

Abstract
Acidic fibroblast growth factor-1 (FGF-1) is an angiogenic protein which requires binding to a polyanion such as heparin for its mitogenic activity and physicochemical stability. To evaluate the extent to which this heparin dependence on solution stability could be reduced or eliminated, the structural integrity and conformational stability of 10 selected FGF-1 mutants were examined as a function of solution pH and temperature by a series of spectroscopic methods including circular dichroism, intrinsic and extrinsic fluorescence spectroscopy and static light scattering. The biophysical data were summarized in the form of colored empirical phase diagrams (EPDs). FGF-1 mutants were identified with stability profiles in the absence of heparin comparable to that of wild-type FGF-1 in the presence of heparin while still retaining their biological activity. In addition, a revised version of the EPD methodology was found to provide an information rich, high throughput approach to compare the effects of mutations on the overall conformational stability of proteins in terms of their response to environmental stresses such as pH and temperature.
Keywords: FGF-1, stability, heparin, EPDs, structure, conformation, circular dichroism, fluorescence, light scattering
Introduction
Acidic fibroblast growth factor-1 (FGF-1) is a potent angiogenic factor being investigated as a pro-angiogenic biopharmaceutical drug candidate for treatment of ischemic disease, including wound healing in diabetic ulcers, peripheral artery disease, and nontreatable (i.e. “no option”) coronary occlusions.1–4 Significant hurdles, however, remain in the successful realization of FGF-1 as a biopharmaceutical drug, principally related to its intrinsically low thermodynamic stability5 in conjunction with three reactive cysteine residues (free thiols) buried within the protein interior.6 These features contribute to irreversible unfolding and aggregation of this 16 kDa protein during production and storage, with associated negative consequences for shelf-life, potency, and immunogenicity.
A substantial number of formulation studies have been performed on FGF-1 with the goal of identifying pharmaceutical excipients that can stabilize the protein during long term storage and administration.7–9 FGF-1 has significant affinity for polyanions such as heparin/heparan sulfate, which as formulation additives can dramatically stabilize FGF-1 against unfolding by both heat and extremes of pH10 as well as against metal catalyzed oxidation of free cysteine residues.7, 9 Based on these observations, FGF-1 for human therapeutic use has been formulated with the addition of both heparin and antioxidants. The use of heparin as an excipient, however, adds considerable complexity including increased expense, its own pharmacological properties (e.g., as an anticoagulant), animal derived material from pig tissues (with potential for infectious contamination), and the possible induction of adverse inflammatory or allergic reactions in a segment of the targeted patient population. Thus, although heparin or related polyanions used as formulation additives solve the FGF-1 stability issue, they introduce other undesirable properties.
An alternative approach to the use of co-solutes to increase protein stability is to directly alter the protein's physical properties by chemical modification or mutagenesis. One approach that has been used to increase the circulating half-life of proteins is “PEGylation” [covalent attachment of polyethylene glycol (PEG), a biocompatible polymer]. This increases the molecular mass of a protein and thereby reduces renal clearance (i.e., glomerular filtration of biomolecules is size dependent) and substantially increases the circulating half-life.11 Furthermore, the attached PEG molecule can mask regions of the protein surface that would otherwise be susceptible to proteolytic attack or immune recognition, increasing the circulating half-life and reducing immunogenicity.12 PEGylation has either little effect or destabilizes the thermodynamic stability of proteins13; thus, the beneficial properties of PEGylation are primarily associated with modulation of renal clearance and reduction of the irreversible pathways associated with degradation and insolubility. One problem with PEGylation is that it can interfere with functional regions on the protein's surface, reducing receptor/ligand affinity by two or more orders of magnitude.12, 14
Mutating proteins to improve properties for human therapeutic application is a practical approach since over 30 mutant proteins have been approved for use as human biopharmaceuticals.15 These include mutations that contribute to increased yields during purification, increased in vivo functional half-life, or improved activity. Examples include mutations of buried free-cysteine residues in beta-interferon (Betaseron®) and interleukin-2 (Proleukin®) as well as other mutations hypothesized to increase thermostability. Thus, a mutational approach to improve the physical properties of proteins is a viable route to develop “second-generation” protein biopharmaceuticals.
In the present report, mutations are introduced into human FGF-1 with the goal of altering the temperature and pH stability profile to more closely resemble that of FGF-1 in complexation with heparin. These mutations target increases in thermostability as well as a reduction in the number of buried free cysteine residues. Furthermore, these mutations select positions with limited surface accessibility. Using empirical phase diagrams (EPDs) to broadly assess the structural integrity of proteins in response to variations in pH and temperature, mutants of FGF-1 are identified to achieve the design goal of matching, or exceeding, the protective effects of heparin as an additive.16, 17 These mutants represent potential “second-generation” forms of FGF-1 that may be successfully formulated for use as a human therapeutic in the absence of heparin.
We also take advantage of the revised EPD approach used in this study to compare the relative conformational stability of FGF-1 mutants to assess the value and utility of this multidimensional vector based approach as a novel analytical comparability tool.18, 19 Comparability assessments in biopharmaceutical development are conducted to determine the similarities (and differences) of various preparations of a biopharmaceutical drug used in clinical development in terms of a protein drug's physicochemical properties as they relate to its safety and efficacy.20, 21 Currently, one of the major analytical challenges in this area is to identify new biophysical approaches that complement in vitro/in vivo potency assays to better assess higher order structure and conformational stability of biopharmaceutical drug candidates. Although the series of FGF-1 mutants evaluated in this study are different molecular entities, the updated EPD data analytical approach used in this study could potentially be applied to biopharmaceutical comparability assessments of different preparations of the same protein drug.
