

Abstract
The phylogenetic structure and community composition were analysed in an existing data set of marine bacterioplankton communities to elucidate the evolutionary and ecological processes dictating the assembly. The communities were sampled from coastal waters at nine locations distributed worldwide and were examined through the use of comprehensive clone libraries of 16S ribosomal RNA genes. The analyses show that the local communities are phylogenetically different from each other and that a majority of them are phylogenetically clustered, i.e. the species (operational taxonomic units) were more related to each other than expected by chance. Accordingly, the local communities were assembled non-randomly from the global pool of available bacterioplankton. Further, the phylogenetic structures of the communities were related to the water temperature at the locations. In agreement with similar studies, including both macroorganisms and bacteria, these results suggest that marine bacterial communities are structured by “habitat filtering”, i.e. through non-random colonization and invasion determined by environmental characteristics. Different bacterial types seem to have different ecological niches that dictate their survival in different habitats. Other eco-evolutionary processes that may contribute to the observed phylogenetic patterns are discussed. The results also imply a mapping between phenotype and phylogenetic relatedness which facilitates the use of community phylogenetic structure analysis to infer ecological and evolutionary assembly processes.
Electronic supplementary material
The online version of this article (doi:10.1007/s00248-011-0005-7) contains supplementary material, which is available to authorized users.
Introduction
Ecological communities are the product of both contemporary biotic and abiotic forces as well as historical (phylogenetic) contingencies [37, 38]. For example, biogeography, local adaptive radiation, intra- and inter-specific interactions together with effects imposed on a community by habitat characteristics, e.g. temperature, salinity and nutrient availability, dictate the community assemblage. These processes often act in concert, and their relative strength varies at different temporal, geographical and phylogenetic scales [5, 36]. Phylogenetic structure analysis (mean relatedness between members of the community) together with the assumption that ecological differences among species map onto phylogenetic relatedness has been used extensively to elucidate community dynamics (reviewed in [3, 25, 34]). The methods have mainly been used and developed to study ecological and evolutionary processes to determine the assembly of plant communities, but they have also been applied to microbial communities [14].
A phylogenetically clustered local community contains species that are, on average, more related to each other than would be expected if assembled randomly from an appropriately defined pool of available species. This pattern has been interpreted as a community largely shaped by the abiotic environment at the site (“habitat”) in which the community is found [36]. This conclusion is based on the notion that habitat characteristics inflict selection pressure on the potential community members in a way such that only a non-random subset of the available (global or regional) species pool is able to colonize or invade and persist. A common term for this abiotic-driven assembly process is “habitat filtering”. In contrast, an overdispersed phylogeny, i.e. that the species are less related than expected, is suggested to imply that inter-specific competition can be an important structuring process. This idea is based on the hypothesis that phylogenetically closely related species cannot coexist because they share critical traits (and therefore niches) whereas more distantly related species do not [11, 36]. A majority of the studies published on phylogenetic structure show communities to be clustered. However, few have considered that the results are scale dependent. The geographical scale influences the result indirectly by dictating the null (or pool) community to which the phylogeny of interest is compared [35]. Also, the dominating ecological and evolutionary processes are different at different geographical scales. For example, phylogenetic structure analysis can reveal signals of competitive exclusion at small (within-habitat) scales and niche conservatism at large (regional) scales [29, 36]. The temporal scale can also have a direct influence as both colonization and speciation are functions of time [5]. Finally, studies of meadow plant communities show that the taxonomic level at which the phylogeny is analysed influences the phylogenetic structure results [28, 29]. The scaling problem gets apparent in our use of the term “habitat”. Here we use the term to indicate a site and its characteristics, without further reference to other biological relevant habitat definitions such as along depth or salinity gradients. Further, interpretations of phylogenetic patterns often lack alternatives to the hypotheses of “habitat filtering” and “competitive exclusion”. For example, local adaptive radiation, in relation to invasion rate, may also explain non-random patterns in phylogenetic structure [5].
Although the use of phylogenetic analyses is particularly rewarding when studying communities where the available phylogenetic information is large, only a few studies have addressed the above questions on microbial communities. For example, bacterial communities sampled in soil, freshwater and saltwater are typically clustered, indicating habitat filtering to be the dominating process [14, 24]. In this study, we do an extensive reanalysis of a large data set on 16S ribosomal RNA gene data from nine globally distributed marine bacterial communities [26]. In contrast to Pommier et al. [26], we focus on phylogenetic analyses to elucidate between-community similarity and structure and within community structure, taking the full evolutionary community history into account. We interpret the phylogenetic patterns according to the traditional dichotomy of “clustering” versus “overdispersion” as signals of “habitat filtering” and “competitive exclusion”, respectively. We discuss eco-evolutionary processes that also may dictate the community assemblages.
