

Abstract
The repeat-associated mysterious proteins (RAMPs) comprise the most abundant family of proteins involved in prokaryotic immunity against invading genetic elements conferred by the clustered regularly interspaced short palindromic repeat (CRISPR) system. Cas6 is one of the first characterized RAMP proteins and is a key enzyme required for CRISPR RNA maturation. Despite a strong structural homology with other RAMP proteins that bind hairpin RNA, Cas6 distinctly recognizes single-stranded RNA. Previous structural and biochemical studies show that Cas6 captures the 5′ end while cleaving the 3′ end of the CRISPR RNA. Here, we describe three structures and complementary biochemical analysis of a noncatalytic Cas6 homolog from Pyrococcus horikoshii bound to CRISPR repeat RNA of different sequences. Our study confirms the specificity of the Cas6 protein for single-stranded RNA and further reveals the importance of the bases at Positions 5–7 in Cas6–RNA interactions. Substitutions of these bases result in structural changes in the protein–RNA complex including its oligomerization state.
Keywords: CRISPR-Cas, RAMP proteins, protein–RNA interactions, RRM fold, RNA-mediated prokaryotic immunity
Introduction
Microbes use extraordinary strategies to combat viral predation and withstand foreign nucleic acids. The clustered regularly interspaced short palindromic repeats (CRISPRs) are found in many bacteria and archaea genomes and are recently demonstrated to confer small RNA-based adaptive immunity against invasive genetic elements.1–8 CRISPR loci typically consist of several to hundreds of short (∼30–40-nucleotide) repeats separated by short (∼30–40-nucleotide) variable sequences (spacers) and are associated with one or more heterogenous families of cas genes that encode Cas proteins.9, 10 The spacer sequences are short DNA segments derived from past infection and they establish the specificity for obstruction of future invasion.1, 2, 11–14 In the CRISPR-facilitated immunity pathway, the repeats and spacers are transcribed into pre-CRISPR RNA (crRNA) that, after being extensively processed and assembled with Cas proteins, participate in the defense process. The molecular process of new DNA incorporation remains completely unknown while the obstruction phase of the pathway has been shown to depend on crRNA.13–16 CRISPR RNAs are predicted to interact and function with an impressive number of repeat-associated mysterious protein (RAMP) proteins found across different CRISPR subtypes.9, 10, 17 Despite their prevalence in CRISPR functional pathways, how RAMP proteins interact with RNA remains partially understood.
RAMP proteins are classified by six distinctive motifs.9, 17 Motifs I–IV and a specific motif are selectively present in different subfamilies. Motif V that contains a C-terminal glycine-rich (G-rich) sequence is the signature motif of RAMPs and universally present in all RAMP subfamilies. Each CRISPR-Cas system has at least one but may have as many as nine RAMP proteins distributed among different Cas families. In addition, RAMP proteins are also found outside the CRISPR loci with no apparent link to CRISPR functions.9 The number of RAMP proteins is strongly correlated to the number of CRISPR units (unique inserts) as well as the variance of insert length, suggesting their potential role in binding crRNA and related RNA.9
The best characterized RAMP protein known so far is the endonuclease required for crRNA biogenesis. Newly transcribed precursor crRNAs (pre-crRNA) are processed through an endoribonucleolytic, and in some cases, additional exo-ribonucleolytic cleavage events leading to mature crRNAs (previously also known as prokaryotic silencing RNA or psiRNA). Cas6 in Pyrococcus furiosus (Pf), Cse3 (also called CasE) in Escherichia coli and Csy4 in Pseudomonas aeruginosa13, 16, 18 are responsible for producing cleaved intermediate of their respective crRNA. The crRNA intermediate cleaved by Cas6 is further trimmed by uncharacterized 3′ exonucleases. Despite a low level of amino acid sequence homology (∼28% between Cas6 and Cse3 and ∼10% between Cas6 and Csy4), these three endonucleases resemble each other in both RNA cleavage activity and structure.13, 16, 18, 19 Each processing endonuclease recognizes a unique CRISPR repeat RNA but similarly cleaves eight nucleotides upstream of the invader-targeting sequence (spacer).13, 16, 18 Despite the similar size, CRISPR repeat RNA vary both in sequence and secondary structures.20 The crRNAs processed by Cas6 are nonstructured while those by Csy4 clearly contain an internal hairpin fold.20 The recently determined co-crystal structures of Cas6, Csy4, and Cse3 indeed revealed two different modes of RNA interactions.18, 21–23 Cas6 specifically binds to the 5′ terminus of its single-stranded substrates21 while Csy4 and Cse3 bind to a stable hairpin immediately preceding the cleavage site.18, 22, 23 These results suggest that CRISPR processing machineries must recognize structurally diverse pre-crRNA and likely coevolves with their CRISPR RNA substrates. The molecular basis for specific interaction between the processing endonucleases and their respective substrates can help us understand the remarkable diversity among RAMP proteins. Of particular interest is how Cas6 family of RAMP proteins confer specificity for nonstructured RNA substrates.
Other known RAMP proteins function differently than the processing endonuclease. The crRNA-associated ribonucleoprotein particle, called Cmr complex, recently isolated from P. furiosus was demonstrated to have crRNA-guided RNA cleavage activity.15 The Cmr complex is comprised of six proteins (Cmr1–Cmr6), four of which (Cmr1, Cmr3, Cmr4, and Cmr6) are RAMP proteins. These RAMP proteins have been shown to be essential to RNA silencing activity. Although no specific functional roles can be assigned to Cmr RAMP proteins, they likely participate in binding or cleaving RNA.