Results
Selection of FGF-1 mutants
A combination of wild-type (WT) and 10 different FGF-1 mutants were chosen for analysis in this study. The mutant proteins were developed via a series of protein stability and folding studies with FGF-1 and were identified experimentally as having enhanced stability, functional half-life, or mitogenic activity.6, 22–28 The previously described thermodynamic stability properties and available in vitro mitogenic activity as well as in vivo half-life values are summarized in Table I. The rationale used for the selection of mutants examined in this study is based on the results in Table I (WT FGF-1 and mutants are labeled A–L). For example, the L26D/H93G (C) mutant (developed as part of a study evaluating the effect of the consensus β-turn motif Asx-Pro-Asx-Gly on FGF-1 stability and folding28) combines a destabilizing mutation (L26D), and an essentially off-setting stabilizing mutation (H93G), resulting in the overall thermostability essentially unchanged in comparison to WT FGF-1 (A) (ΔΔG = 0.9 kJ/mol). This mutant is therefore a thermostability control for the EPD analysis. Two of the FGF-1 mutants [SYM6ΔΔ/K12V/P134V (J) and SYM10ΔΔ (L)] were selected to evaluate the effect of significantly elevated thermostability. These mutants (developed to test the effect of symmetry on FGF-1 protein folding and design22, 29) exhibit diminished mitogenic activity towards 3T3 fibroblasts, which is postulated to be due to an effect of enhanced stability which prevents structure (dynamic) changes essential to the formation of receptor complexes.29 Alternatively, the P134V/C117V (E) mutant has enhanced stability (ΔΔG = −9.3 kJ/mol) yet exhibits similar mitogenic activity in comparison to WT FGF-1 (A). Other examples include K12V/C117V (F) and K12V/C117V/P134V (H) which are stabilizing mutations that, in the absence of heparin, achieve similar mitogenic potency to that of WT formulated in the presence of heparin (mutants E, F, and H were designed to stabilize N- and C-termini β-strand interactions, a region of known structural weakness within FGF-124) The mutants C83T/C117V/K12V (D), C83T/C117V/L44F/F132W (I), and oxidized A66C (G) have eliminated one or more free thiols, in comparison to WT FGF-1 (A). These mutations substantially increase their in vitro functional half-life with varying effects on thermostability (by mutating exclusively at solvent-inaccessible positions, thereby limiting immunogenic potential6, 27). Finally, Symfoil-4P (K), a synthetic ultra stable β-trefoil protein (designed as a hyperthermophile, purely symmetric polypeptide26, 29), was selected to evaluate its EPD profile in the context of its use as a nonfunctional protein engineering “scaffold.”26 For the biophysical characterization work performed in this study, the FGF-1 mutants in Table I were subdivided into two groups (I and II) due to the significant differences in their intrinsic fluorescence behavior (see “Methods section”).
Table I.
Summary of Stability Parameters and Biological Activity Values for WT FGF-1 and 10 FGF-1 Mutants
| FGF-1 protein (reference) | Symbol | ΔΔG (kJ/mol) | EC50 (ng/mL) (−) heparin | EC50 (ng/mL) (+) heparin | Half-life (h) | Heparin Binding |
|---|---|---|---|---|---|---|
| Group I | ||||||
| WT (without and with heparin) | A and B | — | 58.4 ± 25.4 | 0.48 ± 0.08 | 1.0 | Yes |
| L26D/H93G28 | C | −0.9 | N.A. | N.A. | N.A. | Yes |
| C83T/C117V/K12V6 | D | −1.9 | 0.93 ± 0.25 | 0.36 ± 0.12 | 40.4 | Yes |
| P134V/C117V24 | E | −8.8 | 46.8 ± 6.7 | N.A. | N.A. | Yes |
| K12V/C117V24 | F | −9.3 | 4.2 ± 1.7 | N.A. | N.A. | Yes |
| A66C (oxi)27, 46 | G | −10.2 | 5.43 ± 3.9646 | 0.36 ± 0.2046 | 14.246 | Yes |
| K12V/C117V/P134V24 | H | −19.1 | 1.80 ± 0.90 | N.A. | N.A. | Yes |
| Group II | ||||||
| C83T/C117V/L44F/F132W6 | I | −0.4 | 0.74 ± 0.19 | 0.51 ± 0.15 | 42.6 | Yes |
| SYM6ΔΔ/K12V/P134V29 | J | −35 | 741 ± 302 | N.A. | N.A. | No |
| Symfoil-4P26 | K | −44.1 | N.A. | N.A. | N.A. | No |
| SYM10ΔΔ29 | L | −47 | N.D.29 | N.A. | N.A. | No |
Conformational stability (ΔΔG), mitogenic activity (EC50), and in vivo half values are listed from the references provided. FGF-1 proteins in Group I contain a single Trp residue which is quenched at the native state. FGF-1 proteins in Group II contain either an unquenched Trp residue in the native state or do not contain the Trp residue. N.A., not available; N.D., not detected.