Methods
Data
We used an already available data set on marine bacterioplankton sampled by Pommier et al. [26]. The data consist of 16S ribosomal RNA genes from nine different localities around the world. Sampling was conducted: offshore Disko Island, in Baffin Bay; in the Arctic ocean; in the South Atlantic offshore Cape Town, South Africa; in the Sargasso Sea, offshore Bermuda; in the Pacific ocean offshore San Diego, California; offshore Hawaii islands; offshore Sydney, Australia; offshore the Fiji Islands and offshore Concepcion, Chile. All samples were taken at 5 m depth 10 km of the coast and measures were taken to minimize the risk of sampling at extreme sites [26]. The 16S ribosomal RNA genes was analysed by PCR amplification, cloning and Sanger sequencing as described by Pommier et al. [26]. Clone coverage was calculated for each of the nine localities using the Good [9] and the Lee and Chao [18] coverage estimates, showing on average 87% and 74% coverage, respectively [26]. Also, to investigate whether the diversity within localities was captured by sampling, Pommier et al. [26] calculated S Chao1 estimates showing satisfactory results for all but the Hawaii sample. The sequences of the cloned partial 16S rRNA genes can be found in GenBank under the accession numbers DQ668407–DQ672245.
Sequence Analysis
Fourteen thousand three hundred ninety complementary DNA sequences from 7,195 16S rRNA gene clones were included in the data set. This data set was quality-checked and treated by Pommier et al. [26] which resulted in a set of 4,250 sequences that were used for further analysis. To reduce potential remaining errors from the previous analyses, we did an extensive reanalysed sequence data by using state-of-the-art methods for quality check, alignment, data base matching and phylogenetic analysis.
We matched each individual sequence to the Greengenes database which consist of curated, chimera-checked, 16S rRNA gene sequences and aligned them with the NAST algorithm at http://greengenes.lbl.gov [4]. After reviewing the alignments manually, the data were shown to contain some short-length sequences, and to avoid errors, such as short branch attractions in later phylogenetic analysis, we removed all sequences <100 bp. The threshold length of 100 bp was chosen in concordance with the data handling of Pommier et al. [26], and hence, the results are comparative between the studies. In contrast to Pommier et al. [26], who taxonomically assigned operational taxonomic units (OTUs) after clustering, here each sequence in the alignments was taxonomically assigned to the level of phylum with the classification tool of Greengenes. This classification method is based on finding near-neighbours in the Greengenes data base using the Simrank algorithm. Greengenes currently supports three different taxonomies: RDP, NCBI and Hugenholz. If two of these assigned the same taxonomic lineage at the phylum level, the sequence was kept for further analysis. After data base matching, alignment, classification and removal of short sequences, 3,617 sequences were considered high enough quality for further analysis. Note that this is a reduction of about 500 sequences compared to the ones used in Pommier et al. [26] (see Table S1 for details).
Phylogenetic Analysis
For community composition and phylogenetic structure, one phylogenetic tree per phylum from all localities was constructed. Further, trees were also constructed for each of the localities, including all phyla. For analysis of phylogenetic distance between communities one tree was constructed containing the whole data set. For those purposes, the maximum likelihood method in the RAxML 7.0.4 software using default settings and GTRMIX-model [30] was used. This model constructs a tree by approximation with optimization of individual per site substitution rates and classifications of those individual rates into 25 (default) rate categories. The topology found is then evaluated such that it yields stable likelihood values [30]. The phylogenetic trees were used as input to the analyses done on sequence level, e.g. calculations of phylogenetic difference between samples. Other analyses, e.g. Morisita’s index of similarity, require species and species abundance as input. We defined “species” or more appropriate, OTUs, by clustering the sequence-level phylogenetic trees with the RAMI software [27]. This clustering method uses nodes and branch lengths in the phylogenetic tree to construct cluster of sequences, defined by a RAMI threshold of patristic (branch length) distance. As we know little about the correct threshold defining an OTU and the potential effects of the threshold on phylogenetically based results, we did the clustering procedure with threshold values ranging from 0.01 to 0.08. The results were robust over the full range of threshold values; hence, we only report the 0.01 level analysis here. Note also that this approach is different from Pommier et al. [26] who clustered sequences based on nucleotide similarity, not on patristic distances. For each of the clusters, a consensus sequence was created with the RAMI tool rami_consesus. The consensus sequences, belonging to a specific data subset, were aligned and new phylogenetic trees were created with the RAxML software as above. Each consensus sequence in the consensus tree defines an OTU, and the number of sequences within each cluster was treated as abundance in the OTU-level analyses.