Previously, we reported crystal structures of P. furiosus (Pf) Cas6 and its complex with a bound Pf repeat RNA (Pf7 RNA) carrying a 2′-deoxy modification.21 Due to relatively low resolution of the complex and the fact that the cleavage site of the RNA is not bound to Cas6 in the crystal, we searched for other Cas6 homologs that can bind but do not cleave Pf7 RNA. We hypothesized that non catalytic Cas6 homologs would bind RNA similarly as Cas6 but do not require any modification in substrate in order to prevent substrate cleavage during crystallization. Here, we report crystallographic and biochemical studies of a Pyrococcus horikoshii (Ph) Cas6 protein bound with unmodified Pf7 RNA and its variants. P. horikoshii (Ph) genome harbors six CRISPR loci and a set of cas genes that encode the Apern subtype of Cas proteins. The PhCRISPR loci are similar both in number and structure to those of previously studied P. furiosus (Pf). There are three predicted Cas6 proteins in P. horikoshii (Ph1252, Ph0350, and Ph0161) among which Ph1252 has the strongest homology to PfCas6 (65% sequence identity). Ph0350 is homologous to another P. furiosus Cas6 protein, Pf0393 (76% sequence identity), while Ph0161 shows significantly weaker homology to any of the PfCas6 proteins. Both Ph0350 and Pf0393 lack the catalytic residues and are thus believed to be noncatalytic homologs of Cas6. In addition, PhCRISPR repeats share a strong homology to those of Pf. Specifically, the first eight nucleotides of repeats in both genomes are nearly identical except for Position 4 (CRISPRdb, http://crispr.u-psud.fr/crispr/). However, Position 4 is known not to dictate processing by PfCas6,16 suggesting a strong similarity in substrates between Ph and Pf Cas6 proteins. The similarity in both Cas6 proteins and their substrates suggests a closely related processing mechanism between the two organisms.
Our structural studies confirmed the overall structural homology between PfCas6–RNA and PhCas6–RNA complexes. However, we observed structural differences between PfCas6–RNA and PhCas6–RNA complexes that reflect the ability of Cas6 proteins in modulating specificity by varying surface amino acids of the evolutionarily conserved ferredoxin fold. Our study further revealed an unexpected Cas6–RNA interactions that facilitate dimerization of the protein–RNA complex in an RNA sequence-dependent manner.
Results
Identification and characterization of a noncatalytic Cas6
Some archaeal and bacterial genomes encode one or more Cas6 proteins that lack the hypothesized catalytic triad.16 These Cas6 proteins are predicted to bind CRISPR RNA without any nuclease activity. We refer to these proteins as Cas6nc (Cas6 noncatalytic homolog). We expressed and purified both Cas6 proteins (PH1252 and PH0350) from P. horikoshii. The two PhCas6 proteins share 31% sequence identity. We tested cleavage and binding activities of the two PhCas protein on a previously characterized repeat RNA. Pf7 RNA is a Pf repeat RNA and a substrate for PfCas6 both in vitro and in vivo.16 PH1252, which contains the predicted catalytic triad and shares 67% sequence identity with the crRNA processing endoribonuclease PfCas6,16 cleaved Pf7 RNA similarly to PfCas6 [Fig. 1(B)]. PH1252 is then designated as PhCas6. PH0350, on the other hand, lacks the predicted catalytic triad and did not cleave Pf7 RNA [Fig. 1(B)]. It is thus designed PhCas6nc. The fact that a stoichiometric amount of PhCas6nc inhibited RNA cleavage by either PfCas6 or PhCas6 [Fig. 1(B)] indicates that PhCas6nc has the ability to limit the access of the substrate RNA to PhCas6 of PfCas6.
Figure 1.
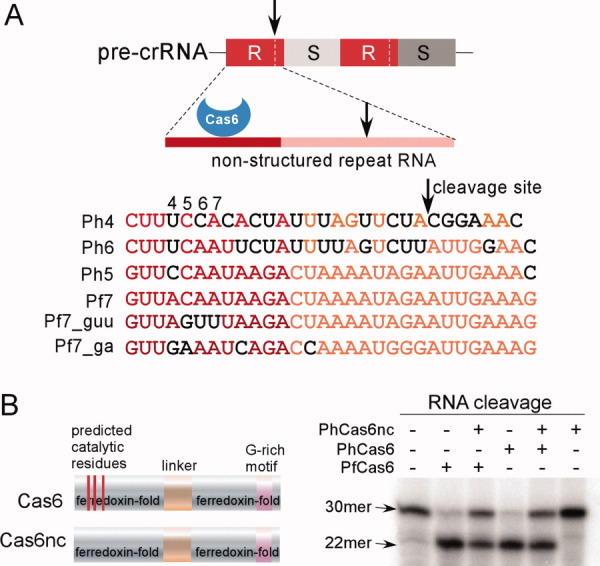
Schematics of the precursor CRISPR RNA (crRNA) and known RAMP proteins. (A) Upper panel shows features of the CRISPR RNA recognized by Cas6 that processes crRNA. “R” denotes repeat and “S” denotes spacer RNA. The darker red line depicts the region recognized by Cas6 and arrows indicate the cleavage site. Lower panel shows the names and sequences of the repeat RNA used in this study. “Ph” refers to P. horikoshii and “Pf” refers to P. furiosus. Black letters designate nucleotides that differ from those of Pf7 RNA. Note the close similarity between Ph5 and Pf7 RNA. (B) Schematic sequence features of the two RAMP proteins, Cas6 and Cas6nc (noncatalytic Cas6), and their cleavage activity on Pf7 RNA. The three red stripes on the schematic sequence indicate the three predicted catalytic residues of Cas6 that are lacking in Cas6nc. The substrate (30mer) and the product (22mer) are indicated by arrows. For each cleavage reaction, the enzyme concentration was 100 nM.
To compare RNA binding properties of PhCas6nc with PfCas6, we studied co-crystal structures of PhCas6nc bound to the previously characterized Pf7 RNA and two of its variants without modification [Fig. 1(A)]. Pf7_guu RNA is a variant of Pf7 RNA containing mutations of CAA to GUU at Positions 5–7. Pf7_ga RNA is a second variant containing mutations of AC to GA mutation at Positions 4–5 and A to C mutation at Position 9 [Fig. 1(A)]. While Pf7 RNA is a substrate for PhCas6, Pf7_guu is not cleaved and Pf7_ga is inefficiently cleaved by PhCas6 (Supporting Information Fig. S1), suggesting a detrimental effect of mutations at Positions 5–7 on processing. PhCas6nc is able to bind with all three RNA, which allowed us to obtain co-crystal structures of PhCas6nc bound with the three RNA, respectively.