Due to the large amount of biophysical stability data generated in this work for WT FGF-1, both with and without heparin, as well as the 10 mutants of FGF-1, one mutant [K12V/C117V/P134V or (H) in Table I] was selected as an example to be compared with WT FGF-1 with heparin (B in Table I) and without heparin (A in Table I) in the main text. The data generated in this study for the other nine FGF-1 mutants are provided in Supporting Information. The biophysical characterization of WT FGF-1 in the presence and absence of heparin by the original EPD methodology has been determined previously,10 but was repeated in this work to provide a direct comparison to the FGF-1 mutants, especially in terms of evaluating of stability profiles using the revised EPD methodology.
Biophysical measurements of FGF-1 and mutants
Circular dichroism
Far-ultraviolet (UV) circular dichroism (CD) analysis was used to characterize the secondary (and tertiary) structure of WT FGF-1 (A in Table I), WT FGF-1 with heparin (B in Table I), and 10 mutants of FGF-1 (C-L in Table I). The mutant K12V/C117V/P134V was selected as an example to represent CD spectra at different solution pH values (pH 3–8) at 10°C (Fig. 1A). The CD spectra of K12V/C117V/P134V from pH 4 to 8 are similar innature, with a broad positive peak at around 228 nm and a strong negative peak near 205 nm. The negative peak near 205 nm is consistent with the expected class II β-protein structure.5, 30 The positive band at 228 nm is presumably a combination of contributions from β-turns, loops and aromatic side chains31–33 and is potentially tertiary structure sensitive. At pH 3, the positive peak at 228 nm is greatly diminished, which suggests an alteration of protein tertiary structure under acidic conditions. Because significant secondary structure still appears preserved, this suggests the possible preserve of a molten globule state.34, 35 WT FGF-1 (with and without heparin) display similar CD spectra from pH 5 to 8 with a positive peak at 228 nm and a negative peak at 205 nm (Supporting Information Figs. 1A and 2A). For WT FGF-1 without heparin at pH 3 as well as WT FGF-1 with heparin at pH 3–4, the CD spectra show a negative peak broadened toward 215 nm, which could be explained by the formation of intermolecular β-sheet structure.5–7, 9 At pH 4, WT FGF-1 without heparin shows a CD spectrum similar to K12V/C117V/P134V at pH 3, with only a negative peak at 205 nm (Supporting Information Fig. 1A).
Figure 1.

Far UV CD analysis of WT FGF-1 and a representative FGF-1 mutant. (A) CD spectra of the K12V/C117V/P134V mutant measured at 10°C at different pH values. (B–D) CD signal at 228 nm as measured as a function of temperature at indicated pH values for (B) K12V/C117V/P134V, (C) WT FGF-1, and (D) WT FGF-1 with heparin. Data points are an average of two independent analyses. Complete CD data sets for the other nine FGF-1 mutants are provided in Supporting Information section. See Table I for a further description of each mutant including K12V/C117V/P134V (H in Table I). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.

Intrinsic fluorescence analysis of WT FGF-1 and a representative FGF-1 mutant. (A) Fluorescence emission spectra (excited at 280 nm) of the K12V/C117V/P134V mutant measured at 10°C at different pH values. (B–D) Fluorescence intensity ratio (I305/I330) as measured as a function of temperature at indicated pH values for (B) K12V/C117V/P134V, (C) WT FGF-1, and (D) WT FGF-1 with heparin. Data points are an average of two separate experiments. Complete intrinsic fluorescence data sets for other nine FGF-1 mutants are provided in Supporting Information section. See Table I for a further description of each mutant including K12V/C117V/P134V (H in Table I). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To monitor structural changes of the proteins, CD signals at 228 nm were collected as a function of temperature from 10 to 90°C. At elevated temperature, the positive peak diminished under all pH conditions examined, whereas the negative peak shifted toward 215 nm (data not shown). As a result, at pH 5–8, the CD signals at 228 nm decreased from positive to negative as the temperature increased, as shown in Figure 1C for mutant K12V/C117V/P134V, Figure 1B for WT FGF-1, and Figure 1D for WT FGF-1 with heparin. This structural transition occurs between 40 and 50°C for WT FGF-1, 50–60°C for K12V/C117V/P134V and 60–70°C for WT FGF-1 in the presence of heparin. Thus, K12V/C117V/P134V shows a significantly higher thermal stability than WT FGF-1, although it is still less stable than WT FGF-1 with heparin.
The CD thermal transitions monitored at 228 nm at pH 3–4 are different when comparing K12V/C117V/P134V, WT FGF-1, and WT FGF-1 with heparin, for example, for K12V/C117V/P134V at pH 4, the CD signal at 228 nm shows a similar positive to negative transition as pH 5–8, albeit at lower temperature (30°C). In contrast, the CD signal at 228 nm for K12V/C117V/P134V at pH 3 does not show a major structural transition. Similarly, WT FGF-1 at pH 3–4 does not show any significant structural transitions. In contrast, WT FGF-1 with heparin at pH 3–4 shows a transition from negative to less negative ellipticity, probably due to the precipitation of protein out of solution.