Comparisons Between Localities
As in Pommier et al. [26], we compared localities by calculating the Morisita’s index of similarity of community composition. In contrast to Pommier et al. [26], where SEQMANII (LASERGENE v.5) was used on sequence similarity, we used our RAMI clusters as in-data for the analysis. Further, we extended the analysis to involve extensive phylogenetic analysis of community similarity and structure. The Unifrac distance matrix tool [20] was used to calculate the pairwise phylogenetic difference between all localities, using one large tree containing all sequences from all localities. Consequently, this analysis is based on sequence level data, not on RAMI cluster as for the Morisita’s index analysis above. This tool calculates a distance metric by comparing common nodes and branch lengths of the sample phylogenies [20]. Two identical communities will have all nodes and branch lengths in common, and the Unifrac distance metric will be zero (0%). In contrast, if two samples differ already in the first node of the phylogenetic tree (e.g. one sample is clustered in one part and the other sample in another part of the total tree), no common nodes or branches exist and the Unifrac metric will be 1 (100% difference). In contrast to the Morisita’s analysis, which compare common species and their abundances, the Unifrac approach takes the full phylogeny, hence the evolutionary history, into account when calculating the distance metrics. As a consequence, the Morisita’s and the Unifrac analyses do not necessarily have to give the same result. For consistency in results, we converted the Unifrac metrics to show similarity instead of distance by subtracting each Unifrac metric value from one (1 − (Unifrac value)). We calculated the significance of the phylogenetic distance between localities using the pairwise Unifrac significance test and P test significance tool. Both tests compare the sample phylogenetic tree to randomly constructed null model trees (1,000 random permutation of environment labels across the sequences). The reported significance value of phylogenetic distances is the fraction of null model trees that have a Unifrac metric value greater or equal to the sampled tree. The P test compares the number of parsimony changes required to explain the distribution of sequences between samples. The reported P value is the fraction of null model trees that require fewer parsimony changes to explain distributions than does the sample tree. Notably, the Unifrac significance test and Unifrac P test are tools primarily suited for analysis of two or a few environments [20]. Consequently, few of the P values reported in the outputs from these tests were significant after doing the Bonferroni correction of multiple comparisons. When showing the relative difference between localities in the full data (one phylogenetic tree containing all sequences from all libraries), we used the Unifrac jackknife environment clustering tool [20]. This tool performs hierarchical clustering analysis of the localities based on the pairwise Unifrac distance metric (pairwise comparisons of libraries) and outputs a dendrogram with coloured nodes denoting the fraction of the random samples that they were recovered in.
Abiotic Factors Affecting Community Dynamics
The surface water temperature and latitude data given by Pommier et al. [26] were used to serve as a proxy for the environmental conditions in the sampled localities. We measured the difference in surface water temperature and latitude between all pairwise sample combinations and related these to pairwise Morisita’s and Unifrac similarity calculation.
Phylogenetic Clustering or Overdispersion
We used Phylocom [35] to analyse clustering or overdispersion patterns in all localities. The net relatedness index (NRI) quantifies the structure of a sample phylogeny derived from the mean phylogenetic distance, consequently capturing the degree of clustering of the phylogeny from root to terminal leaves. The nearest taxa index (NTI) quantifies the terminal structure of the sample phylogeny, hence only captures the clustering of the terminal nodes in the tree. NRI and NTI are defined as:
 |
1 |
 |
2 |
where MPD = mean phylogenetic distance, MNTD = mean nearest phylogenetic taxon distance and sd = standard deviation. The subscript “sample” denotes values derived from the phylogenetic tree analysed and “rndsample” denotes values derived from 999 random phylogenies constructed according to a null model. The null model used here (# 2) constructs a random phylogenetic tree by assigning sequences/OTUs to the localities (localities found in the focal phylogeny) by random draws from the phylogeny pool (all OTUs available in the input data). This maintains the sequence richness of each sample, but the identities of the species occurring in each sample are randomised. Positive NRI/NTI values indicate a clustered phylogeny where coexisting taxa are more related to each other than expected by chance. A negative NRI/NTI value indicates an overdispersed phylogeny where coexisting taxa are less related to each other than would be expected by chance. We calculated the NRI and NTI, using the comstruct Phylocom tool at both community level (including all phyla) and the phylum level (the structure of phylum X in each locality, respectively). As we have little knowledge of the ecological significant scale for bacterial community, the Phylocom analysis was done on both single sequences and sequence clusters (OTUs) as terminal leaves in the phylogenetic trees. Finally, we used a two-tailed significance test for the NRI/NTI results, using the rank high and rank low values in the Phylocom output [35]. The rank values are the number of runs showing the null model to have lower or higher NRI/NTI values than the focal phylogeny. Consequently, positive NRI/NTI values associated with rank low >975 and rank high <25 and negative NRI/NTI associated with rank low <25 and rank high >975 are significant at P < 0.05. The analyses on phylum level aim to investigate both specific phylum/locality combinations and marine bacterial communities in general. Consequently, we Bonferroni-corrected the output from Phylocom by multiplying the P values with 1/n, where n is the number of unique phylum/locality combinations (n = 77).
Results
Community Similarity
The Morisita’s index, based on OTU pairwise similarities and abundances between samples, showed both high and low values (Fig. 1). The highest similarity index was found between Cape Town and Sydney with a joint Morisita’s index of 0.81. This value tells us, given some internal diversity in the compared communities, the probability that two sequences randomly drawn from the two communities, respectively, belong to the same OTU. Consequently, the Cape Town and Sidney samples are 81% similar in terms of OTU composition and abundance. Baffin Bay is relatively dissimilar to all other localities (in some cases 0% similarity), except the Arctic Ocean. Consequently, this sample is to a large extent unique in terms of OTU composition. Note that a majority (26 out of 36) of the pairwise similarity values were found in the two bottom quartiles of the distribution of similarity values. The Unifrac analysis of phylogenetic distance gave somewhat different results; the pairwise Unifrac metric values were on average lower and had less variation (ranging from 0.1 to 0.37). The distribution of Unifrac values was also more evenly distributed; ten out of the 36 pairwise Unifrac values were found in the lower two quartiles. The qualitative pattern among samples was, however, similar between the Unifrac and Morisita’s results. Baffin Bay again stands out, sharing less than 20% of the nodes and branch lengths of any of the other communities (Fig. 1). The significance test and Unifrac P test that were used for analysing the significance of the pairwise distance between communities all showed high P values. A possible explanation for this is the effect of the Bonferroni correction for multiple comparisons; these tests are primarily suited for analysis of two or a few samples [20].