All three structures were determined by the molecular replacement method using the known PfCas6 structure as the search probe (PDBid: 3I4H). The electron density map computed using the experimental amplitudes and phases from refined protein coordinates allowed unambiguous tracing of the bound RNA (Supporting Information Fig. S2). Detailed structure determination procedures are included in the “Materials and Methods” section. The overall structures of the three complexes are shown in Figure 2. In all three cases, PhCas6nc residues 1–239 (wild-type length 239) are included in the refined models. Nucleotides 1–12 of the 30 nucleotide repeat RNAs are included in the Pf7 and Pf7_ga complexes while nucleotides 3–10 are included in the Pf7_guu complex. The fact that the structures lack some of the repeat RNA nucleotides indicates a disorder in these regions, particularly in the 3′ end of the RNA, under our crystallization conditions. The PhCas6nc–RNA co-crystal structure is only the second Cas6–RNA co-crystal structure available and is at a higher resolution than the previously determined PfCas6–RNA crystal structure.21
Figure 2.
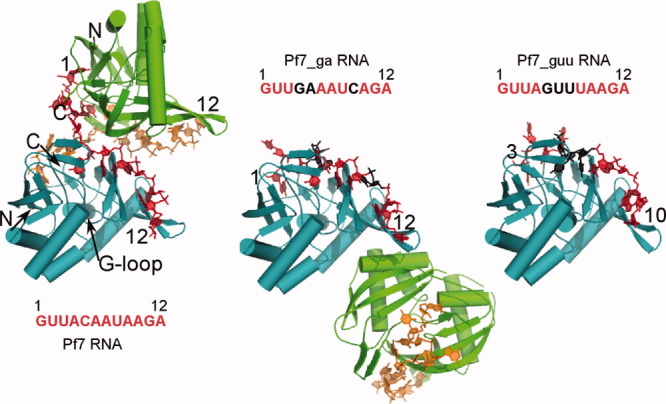
Overview of structures of PhCas6nc bound to three repeat RNAs. In the Pf7 and Pf7_ga complexes, PhCas6nc subunits are colored in teal and green and repeat RNA are colored in red and orange, respectively. Nucleotides that are substituted from Pf7 RNA are highlighted in black. The subunits colored in teal of the three complexes are superimposed and are displayed in the same orientation. The NH2-terminus of the PhCas6nc is labeled as “N” and the COOH-terminus is labeled as “C” on the Pf7 RNA complex. The first and the last nucleotides that are modeled are indicated by numbers.
A cross-subunit protein–RNA dimer
As predicted from protein sequence alignment, PhCas6nc has a nearly identical structure as PfCas6 (PDBcode: 3I4H) [Fig. 3(A)]. The only difference between the two structures is observed in α2 that harbors the putative catalytic residue [Fig. 3(A)]. The α2-helix of PhCas6nc is rotated downward slightly with respect to that of PfCas6 [Fig. 3(A)]. This difference may reflect the different catalytic activities between the two proteins. Despite the similarity in protein structures, the oligomerization state of the PhCas6nc complex in the crystal differs from that of the crystallized PfCas6–Pf7 RNA complex.21 The PhCas6nc–Pf7 RNA complex forms a strong dimer mediated by the repeat RNA (Fig. 2). Each repeat RNA binds across two PhCas6nc subunits and vice versa. The network of RNA–RNA and RNA–protein interactions result in an extensive interface with a total buried solvent accessible area of 2455 Å.2 The central cleft of each PhCas6nc subunit is bound by Nucleotides 1–5 of one RNA and Nucleotides 6–12 of the other RNA molecule (Fig. 2).
Figure 3.
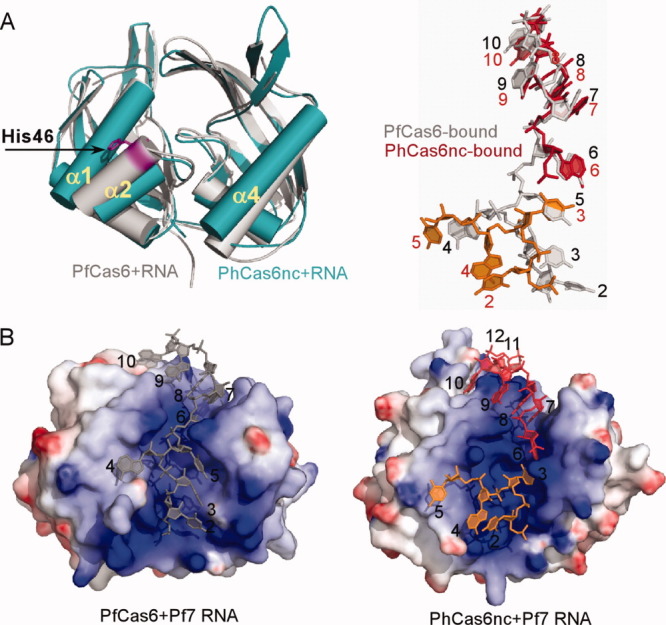
Structural comparison between the PfCas6–RNA (PDBid: 3PKM) and PhCas6nc–RNA complexes. (A) Superimposed protein structures (PfCas6 in gray and PhCas6nc in teal) and comparison of their bound RNA (PfCas6-bound in gray and PhCas6nc-bound in red and orange). (B) Comparison of the PfCas6-RNA (left) and PhCas6nc-RNA (right) complexes in surface representation. Electrostatic potentials were mapped to protein surfaces and are drawn from −10 kT/e (red) to 10 kT/e (blue). The two bound RNA to the same PhCas6nc subunit are distinguished by red and orange.
