulting intergroup bias in inclusion of subjects had a major impact on evaluation of intergroup difference. This highlights the need for great care in subject allocation to eliminate intergroup bias in future studies. However, PM and DM are rare diseases, and clinical studies with extremely limited numbers of patients are inevitably problematic.
GB-0998 was approved last year for cover by the Japanese National Health Insurance system for the indication of decreased muscle strength due to corticosteroid-resistant PM or DM. However, in the light of the present results, we suggest it would be desirable to carry out a postmarketing study with all patients administered GB-0998, in order to obtain clear evidence as to its efficacy. It will be necessary to evaluate the efficacy in accordance with the methods used in clinical studies, in order to assure comparability with the clinical study results. We think that such continuing evaluation will be important to fully characterize the efficacy of this drug in corticosteroid-resistant PM and DM, and also to ensure that the benefit for patients can be maximized.
Acknowledgments
We would like to express our gratitude to the members of the GB-0998 Study Group who are not listed as authors but who provided valuable advice and data for the present study.
Conflict of interest
All the authors have received consulting fees from Mitsubishi Tanabe Pharma Corporation, Osaka, Japan. N. Miyasaka has received research grants from Abbott Japan Co., Ltd., Astellas Pharma Inc., MSD KK, Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eizai Co., Ltd., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical Co., Ltd., and Teijin Pharma Limited. Y. Tanaka has received a research grant from Mitsubishi Tanabe Pharma Corporation. M. Hara is a medical adviser to a clinical trial organized by Toyama Chemical Co., Ltd.
Appendix
This work was supported by Benesis Corporation and Mitsubishi Tanabe Pharma Corporation, Osaka, Japan.
The authors wish to thank the members of this study group, who were as follows: Kazuyoshi Saito (The First Department of Internal Medicine, University of Occupational and Environmental Health, Japan); Tsutomu Takeuchi (Department of Internal Medicine, Saitama Medical Center, Saitama Medical School); Shiro Matsubara (Department of Neurology, Tokyo Metropolitan Neurological Hospital); Hitoshi Kohsaka (Department of Medicine and Rheumatology, Tokyo Medical and Dental University); Yoshinari Takasaki (Division of Rheumatology, Department of Internal Medicine, Juntendo University School of Medicine); Masayuki Matsuda (Third Department of Medicine, Shinshu University School of Medicine); Hiroaki Dobashi (First Department of Internal Medicine, Division of Endocrinology and Metabolism, Hematology, Rheumatology and Respiratory Medicine, Faculty of Medicine, Kagawa University); Takahiko Horiuchi (Department of Medicine and Biosystemic Science, Graduate School of Medical Sciences, Kyushu University); Kiyoshi Migita (Department of General Internal Medicine, NHO National Nagasaki Medical Center); Masataka Kuwana (Section of Rheumatology, Department of Internal Medicine, Keio University School of Medicine); Toshihide Mimura (Department of Rheumatology and Applied Immunology, Faculty of Medicine, Saitama Medical University Hospital); Ryutaro Matsumura (Department of Allergy and Rheumatology, Chiba-East Hospital, National Hospital Organization); Yasuo Suzuki (Division of Rheumatology, School of Medicine, Tokai University); Takeshi Kuroda (Division of Clinical Nephrology and Rheumatology, Niigata University Graduate School of Medical and Dental Sciences); Shunji Yoshida (Section of Rheumatology and Infectious Diseases, Department of Internal Medicine, Fujita Health University School of Medicine); Kazuma Sugie (Department of Neurology, Nara Medical University School of Medicine); Sotoshi Nakano (Department of Neurology, Kansai Medical University); Yuji Yamanishi (Department of Rheumatology, Hiroshima City Hospital); Manabu Osoegawa (Department of Neurology, Neurological Institute, Graduate School of Medical Sciences, Kyushu University); Hirofumi Ochi (Department of Neurology, Neurological Institute, Graduate School of Medical Sciences, Kyushu University); Yoshifumi Tada (Division of Rheumatology, Department of Internal Medicine, Saga University); Masako Hara (Institute of Rheumatology, Tokyo Women’s Medical University); Tatsuya Atsumi (Department of Medicine II, Hokkaido University Graduate School of Medicine); Kazuhiko Yamamoto (Department of Allergy and Rheumatology, University of Tokyo); Atsushi Ihata (Department of Internal Medicine and Clinical Immunology, Yokohama City University Graduate School of Medicine); Hidehiro Yamada (Department of Internal Medicine, St. Marianna University School of Medicine); Takao Fujii (Department of Rheumatology and Clinical Immunology, Graduate School of Medicine, Kyoto University); Yasuhiko Hirabayashi (Department of Rheumatology and Hematology, Tohoku University School of Medicine); Takehiko Ogawa (Department of Rheumatology, Toho University Ohashi Medical Center); Takashi Ogasawara (Division of Rheumatic Diseases, Tokyo Metropolitan Ohtsuka Hospital); Norihiko Watanabe (Department of Allergy and Clinical Immunology‚ Chiba University); Kimihiro Suzuki (Internal Medicine I, National Defence Medical College); Shigeto Tohma (Department of Rheumatology, Sagamihara National Hospital, National Hospital Organization), Yoshinobu Koyama (Department of Collagen Disease and Rheumatism Medicine, Iizuka Hospital)
References
- 1.Bradley WG. Polymyositis. Br J Hosp Med. 1977;17:351–355. [PubMed] [Google Scholar]
- 2.Cherin P. Recognition and management of myositis. Drugs. 1997;54:39–49. doi: 10.2165/00003495-199754010-00003. [DOI] [PubMed] [Google Scholar]
- 3.Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344–347. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 4.Hara M, Kinoshita M, Saito E, Hashimoto H, Miyasaka N, Yoshida T, et al. Prospective study of high-dose intravenous immunoglobulin for the treatment of steroid-resistant polymyositis and dermatomyositis. Mod Rheumatol. 2003;13:319–325. doi: 10.1007/s10165-003-0250-9. [DOI] [PubMed] [Google Scholar]
- 5.Saito E, Koike T, Hashimoto H, Miyasaka N, Ikeda Y, Hara M, et al. Efficacy of high-dose intravenous immunoglobulin therapy in Japanese patients with steroid-resistant polymyositis and dermatomyositis. Mod Rheumatol. 2008;18:34–44. doi: 10.1007/s10165-007-0013-0. [DOI] [PubMed] [Google Scholar]
- 6.Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000. doi: 10.1056/NEJM199312303292704. [DOI] [PubMed] [Google Scholar]
- 7.Roifman CM, Schaffer FM, Wachsmuth SE, Murphy G, Gelfand EW. Reversal of chronic polymyositis following intravenous immune serum globulin therapy. JAMA. 1987;258:513–515. doi: 10.1001/jama.1987.03400040111034. [DOI] [PubMed] [Google Scholar]
- 8.Dalakas MC. Polymyositis, dermatomyositis, and inclusion body myositis. In: Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson JL, et al., editors. Harrison’s principles of internal medicine. 17th ed. New York: McGraw-Hill; 2008. p. 2696−703.
- 9.Aberer W, Bagot M, Braathen L, Chimenti S, Diaz-Perez JL, Hegyi V, et al. Guidelines on the use of high-dose intravenous immunoglobulin in dermatology. European dermatology forum. Available at: http://www.euroderm.org/edf/images/stories/guidelines/guideline_on_ivig-aktuell-neu-2008.pdf. Cited 9 March 2010.
- 10.EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases/EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. Eur J Neurol. 2008;15:893−908. [DOI] [PubMed]
- 11.Maugars YM, Berthelot JM, Abbas AA, Mussini JM, Nguyen JM, Prost AM. Long-term prognosis of 69 patients with dermatomyositis or polymyositis. Clin Exp Rheumatol. 1996;14:263–274. [PubMed] [Google Scholar]
- 12.Marie I, Hachulla E, Hatron PY, Hellot MF, Levesque H, Devulder B, et al. Polymyositis and dermatomyositis:short term and longterm outcome, and predictive factors of prognosis. J Rheumatol. 2001;28:2230–2237. [PubMed] [Google Scholar]
- 13.Oh TH, Brumfield KA, Hoskin TL, Stolp KA, Murray JA, Bassford JR. Dysphagia in inflammatory myopathy:clinical characteristics, treatment strategies, and outcome in 62 patients. Mayo Clin Proc. 2007;82:441–447. doi: 10.4065/82.4.441. [DOI] [PubMed] [Google Scholar]
- 14.Silbert PL, Knezevic WV, Bridge DT. Cerebral infarction complicating intravenous immunoglobulin therapy for polyneuritis cranialis. Neurology. 1992;42:257–258. doi: 10.1212/WNL.42.1.257. [DOI] [PubMed] [Google Scholar]
- 15.Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology. 1994;44:223–226. doi: 10.1212/WNL.44.2.223. [DOI] [PubMed] [Google Scholar]