Figure 1.
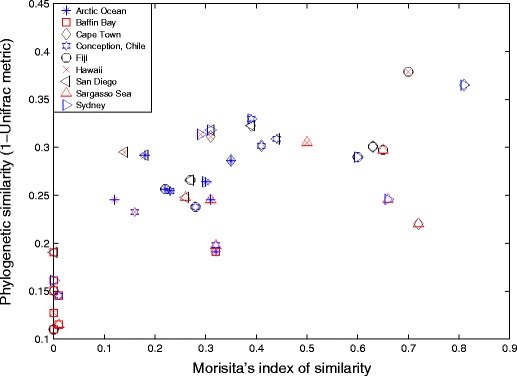
Pairwise comparison of community structure. The Morisita’s index of similarity between communities (x-axis). Converted Unifrac metrics (1 − Unifrac metric) (y-axis). Overlapping points from different communities represents the Morisita’s and Unifrac values for the pairwise comparisons. Similarity is stated in percent where 0 denotes no similarity and 1 denotes identical communities. Correlation coefficient between Morisita’s and Unifrac values, 0.70
Although the pairwise significance tests, above, give high values, the Unifrac jackknife analysis show patterns of differentiation between localities. Two nodes in the Unifrac dendrogram was recovered in >99.9% of the randomised jackknife resampling analysis implying the different localities to be clustered into three distinct clusters (Fig. 2). In addition, four nodes in the dendrogram were recovered in >50% of the resampling procedure, implying further structuring below the three main clusters. Temperature differences were negatively correlated with differences in community composition (Morisita’s index) and phylogeny (Unifrac metric) (Fig. 3a, b). Although weak, the same relationship was also found between latitude and community composition (data not shown).
Figure 2.

Dendrogram illustrating similarity between localities and clustered localities are similar in composition. Hierarchical clustering (UPGMA) of samples has been made with the Unifrac distance matrix as basis. Each node in the dendrogram has been jackknife analysed (jackknife analysis clusters = 75% of smallest sample, 100 permutations). Colour of nodes illustrates the jackknife support value
Figure 3.
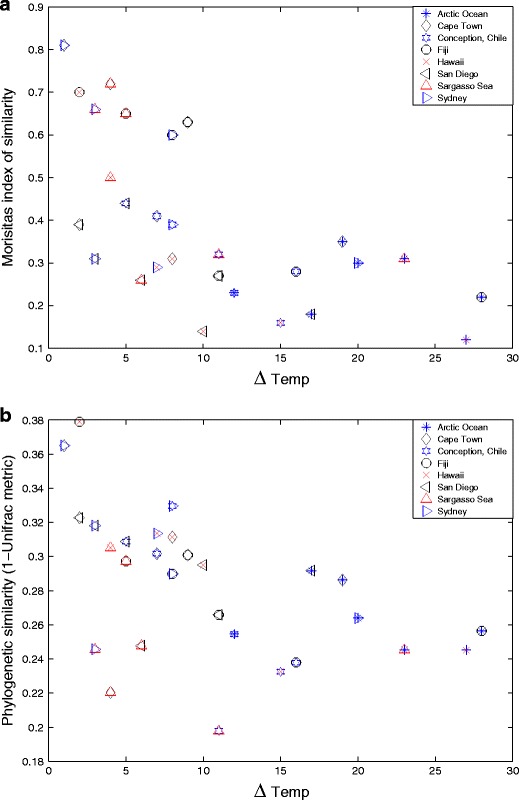
a Morisita’s index of similarity from pairwise comparisons between samples plotted against difference in water temperature at sample site at sampling occasion. Correlation coefficient between Morisita’s index data and difference in water temperature, −0.64. b Phylogenetic similarity from pairwise comparisons between samples plotted against difference in water temperature at sample site at sampling occasion. Correlation coefficient between phylogenetic similarity and difference in water temperature, −0.50
Phylogenetic Clustering and Overdispersion
There were significantly positive NRI and NTI values (clustering) in 5 and 7, respectively, of the nine localities when analysing the phylogenetic structure at the sequence scale (single sequences in the terminal leaves of the analysed phylogenetic tree) (Table 1). The same analysis at the OTU level (clusters of sequences as terminal leaves of the phylogenetic tree) showed similar results with positive NRI and NTI values in 4 and 9, respectively, of the nine localities (Table 1). Consequently, a majority of the communities tend to be phylogenetically clustered. Two and one of the localities had significant negative NRI values in the sequence-level and cluster-level analysis, respectively. Consequently, the members of these communities are less related to each other than expected by chance (i.e. phylogenetically overdispersed). Also, it should be noted that, although in general lower NRI values for the OTU-level analysis, the results were robust for the two analysis approaches (sequence scale and cluster scale) giving similar qualitative NRI and NTI results.