Intrinsic fluorescence spectroscopy
Intrinsic fluorescence spectra were used to monitor the tertiary structure of WT FGF-1 (with and without heparin) as well as the 10 FGF-1 mutants. Because the WT FGF-1 contains eight Tyr and a single Trp residue quenched in the native state, the proteins were excited at 280 nm to simultaneously collect fluorescence from both Tyrosine and Trp residues. The intrinsic fluorescence spectra of K12V/C117V/P134V at 10°C are displayed in Figure 2A. The spectra from the mutant at pH 5–8 have a single peak at 305 nm, which corresponds to the emission of Tyr residues. In contrast, the spectra at pH 3 show a major peak at 305 nm, but a significant broad shoulder at 340 nm, which corresponds to the emission of the previously quenched Trp residues. Trp fluorescence is not observed at pH 5–8 because when the protein is properly folded at near neutral pH conditions, the fluorescence of the single Trp residue is quenched by positive charges from neighboring His and Lys residues.5, 36 At pH 3, the structure of WT FGF-1 is altered, and the nearby His and Lys side chains are moved away from the indole ring of Trp, resulting in dequenching of Trp fluorescence. The intrinsic fluorescence spectrum of the K12V/C117V/P134V mutant at pH 4 show intermediate behavior compared with pH 3 and 5–8. The intrinsic fluorescence spectra of WT FGF-1 and WT FGF-1 with heparin at 10°C are shown in Supporting Information Figures 1C and 2C, respectively. The fluorescence spectra for WT FGF-1 both with and without heparin at pH 3–4 is composed of contributions from Trp and Tyr, whereas Tyr contributions dominate as described above for pH 5–8.
The ratio of the fluorescence intensity at 305 nm and 330 nm (I305/I330) has been shown to sensitively monitor the change in FGF-1 tertiary structure as a function of pH or temperature.10 A higher value (∼ 3) of I305/I330 for proteins in Group I (see Table I) indicates a more native-like, properly folded structure, whereas a lower value (∼1) indicates altered structure. Thus, intrinsic fluorescence spectra were obtained from 10 to 90°C at 2.5°C intervals and I305/I330 values were calculated and plotted as a function of temperature to monitor changes in tertiary structure for the K12V/C117V/P134V mutant and WT FGF-1 with and without heparin (Fig. 2B–D).
As shown in Figure 2B for the K12V/C117V/P134V mutant at pH 3, the fluorescence intensity ratio started at ∼1 and increased gradually as the temperature was increased, without any obvious structural transition, indicating that the higher order structure is already altered at pH 3 at 10°C. In contrast, the fluorescence intensity ratio for pH 5–8 started at about 3 and then decreased to ∼0.7. The fluorescence intensity ratio for the mutant at pH 4 is between the values observed at pH 3 and pH 5–8 (starting at ∼2 and decreasing to ∼1.1 between 25 and 40°C). The thermal transitions for the mutant occurred between 20 and 55°C for pH 4–7, with an apparent bi-phasic transition observed between pH 5 and 8. The first transition is more gradual than the second phase. For WT FGF-1 with and without heparin, the fluorescence intensity ratio at both pH 3 and 4 shows no obvious structural transitions, indicating the expected structural alterations in WT FGF-1 at both pH values. In contrast, for pH 5–8, the ratio quickly decreased from 3 to ∼0.7. By comparing the temperature at the completion of this structural transition for pH 5–8, the transition ended at ∼45°C for WT FGF-1, ∼55°C for the K12V/C117V/P134V, and about 60°C for WT FGF-1 with heparin.
For FGF-1 mutant proteins in Group II (see Table I), however, the ratio of the fluorescence intensity at 305 nm and 330 nm (I305/I330) has a different biophysical meaning because these mutants contain either an unquenched or missing Trp residue. Both a His and Lys residue, which quench the Trp residue in WT FGF, were deleted from SYM6ΔΔ/K12V/P134V (J in Table I) and SYM10ΔΔ (L in Table I). Thus, the I305/I330 value is close to 1 even in the native state of protein (Supporting Information Figs. 4F and 5F). In contrast, C83T/C117V/L44F/F132W (I in Table I) has one extra Trp residue, which is not quenched in the native state. As a result, the I305/I330 value is below 1 across the temperature range from pH 3 to 8 (Supporting Information Fig. 8F). In the case of Symfoil-4P (K in Table I), the protein contains no Trp residues but does have three Tyr residues, so the I305/I330 value is above 3 up to 70–80°C (Supporting Information Fig. 6F). Due to the difference of I305/I330 values in the native state, Symfoil-4P (K in Table I) has a different color scheme in the EPDs (discussed below in Fig. 6) even in the low temperature region compared with the other mutants in Group II.
Figure 4.