The primary difference in protein–RNA interactions between Pf7 RNA–PhCas6nc and Pf7 RNA–PfCas6 complexes is the way in which the 5′ region of the Pf7 RNA interacts with the proteins (Fig. 3). The conformations of the bound RNA are compared in Figure 3(A) where both proteins are superimposed. The root mean square deviation for Nucleotides 1–5 between the two structures is 7.5 Å (total aligned atoms: 46). In the PhCas6nc complex, Nucleotides 1–5 of the bound Pf7 RNA splay in different directions on the surface of the protein without establishing extensive interactions with the protein [Fig. 3(B)]. The phosphate backbone within this segment makes two sharp bends (at U2 and U3, respectively) and only nucleotide U3 forms specific interaction with the protein [Fig. 4(A) and Supporting Information Fig. S3]. U2 has no contact with the protein and is stabilized by base stacking interactions with A4 that in turn stacks with the C-terminus of the protein [Fig. 4(A)]. C5 initiates the interaction of the RNA with the other subunit and interacts with residues from both subunits [Fig. 4(A) and Supporting Information Fig. S3]. Nucleotide A6 from the other repeat RNA is placed in the narrowest part of the binding cleft and forms a Reverse Hoogsteen pair with U3 [Fig. 4(A) and Supporting Information Fig. S3]. In addition, the well-conserved residue Arg60 stabilizes the sugar phosphate backbone of A6. Therefore, U3 and A6 (of the other bound RNA) together form an impressive network of interactions with the protein and with each other via both base and backbone functional groups [Fig. 4(A) and Supporting Information Fig. S3].
Figure 4.
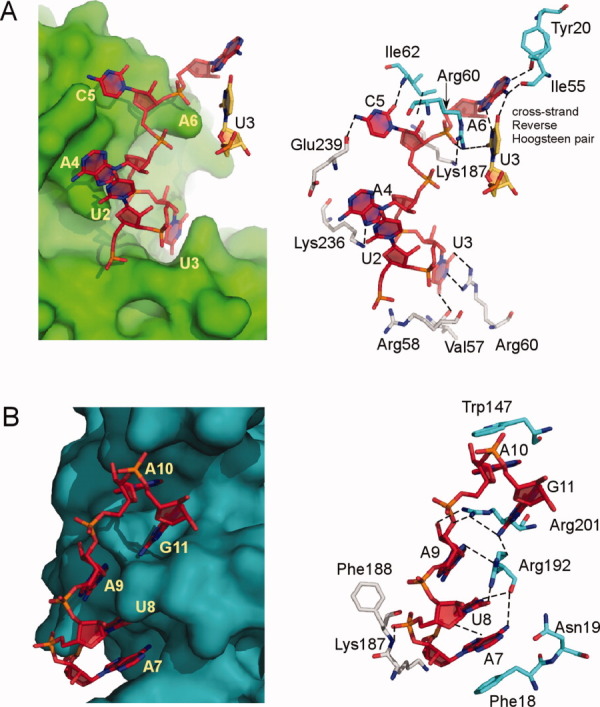
Key interaction features of the PhCas6nc–Pf7 RNA complex. The two subunits of PhCas6nc are represented by surface (left) or stick models (right) and the RNA are shown by red (or orange) stick models. The same repeat RNA interacts with two different PhCas6nc subunits (A and B, respectively) that are distinguished by teal surface (or grey–blue–red stick models) and green surface (or cyan–blue–red stick models), respectively. Contacts between RNA and protein atoms of shorter than 3.4 Å are indicated by dashed lines. The reverse Hoogsteen pair between A6 of one RNA and U3 of the other RNA (orange) is also depicted.
The interactions between Nucleotides 6–10 and PhCas6nc are similar to what were observed between the same region of the RNA and PfCas6 despite the difference in amino acids between the two proteins. Nucleotides 7–9 form continuous base stacking (Base 9 contributes to partial stacking) that is interrupted by the insertion of Arg201 between Nucleotide A9 and A10 [Fig. 4(B) and Supporting Information Fig. S3]. A10 is rotated away from the stacking and its nucleobase is sandwiched by protein residues Arg201 and Trp147 [Fig. 4(B) and Supporting Information Fig. S3]. The phosphate backbone of Nucleotides 11 and 12 makes two sharp kinks that place the three nucleobases (10, 11, and 12) close together in space for their mutual stabilization. The location of Nucleotide 12 allows prediction of the binding path of the disordered region (Nucleotides 13–28) in our structure (Fig. 2). This predicted path is near the highly conserved G-rich loop (Fig. 2), thereby suggesting a possible role of the G-rich loop in interacting with CRISPR RNAs. Consistently, we have shown previously that mutations in the G-rich loop of PfCas6 abolished its RNA cleavage activity.21
Dimerization of the protein–RNA complex provides a means to stabilize the complex via cross-subunit interactions. It is evident from Figure 4 and Supporting Information Figure S3 that Nucleotides 5–7 interact with residues from both subunits. For instance, the extensive interaction between the nucleobase of C5 and one subunit is further enhanced by the electrostatic interaction between its phosphate backbone and the other subunit (Fig. 4 and Supporting Information Fig. S3). Similar principles of interactions also apply to Nucleotides U8 (Fig. 4 and Supporting Information Fig. S3). This mode of RNA binding significantly enhances RNA binding to PhCas6nc.
Structural evidence for RNA sequence-dependent oligomerization
The structures of PhCas6nc bound with the two variants of the Pf7 RNA, Pf7_guu and Pf7_ga, reveal the protein's plasticity in binding different repeat RNAs and suggest a critical role of RNA sequences in oligomerization. The complex between PhCas6nc and Pf7_guu or Pf_ga no longer forms the cross-subunit dimer. Rather, both complexes form a 1:1 stoichiometric protein–RNA complex [Figs. 2 and 5]. The Pf7_guu complex crystal contains a single protein–RNA complex in the asymmetric unit (Fig. 2). Although the Pf7_ga complex contains two molecules in the asymmetric unit, the interface is relatively small (∼600 Å2) and made exclusively of proteins, unlike the Pf7 complex whose interface is made of RNA (Fig. 2).
Figure 5.

Comparison of the RNA–protein interaction features of the three complexes. Upper panel, comparison of the bound RNA. For each compared pairs, the two protein molecules are superimposed and RNA molecules follow. The RNA molecules are distinguished by grey and red colors and are identified by the labels of the corresponding colors. Lower panel, comparison of protein residues (cyan–blue–red stick models) that interact with the three RNA (grey or red). Each RNA is identified by the sequence shown directly below and the nucleotides different from Pf7 RNA are identified by underlined numbers.