Table 1.
NRI and NTI for globally distributed bacterial communities
| Sequence analysis | Cluster analysis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | NRI | RGR | NTI | RGR | N | NRI | RGR | NTI | RGR | |
| Arctic Ocean | 286 | 0.92 | 813 | 3.41** | 999 | 143 | 0.35 | 620 | 4.54** | 999 |
| Baffin Bay | 253 | 29.03** | 999 | 7.33** | 999 | 62 | 6.52** | 999 | 5.34** | 999 |
| Cape Town | 211 | 1.09 | 863 | 2.73** | 996 | 135 | −0.11 | 457 | 3.64** | 999 |
| Conception, Chile | 677 | −2.34** | 8 | 6.02** | 999 | 248 | 2.18** | 987 | 5.19** | 999 |
| Fiji | 542 | 6.52** | 999 | 1.32 | 913 | 272 | 2.00* | 965 | 2.01** | 974 |
| Hawaii | 402 | 4.48** | 999 | 1.67* | 954 | 232 | 1.13 | 873 | 4.12** | 999 |
| San Diego | 438 | −4.70** | 0 | 5.42** | 999 | 142 | −4.01** | 0 | 3.43** | 998 |
| Sargasso Sea | 507 | 13.31** | 999 | 3.87** | 998 | 257 | 5.89** | 999 | 5.50** | 999 |
| Sidney | 301 | 2.62** | 991 | 0.86 | 803 | 197 | 0.17 | 575 | 3.49** | 999 |
NRI net relatedness index, NTI nearest taxa index, N number of taxa in community, RGR number of runs where the observed NRI/NTI values are greater than in the randomly drawn null community, left results from sequence-level analysis, right results from cluster-level analysis
Stars indicate statistically significant (* - P < 0.01; ** - P < 0.05) community structuring
When testing the structure of individual phyla within the different localities, once again a large proportion of the data subsets were clustered (Fig. 4). If single sequence-level data were used, significant phylogenetic structure was found in 34 out of the 77 phylum-by-locality data sets having sufficient data for Phylocom calculations (Fig. 4a). Only seven of the NRI or NTI values were found to be significantly negative. In the OTU-level analyses, significant phylogenetic structure was found in 29 out of the 77 phylum-by-locality combinations containing sufficient data for Phylocom calculations (Fig. 4b). Only two of the NRI or NTI values were found to be significantly negative. After Bonferroni correction, no significant negative results remained; however, 16 and 9 significant positive NRI or NTI values remained in the sequence-level and cluster-level analysis, respectively. Notably, 28 out of the 41 significant NRI and NTI values at the OTU level were recovered in the single sequence-level analysis. Although the two approaches give qualitatively similar results, the discrepancy between the two possibly is a result of reduction in noise, especially in the terminal parts of the phylogenetic trees, as individual sequences are clustered together.
Figure 4.
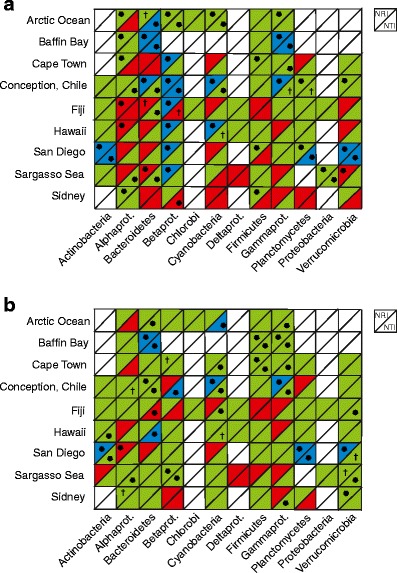
Net relatedness index (NRI) and nearest taxa index (NTI) for all phyla represented in each of the localities. One square per phylum and locality combination, upper left part represents NRI and the lower right part represents NTI. Colour code green denotes positive values (clustered community). Red denotes negative values (overdispersed community). *P < 0.05 and †P < 0.10 denote significant results. Blue denotes positive results, P < 0.05 with Bonferroni correction. Note that after Bonferroni correction, no significant negative results remain. a Analysis made on sequence level, terminal leaves in the phylogenetic tree consists of one single sequence. b Analysis made on OTU level, terminal leaves in phylogenetic tree consist of sequences clustered together based on phylogenetic similarity
Discussion
In this study, we have used a combination of phylogenetic structure analysis and data on abundance, geographical distribution and habitat characteristics of marine bacterial communities to elucidate potential ecological and evolutionary processes dictating the assemblages. In addition to an extensive reanalysis of the raw data, sampled by Pommier et al. [26], we include phylogenetic analyses to investigate community similarity and structure. Consequently, in contrast to Pommier et al. [26], we do analyses on both community composition and the community phylogeny, hence taking the full evolutionary history into account. Results from our Morisita’s index analysis show the marine bacterioplankton communities to be different from each other in terms of OTU composition and relative abundance. These results are different from the results presented by Pommier et al. [26] who stated that most communities were similar. Our phylogenetic analysis shows that the communities are different also in an evolutionary perspective. We found a relationship between-community structure and temperature at sample site (Fig. 3). Finally, we show that the marine bacterial communities across the globe studied here typically are phylogenetically clustered. From the available pool of bacterial types, a distinct subset is non-randomly recruited into the local marine bacterioplankton communities. As suggested by Webb et al. [36], this can be interpreted such that the members of those subsets share traits that make them suitable for the particular environmental conditions at the specific localities. That is, habitat characteristics dictate whether a species, with specific phenotypic and genotypic characteristics, can colonize a novel habitat or invade a community. We have limited information about the specific environmental characteristics at spatial and temporal scales relevant to bacterial communities. We did find a relationship between water temperature at the site (and sampling occasion) and community composition. Whether that relationship says something about the environmental characteristics responsible for “habitat filtering” or not is, however, an open question. A correlation between the phylogenetic signal and whatever environmental variables is only truly meaningful if the abiotic requirements of bacterial taxa is fully known.