Static light scattering analysis of WT FGF-1 and a representative FGF-1 mutant as a function of temperature at the indicated pH values. (A) K12V/C117V/P134V, (B) WT FGF-1, and (C) WT FGF-1 with heparin. Data points are an average of two independent experiments. The complete static light scattering data set for the other nine FGF-1 mutants is provided in Supporting Information section. See Table I for a further description of each mutant including K12V/C117V/P134V (H in Table I). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
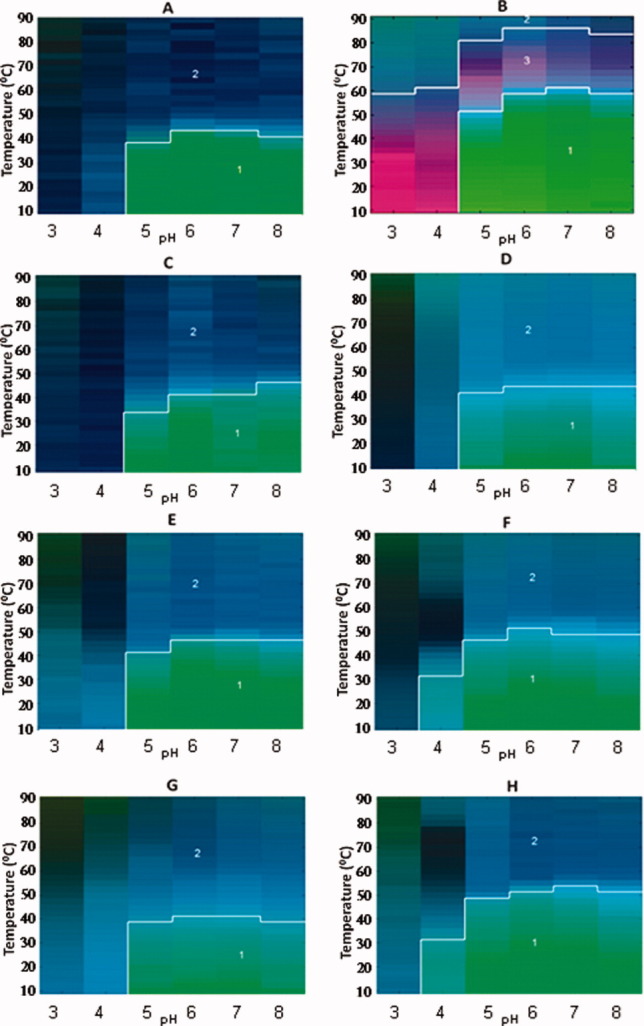
Empirical Phase Diagrams (EPDs) for WT FGF-1 (A), WT FGF-1 with heparin (B), and 6 different FGF-1 mutants without heparin (C–H). The EPDs were constructed based on intrinsic fluorescence intensity ratio at two wavelengths (I305/I330 nm), intrinsic fluorescence intensity, CD at 228 nm, static light scattering (SLS) and ANS fluorescence intensity at 480 nm. All FGF-1 mutants in this Figure contain a single Trp residue which is quenched at the native state. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 6.
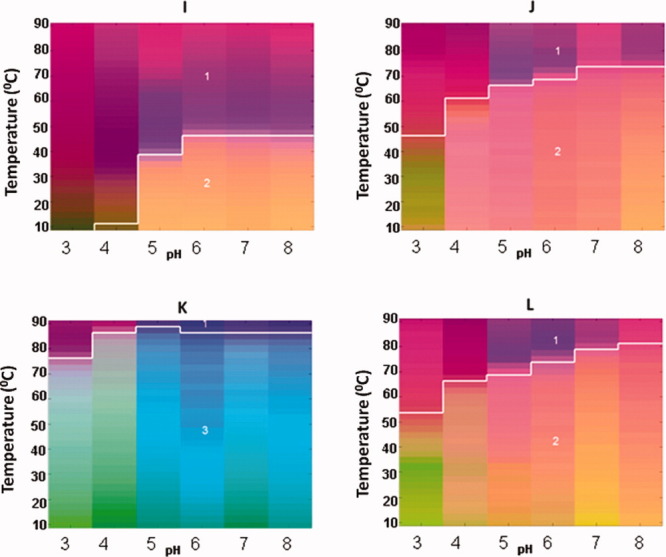
Empirical Phase Diagrams (EPDs) for four different FGF-1 mutants without heparin (I–L). The EPDs were constructed based on intrinsic fluorescence intensity ratio at two wavelengths (I305/I330 nm), intrinsic fluorescence intensity, CD at 228 nm, static light scattering (SLS) and ANS fluorescence intensity at 480 nm. FGF-1 mutants in this Figure contain either an unquenched Trp residue in the native state or do not contain the Trp residue. See Table I for description of each mutant. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
1-Anilino-8-naphthalnesulfonate fluorescence
1-Anilino-8-naphthalnesulfonate (ANS) has been extensively used to detect the exposure of apolar binding sites on the surface of proteins and to detect the presence of molten globule-like states.30, 37, 38 At pH 3 and 4, substantial ANS fluorescence was observed for K12V/C117V/P134V (Fig. 3A), WT FGF-1 (Supporting Information Fig. 1H), and WT FGF-1 with heparin (Supporting Information Fig. 2H) at 10°C. Above pH 5, however, much less fluorescence is observed in the presence of these samples at 10°C. Thus, ANS fluorescence spectra indicate that the tertiary structure of FGF-1 is altered in acidic condition even at low temperature, consistent with CD and intrinsic fluorescence results.
Figure 3.