Since the two variant complexes share a common change in nucleotide at Position 5: both Pf7_guu and Pf_ga RNA contain a purine while Pf7 RNA contains a cytosine at Position 5, this substitution may be critical for the observed structural changes in oligomerization. This hypothesis is supported by the observed interactions of Nucleotide 5. In the Pf7 complex, C5 is splayed out and thus able to form stable interactions with both PhCas6nc subunits [Fig. 6(A)]. On the other hand, A5 of Pf7_ga or G5 of Pf7_guu is placed in a deep pocket of a single subunit of PhCas6nc [Fig. 6(A)]. The fact that both adenine and guanine fit into the same binding pocket suggests that it is the size rather than the functional groups of the nucleotide that is important for binding. The Nucleotide 5 binding pocket (for purine) comprises a combination of hydrophobic and positively charged residues. The backbone atoms of residues 204–206 line the bottom of the pocket while positively charged residues Arg54, Lys187, and Lys189 surround the pocket [Fig. 6(A)]. The O6 of G5 and N6 of A5 atoms contact the same set of backbone atoms of Residues 204–206. The N2 atom of G5 makes additional contacts with the side chains of Lys187 and Lys189. As a result, the two lysine residues in the Pf7_ga structure are less tightly packed against the purine nucleobase than those in the Pf7_guu structure [Fig. 6(A)]. Thus, PhCas6nc is able to fine-tune this purine binding pocket. Consistent with the hypothesis that C5 is crucial to the formation of the cross-subunit dimer, we mutated C5 of Pf7 RNA to adenine (Pf7_C5A) and found that the C5 change alone reduced dimer formation mediated by Pf7 RNA [Fig. 6(B)].
Figure 6.
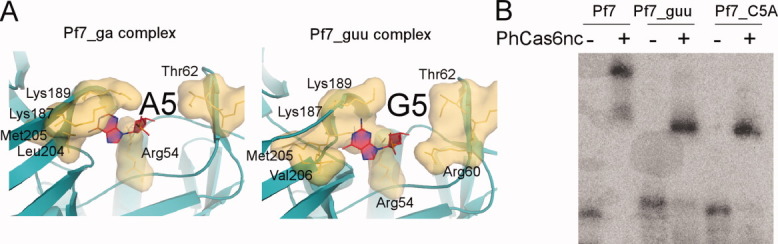
The binding pocket for Nucleotide 5 and mutational result. (A) Close-up view of Nucleotide 5 binding pocket. Residues that interact with Nucleotide 5 are in surface representation and are indicated by labels. (B) Gel mobility shift result of PhCas6nc (1 μM) binding to Pf7, Pf7_guu, and Pf7_C5A RNAs. The upper shifted band is interpreted as the protein–RNA dimer and the lower shifted band is the protein–RNA monomer.
The Pf7_guu structure also differs from the other two structures in that its RNA Nucleotides 6 and 7 are uridine instead of adenosine. The two uridine nucleotides are able to maintain the same conformation as the two adenine nucleotides in the other two structures (Fig. 2). As a result, the structures in this region do not deviate one from another significantly (Fig. 5). N6 of adenine is at a nearly identical position as O4 of uridine. In all cases, regardless of base identity, nucleobase-amino acid stacking interactions are observed at these positions.
Highly related species have different RNA specificities
Despite that P. furiosus and P. horikoshii are closely related species, PhCas6nc binds to Pf7 RNA with much higher affinity than PfCas6 (Supporting Information Fig. S4). In addition, substitution of CAA in the Pf7 RNA to GUU at Positions 5–7 is not tolerated by PfCas6 because the enzyme failed to bind to and cleave the Pf7_guu RNA21 while PhCas6nc is able to bind the same RNA. These data suggest that PhCas6nc has a different protein surface between its ferredoxin domains than that of PfCas6. Comparison of PfCas6 and PhCas6nc structures indicates that Nucleotides 1–5 of Pf7 RNA are bound quite differently in the two complexes [Fig. 3(A)]. Nucleotides 1–5 bound to PfCas6 are shifted toward in the 5′ direction with respect to those bound to PhCas6nc. Residues interacting with these nucleotides are not conserved between PhCas6nc and PfCas6 (Supporting Information Fig. S5). For instance, Lys187, Arg54, and Leu204 of PhCas6nc are substituted by Ile192, Leu64, and His210 of PfCas6, respectively. These differences likely account for the fact that PfCas6 did not tolerate mutations of Nucleotides 5–7. This comparison suggests that RAMP proteins can achieve sequence specificity by varying surface amino acids.
Biochemical evidence for RNA sequence-dependent oligomerization
To substantiate structural observations, we further mutagenized Pf7 and carried out in vitro binding assays. Pf7 RNA forms two slowly migrating species on a native polyacrylamide gel upon mixing with PhCas6nc. We interpret the slower species as the strong dimer observed in the crystal (Figs. 6 and 7). Consistently mutation of 5–7 from UAA to GUU (Pf7_guu RNA) greatly reduced formation of the slower but not the faster migrating species (Fig. 7). Mutations or deletions of the 5′ end of the repeat RNA seemed to have little effect on PhCas6nc binding. Specifically, removal of Nucleotides 1, 1–2, 1–3 did not disrupt binding by PhCas6nc (Fig. 7). Mutation of U2U3 to A2A3 also did not affect PhCas6nc binding, suggesting a minor role played by the cross-strand reverse Hoogsteen pairing between U3 and A6 in dimerization. These results confirm the importance of the central region of Pf7 in its interaction with PhCas6nc and, furthermore, reveal a weak interaction between PhCas6nc and the 5′ end of the repeat RNA.
Figure 7.