Although our results suggest that marine bacterial communities are structured by “habitat filtering” processes, we recognize other possible explanations for the patterns of phylogenetic clustering. In addition to habitat characteristics, the recruitment of species between habitats is affected by, e.g. the organism propensity to disperse, whether the organism are resilient to novel conditions and the distance between habitats. Further, the rate of local adaptive radiation affects the community structure [5, 14]. Local adaptive radiation, in turn, is affected by various factors such as temporal and spatial habitat heterogeneity and niche breadth of the organisms. The pattern of phylogenetic clustering, often observed in macroorganisms, seems to be largely shared by marine bacteria. However, effects from ecological interactions should not be ruled out. The results have been shown to be scale dependent, and a community filtered by its habitat can either be neutral [15, 39, 40] or retain strong niche partitioning [23, 31, 32]. It has been suggested that spatial and temporal heterogeneity in most environments where bacteria are abundant could potentially allow for a huge neutral diversity as we typically measure it at intermediate and large scales [10, 19, 33]. In contrast, these organisms may be similar in its resource requirements potentially causing selection for niche partitioning between competing strains [12, 21]. Consequently, to understand the full range of processes and the relative strength between them, signals hidden below dominating patterns of phylogenetic structure and various scales need to be investigated.
We have considered the phylogenetic structure at various scales by doing the analysis on sequence level (used here as a proxy for individuals) and OTU level (proxy for species). We did the data analysis on different sets (each community including all phyla) and subsets (each phylum within each community). In addition, we made efforts to find signals below the results of phylogenetic clustering. It has, e.g. been suggested that analysis of the distribution of relative abundances (fractional abundances) between closely and distantly related pairs of species can reveal the presence of overall niche structure [16, 17]. Discrepancy between the fractional abundance distribution of related pairs and the distribution of random pairs might indicate niche separation between closely related taxa. In contrast, if the patterns in abundance are indistinguishable from random, the community members are assumed to be neutral and interchangeable entities. The mechanisms behind these patterns are, as for the phylogenetic structure analysis, based on the assumption of close mapping between phylogeny and traits. Given this mapping, related species share traits to a larger extent than nonrelated species. According to this hypothesis [16, 17], similar traits result in similar niches which may give rise to high competition between closely related species. Ultimately these interactions may be manifested in the relative species abundances. We conducted fractional abundance analysis on our data by comparing abundances of closely related and randomly selected OTUs in the phylogenetic tree, respectively. Unfortunately, this data set does not allow for any conclusive answers because of the small sample size. We do, however, hope that our efforts may inspire future studies to use fractional abundance analysis on data sets containing enough sequences to get resolution on the level of OTU abundance.
The Morisita’s index results, shown here, suggest that our methodological approach is more sensitive in detecting community differences than, e.g. the approach used in Pommier et al. [26]. This may be due to the RAMI clusters which most likely have a larger biological significance than clusters that are defined from sequence similarity only. It should, however, also be noted that the results may be affected by threshold values in the clustering algorithm. However, we found the results to be robust over the full range of RAMI thresholds (0.01–0.08) and taxonomic scale. In addition, the Unifrac results support the Morisita’s index patterns. Interestingly, there are, however, some differences between the two analyses (Fig. 1). This is possible because two communities that differ extensively in species composition and abundance may not be totally evolutionary different, especially if they share ancestry but have experienced resent isolation and disruptive selection. On the other hand, communities with similar species abundance distributions may still have different evolutionary history. From this, we conclude that both community composition and phylogeny structure patterns need to be considered; the patterns may be different and answer, somewhat, different questions.
As stated above, the relationship between water temperature at sample site and community structure suggest that habitat characteristics may affect the assembly. However, further experiments are needed to more specifically identify the habitat characteristics that drive the structuring process; temperature may be a direct physiological effect on the bacteria or indirect effects via secondary environmental effects. Also, temperature is only one of many habitat characteristics. It would be interesting to investigate the relationship between other factors such as salinity and resource availability and community structure (see, e.g. [6]).
Studies on natural bacterial communities have shown both spatial and temporal variation in community composition [1, 7, 8, 13, 22, 41]. This data set is sampled at different geographical sites and at different temporal occasions. Consequently, the sampling scheme may affect the results. However, the consistency in our results lead us to speculate that, despite plausible temporal variation in community composition, the dominating assembly processes may be more or less similar. Nevertheless, analyses on scales different from the ones investigated in this study are desirable to fully understand temporal dynamics and geographical scale dependencies.