ANS fluorescence analysis of WT FGF-1 and a representative FGF-1 mutant. (A) ANS fluorescence spectra of K12V/C117V/P134V measured at 10°C at different pH values. (B–D) ANS fluorescence intensity ratio as measured as a function of temperature at indicated pH values for (B) K12V/C117V/P134V, (C) WT FGF-1, and (D) WT FGF-1 with heparin. Data points are an average of two independent experiments. The complete ANS fluorescence data sets for the other nine FGF-1 mutants are provided in Supporting Information section. See Table I for a further description of each mutant including K12V/C117V/P134V (H in Table I). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The ANS fluorescence intensity at 480 nm was monitored from 10 to 90°C at 2.5°C intervals. K12V/C117V/P134V manifested an increase in ANS fluorescence intensity at ∼40°C for pH 5 and ∼50°C at higher pH values (Fig. 3B). As shown in Figure 3C, at pH 5–8, WT FGF-1 shows an increase in ANS fluorescence intensity starting at 20°C. By including heparin with WT FGF-1, the onset temperature of ANS binding was delayed to 55°C for pH 5–8 (Fig. 3D). At pH 3–4, the major trend is a decrease in ANS fluorescence intensity as temperature increases for all three proteins. At pH 4, the K12V/C117V/P134V mutant shows slightly more neutral pH-like structural transitions (compared with WT FGF-1 with and without heparin), probably because the K12V/C117V/P134V mutant has more native-like structure at pH 4.
Static light scattering
Both acidic pH conditions and elevated temperatures cause aggregation/precipitation of WT FGF-1.2, 3 Due to the low protein concentration used during dialysis, however, no gross precipitation was observed after dialysis at pH 3–4. The temperature induced aggregation of FGF-1 was followed by the static light scattering (SLS) signal at 280 nm. As shown in Figure 4A, very little scattering was observed at low temperatures for the K12V/C117V/P134V mutant under all pH conditions. An increase in light scattering intensity started at ∼45°C for pH 5–8 and ∼55°C for pH 4. No significant aggregation could be detected by light scattering for pH 3 up to 90°C. A decrease in light scattering signal was observed for pH 5–8 above ∼60°C due to the precipitation of protein. For WT FGF-1 at pH 3, light scattering initiated at a higher level than other pH conditions, probably due to acidic buffer induced aggregation, although no obvious visible precipitation was observed. The onset of WT FGF-1 aggregation at pH 4–8 started at ∼40°C. For WT FGF-1 with heparin, the light scattering signal for both pH 3 and 4 started at higher values than all other pH conditions, immediately followed by a gradual increase and then decrease. This aggregation started at ∼45°C for pH 5 and ∼60°C for all other pH values. At a solution pH between 6 and 8, protein aggregation started at 40°C, 45°C, and 60°C for WT FGF-1, K12V/C117V/P134V, and WT FGF-1 with heparin, respectively. Again, the K12V/C117V/P134V mutant displayed improved stability under acidic conditions.
Empirical phase diagrams
The EPD approach has been developed to combine large amounts of data from multiple measurements to evaluate the overall conformational stability of proteins and other macromolecular systems under different environmental stresses.16, 17, 39 As shown in Figures 5 and 6, EPDs were generated for WT FGF-1 (with and without heparin) and for the 10 different FGF-1 mutants. All of the measurements as a function of temperature and solution pH (as described above and in Supporting Information section) were summarized into RGB color plots in a two-dimensional space of pH and temperature using the revised EPD approach described in “Method section.” As explained in more detail in “Method section,” the EPDs for the FGF-1 mutants were generated in two groups (I and II). Group I (Fig. 5) includes proteins A–H in Table I and Group II (Fig. 6) includes proteins I–L in Table I. Colors can be compared directly within each group. A clustering method was then used to generate three distinct regions for each group of EPDs (not each EPD). The location of boundary of the individual regions can be used to compare the conformational stability among and between the two groups.
As shown in Figure 5, the EPD for WT FGF-1 (A) has two distinct regions, a blue area (Region 2) and a green area (Region 1). The green region (Region 1) covers the low temperature and neutral pH range, which represents the native, most stable form of the protein. A structural transition occurs at ∼44°C for pH 6 and 7, which are the most stable pH conditions for WT FGF-1 (A). Although a third light blue region is present around the boundary between Regions 1 and 2, which represents molten globule-like behavior,34, 35 this subtle color difference was not defined as a separate region by this clustering analysis. On the addition of heparin to WT FGF-1 (B), a third region (pink) appears between Regions 1 and 2 as well as at low temperatures at pH 3 and 4. The formation of this third region is primarily due to the strong negative CD signal at low temperatures for pH 3 and 4 and at high temperatures at pH 5–8 (Fig. 1D). From inspection of the CD spectra of WT FGF-1 with heparin (B) (Supporting Information Fig. 2A), it is apparent that intermolecular β-sheet structure has formed at pH 3 and 4 at 10°C and pH 5–8 at higher temperatures. As the temperature increased, protein aggregates started to form as indicated by increased light scattering (Fig. 4C), which is followed by a slight decrease presumably due to precipitation of protein. This third region in the EPD appears to be unique to WT FGF-1 with heparin (B). The structural transition between Regions 2 and 3 for WT FGF-1 with heparin occurred at 60°C, which is about 16°C more stable than WT FGF-1 without heparin.