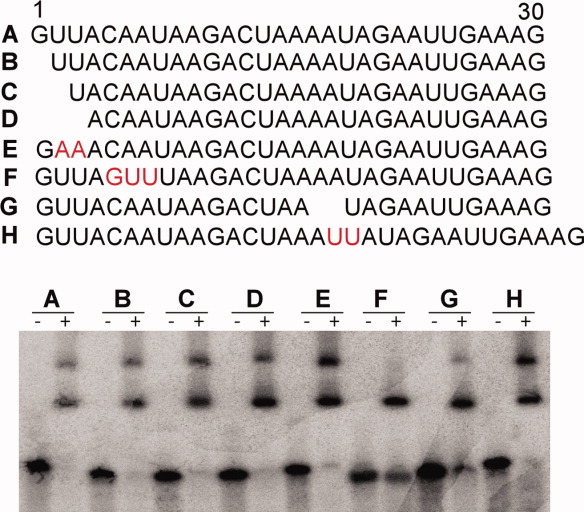
RNA mutagenesis and binding assay results. Upper panel, sequences of the wild-type and mutant repeat RNA constructs used in RNA-binding studies and their results are shown below. Mutated or modified sequences are highlighted red with exception of deletions (spaces). Lower panel, gel mobility shift assay results of the mutant RNA by PhCas6nc. The concentration of PhCas6nc was 1.0 μM in all cases. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
We measured the fraction of bound for all three RNA as a function of PhCas6nc concentrations. The binding profiles all show sigmoid features that cannot be accounted for by the simple single-site binding model, suggesting certain degrees of cooperativity in binding [Fig. 8(A)]. Since we were unable to separate the two slowly migrating protein–RNA species in obtaining the fraction of RNA bound, it is not possible to model the binding reactions quantitatively. However, it is clear that the sigmoid feature is the most pronounced for the Pf7 RNA. This result is consistent with the structural data that Pf7 RNA mediates cross-subunit of the PhCas6nc-RNA complex. The presence of sigmoid features in both Pf7_ga and Pf7_guu RNA binding is also consistent with the tendency of formation of protein:RNA dimers at high concentrations [Fig. 8(A)].
Figure 8.

Binding analyses of the three RNA to PhCas6nc. (A) Native gel mobility shift assay of Pf7, Pf7_ga, and Pf7_guu RNA binding to PhCas6nc. Left, plots of fraction of RNA bound versus protein concentrations. Solid lines are traces that follow experimental data points. Error bars were computed from averaging of at least two duplicated measurements. Right, representative radiogram images for each RNA–protein complex. (B) Analytic size-exclusion chromatography results of Pf7, Pf7_ga, and Pf7_guu RNA binding to PhCas6nc. For each elution profile, peaks of RNA or protein in isolation (thin dash lines) are included for comparison. The peak position for isolated PhCas6nc is consistent with it being a monomer. Both the monomer and dimer peaks of the RNA–protein complexes were verified to contain both RNA and protein (Supporting Information Fig. S6).
To independently compare oligomerization of the three complexes, we performed analytic size-exclusion chromatography. As shown in Figure 8(B), PhCas6nc alone clearly migrates as a monomer while its complexes with Pf7 RNA and Pf7_ga RNA exist at an equilibrium of dimer and monomer and that with Pf7_guu is primarily monomeric [Fig. 8(B) and Supporting Information Fig. S6]. The apparent molecular weights of PhCas6nc–Pf7 complexes are 41.6 kDa (theoretical MW: 38.1 kDa) and 68.8 kDa (theoretical MW: 76.1 kDa) for the monomer and dimer, respectively, those of PhCas6nc-Pf7_ga complex are 41.6 kDa (theoretical MW: 38.1 kDa) and 70.7 kDa (theoretical MW: 76.1 kDa) for the monomer and dimer, respectively, and those of PhCas6nc–Pf7_guu complex are 39.7 kDa (theoretical MW: 38.1 kDa) and 61.6 kDa (theoretical MW: 76.1 kDa) for the monomer and dimer, respectively [Fig. 8(B)]. Thus, Pf7 and Pf7_ga RNA have the ability to induce dimerization of the protein–RNA complex, consistent with the structural and gel mobility shift data. Pf7 RNA is able to form a cross-subunit interaction with PhCas6, thereby bringing two otherwise monomeric PhCas6nc together in space. Although Pf7_ga RNA complex has a small dimerization interface, the additional RNA nucleotides that are disordered in the crystal may further extend this interface to form stable dimers. Variations in the apparent molecular weights of the dimer peaks of the three complexes may reflect the different hydrodynamic shapes of the dimers.
The question arises whether native P. horikoshii repeat RNA have an intrinsic ability to mediate dimerization of the PhCas6nc–RNA complex. We performed both gel mobility shift and analytic size-exclusion chromatography analyses on two P. horikoshii repeat RNAs that are least related in sequence to Pf7 RNA (Fig. 1A). Interestingly, we found that one of these repeats, Ph6 RNA, is able to form a dimer while the other, Ph4 RNA, did not (Supporting Information Fig. S7). We note that Ph6 RNA has an identical set of trinucleotides at Positions 5–7 as Pf7 while Ph4 RNA has a different Nucleotide 6 (cytidine instead of adenosine) despite the same cytidine residue at Position 5 Fig. 1A (Supporting Information Fig. S7). This result again confirms the important role observed for Nucleotide 6 in PhCas6nc–RNA interaction. In addition, both Ph6 and Ph4 RNA have a pyrimidine while Pf7 has a purine nucleotide at Position 4, consistent with the observation that Position 4 is not essential for dimer formation. However, this result emphasizes that it is the combination of Nucleotides 5–7 rather than individual nucleotides that dictate the oligomerization state of the complex.
Discussions
RAMP proteins are a new class of RNA-binding proteins involved in the CRISPR-mediated prokaryotic immunity against invaders. RAMP protein binds CRISPR repeat RNA specifically and may further enhance binding by dimerization. Similar to the RRM proteins that may contain one or more ferredoxin folds, the RAMP protein studied here binds single-stranded RNA with sequence selectivity.24 However, unlike RRM proteins that act as independent RNA-binding motifs,24 RAMP use two ferredoxin folds to bind single-stranded RNA and confer sequence specificity by residues at the central cleft between the ferredoxin folds. Therefore, RAMP proteins comprise a unique class of RNA-binding proteins that are evolved from the well-conserved ferredoxin fold.