Finally, the link between phylogenetic patterns and ecological processes hinges on a relationship between phenotypic characteristics and “species” relatedness. Several studies [2, 14, 24] suggest that such a relationship exists at least for some taxa. In addition, we argue that the non-random phylogenetic patterns found in this study imply a link between ecological assembly processes and patterns. This link likely is due to niche conservatism, i.e. a mapping between bacterial phenotype and relatedness. However, as the ecology of a great majority of the bacteria in natural communities is largely unknown, this assumption may not be valid for all taxa and traits. Consequently, we invite further investigations of the distribution of traits over the phylogeny of bacteria to strengthen the implications of phylogenetic signal analysis.
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
(DOCX 22 kb)
Acknowledgements
This study was financially supported by grants (to Per Lundberg and Anders Tunlid) from the Swedish Research Council and the Swedish research council Formas. The comments from two anonymous reviewers greatly improved the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Andersson AF, Riemann L, Bertilsson S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J. 2010;4:171–181. doi: 10.1038/ismej.2009.108. [DOI] [PubMed] [Google Scholar]
- 2.Boyd E, Hamilton T, Spear J, Lavin M, Peters J. [FeFe]-hydrogenase in Yellowstone National Park: evidence for dispersal limitation and phylogenetic niche conservatism. ISME J. 2010;4:1485–1495. doi: 10.1038/ismej.2010.76. [DOI] [PubMed] [Google Scholar]
- 3.Cavender-Bares J, Kozak KH, Fine PVA, Kembel SW. The merging of community ecology and phylogenetic biology. Eco Lett. 2009;12:693–715. doi: 10.1111/j.1461-0248.2009.01314.x. [DOI] [PubMed] [Google Scholar]
- 4.DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006;34:394–399. doi: 10.1093/nar/gkl244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emerson BC, Gillespie RG. Phylogenetic analysis of community assembly and structure over space and time. Trends in Ecol Evol. 2008;23:619–630. doi: 10.1016/j.tree.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Fuhrman J, Steele J. Community structure of marine bacterioplankton: patterns, networks, and relationships to function. Aquat Microbial Ecol. 2008;53:69–81. doi: 10.3354/ame01222. [DOI] [Google Scholar]
- 7.Fuhrman J, Steele J, Hewson I, Schwalbach MS, Brown MV, Green JL, Brown JH. A latitudinal diversity gradient in planktonic marine bacteria. Proc Nat Acad Sci. 2008;105:7774–7778. doi: 10.1073/pnas.0803070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giovannoni SJ, Stingl U. Molecular diversity and ecology of microbial plankton. Nature. 2005;437:343–348. doi: 10.1038/nature04158. [DOI] [PubMed] [Google Scholar]
- 9.Good IJ. The population frequencies of species and the estimation of population parameters. Biometrika. 1953;40:237–264. [Google Scholar]
- 10.Gooday AJ, Bett BJ, Escobar E, Ingle B, Levin LA, Neira C, Raman AV, Sellanes J. Habitat heterogeneity and its influence on benthic biodiversity in oxygen minimum zones. Mar Ecol -Evol. 2010;31:125–147. [Google Scholar]
- 11.Graves GR, Gotelli NJ. Assembly of avian mixed-species flocks in Amazonia. Proc Nat Acad Sci. 1993;90:1388–1391. doi: 10.1073/pnas.90.4.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grover JP. Resource competition and community structure in aquatic microorganisms: experimental studies of algae and bacteria along a gradient of organic carbon to inorganic phosphorus supply. J Plankton Res. 2000;22:1591–1610. doi: 10.1093/plankt/22.8.1591. [DOI] [Google Scholar]
- 13.Hewson I, Steele J, Capone D, Fuhrman J. Temporal and spatial scales of variation in bacterioplankton assemblages of oligotrophic surface waters. Mar Ecol Prog. 2006;311:67–77. doi: 10.3354/meps311067. [DOI] [Google Scholar]
- 14.Horner-Devine MC, Bohannan BJM. Phylogenetic clustering and overdispersion in bacterial communities. Ecology. 2006;87:100–108. doi: 10.1890/0012-9658(2006)87[100:PCAOIB]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 15.Hubbell SP. The unified neutral theory of biodiversity and biogeography (MPB-32) Princeton: Princeton University Press; 2001. [Google Scholar]
- 16.Kelly CK, Bowler MG, Pybus O, Harvey PH. Phylogeny, niches, and relative abundance in natural communities. Ecology. 2008;89:962–970. doi: 10.1890/07-0322.1. [DOI] [PubMed] [Google Scholar]
- 17.Kelly CK, Bowler MG. Temporal niche dynamics, relative abundance and phylogenetic signal of coexisting species. Theor Ecol. 2009;2:161–169. doi: 10.1007/s12080-008-0037-x. [DOI] [Google Scholar]
- 18.Lee SM, Chao A. Estimating population-size via sample coverage for closed capture-recapture models. Biometrics. 1994;50:88–97. doi: 10.2307/2533199. [DOI] [PubMed] [Google Scholar]