The EPDs for 6 of the FGF-1 mutants are also shown in Figure 5. The EPDs for the mutants show 2 regions (green for Region 1 and blue for Region 2). The K12V/C117V/P134V (H) mutant is most stable at pH 6 and 7, with a transition at 52°C, which is ∼8°C higher than WT FGF-1. At pH 4, structural transitions are present at ∼30°C, which differs from WT FGF-1 and most of the other mutants where transitions at pH 4 cluster with pH 3. Thus, K12V/C117V/P134V (H) has enhanced resistance to acidic unfolding. The K12V/C117V (F) mutant is similar to K12V/C117V/P134V (H), except for a lower overall thermal stability (∼4°C less).
The EPDs for the other four mutants of FGF-1 are shown in Figure 6. The EPDs of C83T/C117V/L44F/F132W (I), SYM6ΔΔ/K12V/P134V (J), and SYM10ΔΔ (L) have Regions 1 (purple) and 2 (orange). Region 2 is at low temperature and in the neutral pH range. It thus represents the native, stable region. Both SYM6ΔΔ/K12V/P134V (J) and SYM10ΔΔ (L) show a very high degree of enhanced physical stability. In contrast, the EPD of Symfoil-4P (K) manifests a Regions 1 (purple) and 3 (light blue/green) due to the difference in the I305/I330 value, as explained above, where Region 3 represents the native stable region. As expected, SYM10ΔΔ (L) and Symfoil-4P (K), a synthetic ultra stable β-trefoil protein, are the most conformationally stable mutants observed by the EPD analysis. The EPDs and clustering of the FGF-1 mutants are consistent with the thermodynamic conformational stability data previously obtained for the mutants (see Table I).
Discussion
The stability of FGF-1 is strongly dependent on complexation with polyanions such as heparin.7, 8 This stabilization is important endogenously where the biological activity of this (and many other) growth factors requires interaction with polyanionic proteoglycans and protein receptors on the cell surface. In addition, the use of FGF-1 as a therapeutic agent requires the presence of heparin or other polyanions because the protein is unstable during storage and administration in its absence. In this work, we have analyzed a series of FGF-1 mutants with the goal of reducing or eliminating the need for exogenous heparin by enhancing the protein's intrinsic stability. Refer to Table I for the description of each FGF-1 mutant and Figures 5 and 6 for the summary of the biophysical stability results which will be the focus of the rest of this discussion. Rather than the usual reliance on evaluating protein stability from a single, unique biophysical technique (e.g., CD, intrinsic and ANS fluorescence, light scattering, etc.), we have evaluated the protein stability data from multiple experimental methods holistically in the form of a multivariable vector-based approach designated the EPD to provide a broad picture of the physical behavior of the FGF-1 mutants as a function of temperature and pH.17
Increases in stability in this context can be viewed as an expansion of the apparent phase boundaries obtained in the EPD (i.e., the region of abrupt color change). For example, when comparing WT FGF-1 in the absence and presence of heparin, this boundary is moved to significantly higher temperature (Fig. 5) with an accompanying dramatic increase in biological activity (the EC50 is approximately 120× lower as shown in Table I). Several of the mutants [e.g., Symfoil-4P (K), SYM6ΔΔ/K12V/P134V (J), and SYM10ΔΔ (L)] display even greater increase in conformational stability, but demonstrate little biological activity. In contrast, a number of the FGF-1 mutants [K12V/C117V (F), C83T/C117V/L44F/F132W (I), A66C (G), and K12V/C117/P134V (H)] display improved free energies of unfolding relative to WT, major increases in mitogenic activity in the absence of heparin (Table I), with EPDs similar to those of WT in the presence of heparin (Fig. 5). Thus, these FGF-1 mutants seem to be good candidates for future therapeutic applications from a stability and potency point of view.
Because the biophysical data for WT FGF-1 (with and without heparin) and the other six mutants (Table I and Fig. 5) were mathematically processed together as the same data set, the colors from these EPDs can be directly compared with each other. Different color regions within the same EPD reflect different physical states of protein under the indicated pH and temperature condition. Color changes indicate transitions in these states. Note that these are not necessarily thermodynamic states because reversibility between them is frequently not present. To facilitate the visualization of different color regions in EPDs, a clustering method was used to divide each EPD into different regions based on the input data (see below for further discussion).
The FGF-1 mutants in Figure 6 have different fluorescence ratios (I305/I330 nm) in their native states. This effect is due to the deletion of a lysine, and thus the Trp fluorescence in SYM6ΔΔ/K12V/P134V (J) and SYM10ΔΔ mutants (L) (see Table I) is not quenched in the native state. Thus, the I305/I330 profile for these two mutants is quite different from the others (see Supporting Information Figs. 4F and 5F). C83T/C117V/L44F/F132W (I in Table I) has an additional Trp residue which is not quenched in the native state. Symfoil-4P (L in Table I) does not have any Trp residues. As a result, EPDs for these SYM6ΔΔ/K12V/P134V (J) and SYM10ΔΔ (L) mutants were generated separately (Fig. 6), and their colors are compared with each mutant within this group. Nonetheless, the location of the cluster boundaries for the phase transitions shown in the EPDs for the various mutants can be compared with each other.