In addition to the unique ability in recognizing single-stranded RNA, the RAMP protein studied here, PhCas6nc, exhibits a previously uncharacterized propensity for RNA sequence-dependent oligomerization. Isolated PhCas6nc is a monomer whereas its complex with Pf7 RNA or Pf7_ga RNA has a strong tendency to form a dimer. Interestingly, the two types of dimers are different in their interfaces. Pf7 RNA is able to mediate dimer formation through an exclusive RNA interface. Pf7_ga RNA most likely mediates dimerization by enhancing the existing protein–protein interface. The RNA that has the weakest interaction with PhCas6nc, Pf7_guu, forms predominately monomer, suggesting the importance of protein–RNA interaction to dimerization. Finally, our biochemical studies reveal a difference in oligomerization of PhCas6nc when binding its likely physiological substrates. While Ph6 RNA induces dimer formation, Ph4 RNA clearly forms a monomer. The intrinsic ability for RAMP proteins to form RNA-dependent oligomers may have important implications in regulation of CRISPR function.
The RNA sequence-dependent dimerization observed in PhCas6nc may apply to other Cas6 proteins including the processing endonuclease. This may have a regulatory role in CRISPR RNA processing. Unprocessed CRISPR RNA contain multiple repeats that may interact with many RAMP proteins. The potential for PhCas6nc to bind CRISPR RNA as a dimer may allow it to interact with unprocessed CRISPR RNA cooperatively. The structural and sequence similarity between PhCas6nc and the catalytic Cas6 proteins suggest a potential for the endoribonuclease to interact with unprocessed CRISPR RNA cooperatively for the benefit of efficiency and accuracy.
Materials and Methods
Protein preparation
The DNA encoding P. horikoshii PH1250 (PhCas6) and PH0350 (PhCas6nc) proteins were cloned into vector pET28a (Novagen). The N-terminal histidine-tagged PhCas6 and PhCas6nc proteins were over-expressed in Escherichia coli BL21 (DE3) cells and purified by a similar method as that described for PfCas6.16 Briefly, the transformed BL21 (DE3) cells were induced with 0.2 mM isopropyl-β-D-thiogalactopyranoside (IPTG) when optical density reached 0.6–0.7 (600 nm). The protein-expressing cells continued to grow for 16 h at 25°C before being harvested and disrupted by ultrasonication in a buffer containing 25 mM sodium phosphate (pH7.5), 0.1 mM phenylmethlsulfonyl fluoride, 10% glycerol, 1M NaCl, and 14.2 mM β-mercaptoethanol. The cell lysate was then heated at 70°C for 15 min to denature bacterial proteins and cleared by centrifugation. The supernatant was loaded onto a Ni2+-NTA (Qiagen) affinity column and the elutant was loaded onto the Superdex 200 column (GE Healthcare). Peak fractions of the protein were concentrated to 80–100 mg/ml in a buffer containing 20 mM Tris (pH 7.5), 5% glycerol, 1M NaCl, 5 mM β-mercaptoethanol (βME), and 0.5 mM ethylenediamine tetra acetic acid and stored at −80°C.
Crystallization
All RNA oligos (Fig. 1) were ordered from Integrated DNA Technology (Coralville, IA). The PhCas6nc–RNA complexes were prepared by mixing 1:1.2 molar ratio of protein:RNA before crystallization. Sequences and names of the repeat RNA used are described in Figure 1. The Pf7 and Pf_ga RNA-containing complexes were crystallized in reservoir solutions containing 0.005M MgSO4, 0.05M MES (pH 6.0), 3–8% PEG4000, 0.2M NaCl, and 0.1M KCl. The Pf7_guu RNA-containing complex was crystallized in a reservoir solution containing 0.01M MgSO4, 0.05M Na Cacodylate (pH 6.0), and 1.8M Lithium Sulfate. To cryo-protect the crystals for data collection, crystals were briefly soaked in a solution containing the reservoir solution supplemented with 10% ethylene glycol. The data sets were processed with HKL2000.25 The Pf7 RNA-containing complex crystals are in space group P43212 with unit cell dimensions a = b = 88.8 Å and c = 257.6 Å. The Pf7_ga RNA-containing crystals are also in space group P43212 with unit cell dimensions a = b = 75.3 Å and c = 257.6 Å. The Pf7_guu RNA-containing crystals are in space group P41212 with unit cell dimensions a = b = 76.8 Å and c = 193.1 Å. For the Pf7 RNA crystals, there are two molecules in the asymmetric unit and the Matthews coefficient is 3.2 A3/Da indicating a solvent content of 61.4%. For the Pf7_ga RNA crystals, there are also two molecules in the asymmetric unit and the Matthews coefficient is 2.3 A3/Da indicating a solvent content of 51.8%. For the Pf_guu RNA crystals, there is one molecule in the asymmetric unit and the Matthews coefficient is 3.6 A3/Da indicating a solvent content of 65.4%.
Structure determination
The structures of PhCas6nc–RNA complexes were solved by molecular replacement methods using the approach implemented in PHENIX.26 The previously determined crystal structure of PfCas6 protein (PDBid: 3I4H) was used as the search model. Iterative model building was performed to model the structure of Phcas6nc using the autobuild function of PHENIX26 and the graphic programs COOT27 and O.28 RNA nucleotides were built in COOT27 and the final model was refined using PHENIX.26 For both Pf7 and Pf7_ga complexes, non-crystallographic symmetry restraint between the two protomers was enforced throughout the refinement. The Pf7_guu complex has a relatively high Wilson thermal factor as a result of large static disorder, which resulted in high average B-factors (Table I). For Pf7_ga and Pf7_guu complexes, no ordered solvent molecules were modeled due to their modest resolution. For refinement of all three complexes, bulk solvent correction, anisotropic scaling, and individual isotropic atomic displacement refinement were used. The final refinement parameters for the three structures are shown in Table I or are included in the deposited protein data bank files.
Table I.