- 19.Levin LA, Sibuet M, Gooday AJ, Smith CR, Vanreusel A. The roles of habitat heterogeneity in generating and maintaining biodiversity on continental margins: an introduction. Mar Ecol Evol Persp. 2010;31:1–5. [Google Scholar]
- 20.Lozupone C, Knight R. Unifrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Makino W, Cotner JB. Elemental stoichiometry of a heterotrophic bacterial community in a freshwater lake: implications for growth- and resource-dependent variations. Aquat Microbial Ecol. 2004;34:33–41. doi: 10.3354/ame034033. [DOI] [Google Scholar]
- 22.Martiny JBH, Bohannan BJM, Brown JH, et al. Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol. 2006;4:102–112. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 23.Nee S, Harvey PH, May RM. Lifting the veil on abundance patterns. PBioS. 1991;243:161–163. [Google Scholar]
- 24.Newton RJ, Jones SE, Helmus MR, McMahon KD. Phylogenetic ecology of the freshwater actinobacteria acI lineage. Appl Environ Microbiol. 2007;73:7169–7176. doi: 10.1128/AEM.00794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pausas JG, Verdú M. The jungle of methods for evaluating phenotypic and phylogenetic structure of communities. BioScience. 2010;60:614–625. doi: 10.1525/bio.2010.60.8.7. [DOI] [Google Scholar]
- 26.Pommier T, Canbäck B, Riemann L, Boström KH, Simk K, Lundberg P, Tunlid A, Hagström Å. Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol. 2007;16:867–880. doi: 10.1111/j.1365-294X.2006.03189.x. [DOI] [PubMed] [Google Scholar]
- 27.Pommier T, Canbäck B, Lundberg P, Hagström Å, Tunlid A. RAMI: a tool for identification and characterization of phylogenetic clusters in microbial communities. Bioinformatics. 2009;25:736–742. doi: 10.1093/bioinformatics/btp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Silvertown J, Dodd M, Gowing D. Phylogeny and the niche structure of meadow plant communities. J Ecol. 2001;89:428–435. doi: 10.1046/j.1365-2745.2001.00553.x. [DOI] [Google Scholar]
- 29.Silvertown J, Dodd M, Gowing D, Lawson C, McConway K. Phylogeny and the hierarchical organization of plant diversity. Ecology. 2006;87:39–49. doi: 10.1890/0012-9658(2006)87[39:PATHOO]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 30.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 31.Sugihara G, Bersier LF, Southwood TRE, Pimm SL, May RM. Predicted correspondence between species abundances and dendrograms of niche similarities. Proc Nat Acad USA. 2003;100:5246–5251. doi: 10.1073/pnas.0831096100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugihara G. Minimal community structure: an explanation of species abundance patterns. Am Nat. 1980;116:770–787. doi: 10.1086/283669. [DOI] [PubMed] [Google Scholar]
- 33.Therriault TW, Kolasa J. Explicit links among physical stress, habitat heterogeneity and biodiversity. Oikos. 2000;89:387–391. doi: 10.1034/j.1600-0706.2000.890220.x. [DOI] [Google Scholar]
- 34.Vamosi SM, Heard SB, Vamosi JC, Webb CO. Emerging patterns in the comparative analysis of phylogenetic community structure. Mol Ecol. 2009;18:572–592. doi: 10.1111/j.1365-294X.2008.04001.x. [DOI] [PubMed] [Google Scholar]
- 35.Webb CO, Ackerly DD, Kembel SW. Phylocom: software for the analysis of phylogenetic community structure and trait evolution. Bioinformatics. 2008;24:2098–2100. doi: 10.1093/bioinformatics/btn358. [DOI] [PubMed] [Google Scholar]
- 36.Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. Phylogenies and community ecology. Annu Rev Ecol Syst. 2002;33:475–505. doi: 10.1146/annurev.ecolsys.33.010802.150448. [DOI] [Google Scholar]
- 37.Westoby M, Leishman MR, Lord JM. On misinterpreting the ‘phylogenetic correction’. J Ecol. 1995;83:531–534. doi: 10.2307/2261605. [DOI] [Google Scholar]
- 38.Westoby M. Phylogenetic ecology at world scale, a new fusion between ecology and evolution. Ecology. 2006;87:163–165. doi: 10.1890/0012-9658(2006)87[163:PEAWSA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 39.Volkov I, Banavar JR, He F, Hubbell SP, Maritan A. Density dependence explains tree species abundance and diversity in tropical forests. Nature. 2005;438:658–661. doi: 10.1038/nature04030. [DOI] [PubMed] [Google Scholar]
- 40.Volkov I, Banavar JR, Hubbell SP, Maritan A. Neutral theory and relative species abundance in ecology. Nature. 2003;424:1035–1037. doi: 10.1038/nature01883. [DOI] [PubMed] [Google Scholar]
- 41.Yutin N, Suzuki MT, Teeling H, Weber M, Venter JC, Rusch DB, Béjà O. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific oceans using the global ocean sampling expedition metagenomes. Environ Microbiol. 2007;9:1464–1475. doi: 10.1111/j.1462-2920.2007.01265.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 22 kb)