Data Collection and Refinement Statistics (Values in Parenthesis Refer to Those of the Highest Resolution Shell)
| PhCas6nc-Pf7 (PDBid: 3QJL) | PhCas6nc-Pf7_ga (PDBid:3QJJ) | PhCas6nc-Pf7_guu (PDBid:3QJP) | |
|---|---|---|---|
| Diffraction data | |||
| Space group | P43212 | P43212 | P41212 |
| Unit-cell parameters (Å) | |||
| a | 88.77 | 75.27 | 76.78 |
| b | 88.77 | 75.27 | 76.78 |
| c | 257.63 | 257.35 | 193.08 |
| Resolution range (Å) | 39.7–2.7 (2.8–2.7) | 37.3–2.8 (2.9–2.8) | 30.3–3.3 (3.5–3.3) |
| No. of unique reflections | 28540 (1098) | 19069 (904) | 9304 (440) |
| Redundancy | 24.6 (12.6) | 20.5 (15.4) | 15.0 (11.3) |
| Completeness (%) | 97.3 (77.3) | 99.9 (100) | 99.6 (97.1) |
| I/σ (I) | 33.8 (2.2) | 62.3 (7.8) | 39.2 (2.1) |
| Rsym(%) | 11.2 (48.1) | 11.8 (55.2) | 8.1 (52.5) |
| Refinement statistics | |||
| Resolution range (Å) | 50.0–2.7 (2.8–2.7) | 50.0–2.8 (2.9–2.8) | 50.0–3.3 (3.5–3.3) |
| Rwork(%) | 23.1 (38.7) | 19.4 (33.1) | 23.1 (33.1) |
| Rfree(%) | 27.7 (47.9) | 24.9 (44.0) | 27.7 (39.7) |
| Model information | |||
| No. of protein-RNA complexes | 2 | 2 | 1 |
| No. of amino-acid/nucleotide | 480/24 | 478/24 | 239/10 |
| No. of protein/RNA atoms | 3892/508 | 3890/512 | 1945/168 |
| No. of water molecules | 39 | 0 | 0 |
| R.m.s. deviation of the model | |||
| Bond length (Å) | 0.003 | 0.007 | 0.007 |
| Bond angle (°) | 0.7 | 1.1 | 1.1 |
| Average B-factors of protein/RNA (Å2) | 59.2/63.4 | 60.3/72.7 | 103/134 |
| Average B-factor of water (Å2) | 48.6 | NA | NA |
| Ramachandran plot | |||
| Residues in favored region | 467 (97.3%) | 470 (97.9%) | 228 (95.4%) |
| Residues in additionally allowed region | 13 (2.7%) | 10 (2.1%) | 11(4.6%) |
| Residues in disallowed region | 0 (0%) | 0(0%) | 0(0%) |
RNA-binding and cleavage assays
RNA-binding and cleavage reactions were carried out by addition of PhCas6nc or PfCas6 at concentrations of 0.1–3 μM to a 20 μL solution containing trace amount of 5′ end 32P-radiolabeled RNA, 0.02M HEPES–KOH (pH 7.0), 0.25M KCl, 0.075M DTT, 0.0015M MgCl2, 5 μg of E. coli tRNA, and 10% glycerol at 70°C. For cleavage assays, the reaction mixture was loaded on a denaturing 15% polyacrylamide gel. For binding assays, the reaction mixture was directly loaded on a non-denaturing 10% polyacrylamide gel to assess RNA binding. 5′-radiolabeled RNAs were visualized by phosphorimaging.
Analytic size-exclusion chromatography analysis
50 μl protein, RNA, and protein–RNA complexes samples were prepared at equal concentrations in buffer A [20 mM Tris pH 7.5, 300 mM NaCl, 5% glycerol (v/v)]. Each sample was loaded onto a Superdex 200 PC 3.2/30 column (GE HealthCare) equilibrated with buffer A at a running rate of 0.05 ml/min. Peak fractions detected by 280 nm absorption were analyzed by silver-stained SDS-PAGE gels (Supporting Information Fig. S6). The apparent molecular weight of each peak was estimated based on the elution volumes of four standard molecular weight proteins under the identical experimental condition and on the assumption that molecular weights extrapolate linearly throughout the elution volume (Supporting Information Fig. S6).
Accession Numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 3QJL (PhCas6nc-Pf7RNA), 3QJJ (PhCas6nc-Pf7_ga), and 3QJP (PhCas6nc_guu).
Acknowledgments
X-ray diffraction data were collected from the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions for APS beamlines may be found at http://necat.chem.cornell.edu/ and www.ser-cat.org/members.html.
Glossary
Abbreviations:
- CRISPR
clustered regularly interspaced short palindromic repeats
- RAMP
repeat-associated mysterious proteins.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 2.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 3.Sorek R, Kunin V, Hugenholtz P. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol. 2008;6:181–186. doi: 10.1038/nrmicro1793. [DOI] [PubMed] [Google Scholar]
- 4.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–407. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 8.Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 12.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 13.Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hale CR, Zhao P, Olson S, Duff MO, Graveley BR, Wells L, Terns RM, Terns MP. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carte J, Wang R, Li H, Terns RM, Terns MP. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makarova KS, Aravind L, Grishin NV, Rogozin IB, Koonin EV. A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis. Nucleic Acids Res. 2002;30:482–496. doi: 10.1093/nar/30.2.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebihara A, Yao M, Masui R, Tanaka I, Yokoyama S, Kuramitsu S. Crystal structure of hypothetical protein TTHB192 from Thermus thermophilus HB8 reveals a new protein family with an RNA recognition motif-like domain. Protein Sci. 2006;15:1494–1499. doi: 10.1110/ps.062131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunin V, Sorek R, Hugenholtz P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007;8:R61. doi: 10.1186/gb-2007-8-4-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang R, Preamplume G, Terns MP, Terns RM, Li H. Interaction of the Cas6 Riboendonuclease with CRISPR RNAs: Recognition and Cleavage. Structure. 2011;19:257–264. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gesner EM, Schellenberg MJ, Garside EL, George MM, Macmillan AM. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat Struct Mol Biol. 2011;18:688–692. doi: 10.1038/nsmb.2042. [DOI] [PubMed] [Google Scholar]
- 23.Sashital DG, Jinek M, Doudna JA. An RNA-induced conformational change required for CRISPR RNA cleavage by the endoribonuclease Cse3. Nat Struct Mol Biol. 2011;18:680–687. doi: 10.1038/nsmb.2043. [DOI] [PubMed] [Google Scholar]
- 24.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. San Diego: Academic Press; 1997. [DOI] [PubMed] [Google Scholar]
- 26.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.