FGF-1 is known to form molten globule-like states under conditions of acidic pH as well as at moderately elevated temperatures at higher pH. This molten globule conformation is visually identified as a light blue region between the dark blue (Region 2) and green (Region 1) areas identified by the clustering boundary for the WT FGF and many of the mutants’ EPDs in Figure 5. This observation of the formation of a molten globule state for FGF-1 under environmental stress is consistent with previous work.10 It should be noted, however, that the clustering methodology used in this work was not able to identify this subtle color change. Thus, the current version of the clustering analysis identifies the transition region between large conformational changes (e.g., between the native and unfolded, aggregated states of the FGF-1 mutants) but is not currently able to consistently identify the more subtle color changes represented by the molten globule state. The clustering methodology used in this work is a prototype version, and by exploring different clustering methods (work in progress), automatic clustering analysis might be able to identify the more subtle color differences (e.g., the molten globule state for FGF-1) that are observed visually by human sight in the future. Nonetheless, this version of the clustering analysis was able to identify several FGF-1 mutants, for example K12V/C117V/P134V (H), which display an expanded native state (Region 1 in Fig. 5). These FGF-1 mutants are stable, bioactive forms of the growth factor that are not heparin dependent.
A second purpose of this study was to test the ability of EPDs to distinguish the conformational stability of a series of mutant proteins. We have previously successfully utilized the EPD method for the development of stabilized pharmaceutical formulations of macromolecular systems such as biopharmaceutical drugs and vaccines.17, 39 Instead of relying on one or only a few biophysical parameters, EPDs use a two-dimensional “stimulation/response” color pattern diagram. As is evident from Figures 5 and 6, both dramatically different and highly similar EPDs are produced with only a few mutational differences between proteins. Thus, this approach offers a unique methodology to compare proteins that is not based on structure alone, but rather uses the way a protein responds to environmental stress (e.g., pH and temperature) as a comparative principle. The EPD approach is not limited to temperature and pH but can also be used to evaluate the effect of protein concentration as well as solute and ionic strength among other solution variables.17, 39 We have recently shown that use of methods sensitive to the internal motions of proteins (e.g., isotope exchange, ultrasonic spectroscopy, solute spectral quenching, etc.) in the context of EPDs can also provide insight into more dynamic aspects of a protein behavior. Thus, the EPD approach would seem to offer an improved method to analyze the effect of mutation on protein structure.
In addition, it should be possible in the future to evaluate the revised EPD methodology developed in this work to compare stability of different mutants, as a new analytical approach to assess comparability.18, 19 During the pharmaceutical development of a protein therapeutic drug candidate or vaccine, comparability assessments are performed to determine if a protein's key physicochemical properties have changed (or not) across different preparations used during clinical trials. These analytical assessments typically involve a combination of routine quality control tests (e.g., sodium dodecyl sulfate polyacrylamide gel electrophoresis, size exclusion-high performance liquid chromatography, isoelectric focusing, etc.), more sophisticated analysis using mass spectrometry and biophysical methods (e.g., intact MW analysis, peptide and oligosaccharide maps, differential scanning calorimetry, analytical ultracentrifugation, etc.), and stability profiles under accelerated and storage temperature conditions.20, 21 It has recently been shown that comparison of accelerated temperature stability profiles can be an effective and sensitive way to perform comparability assessments.20 Because the EPD methodology provides a rapid, high throughput approach to collect and evaluate conformational stability data under accelerated conditions of temperature and pH, this approach could potentially be useful as a comparability assessment tool. The EPD method is currently being evaluated in our laboratories for its utility in examining the conformational stability of different preparations of the same protein with modest changes in post translational modifications (e.g., glycosylation patterns).
In summary, the conformational stability of WT and 10 different FGF-1 mutants was examined as a function of temperature and solution pH by a series of biophysical techniques, and the resulting data sets were analyzed with an EPD methodology. The revised version of the EPD methodology used in this work resulted in effective and convenient comparisons of the effect of temperature and solution pH on the structural properties of a wide range of FGF-1 mutants. The conformational stability profiles of several of the FGF-1 mutants were comparable to the WT FGF-1 in the presence of heparin. Thus, candidate FGF-1 mutants were identified with enhanced stability profiles in the absence of heparin and can be considered promising second-generation therapeutic FGF-1 candidates. These mutants are currently undergoing preclinical pharmacokinetic studies in animals.
Materials and Methods
Materials
Recombinant FGF-1 proteins (WT and mutants, except for Symfoil-4P) utilized a synthetic gene for the 140 amino acid form of human FGF-140–43 containing an additional amino-terminal six His tag as previously described.44 The design strategy of the Symfoil-4P mutant used complete gene synthesis and was reported previously.26 The FGF-1 protein mutants used in this study are listed in Table I. The QuikChange™ site directed mutagenesis protocol (Agilent Technologies, Santa Clara, CA) was used to introduce all point mutations and was confirmed by nucleic acid sequence analysis (Biomolecular Analysis Synthesis and Sequencing Laboratory, Florida State University). All expression and purification protocols followed previously published procedures involving sequential chromatographic steps using Ni-NTA resin and heparin Sepharose CL-6B affinity resin.44 Three of the mutant proteins, SYM6ΔΔ/K12V/P134V, SYM10ΔΔ (=SYM7ΔΔ/K12V/P134V/H93G) and Symfoil-4P, which have no affinity for heparin, were purified by Ni-NTA chelation and Superdex 75 size exclusion chromatography as reported previously.6, 26 The purified protein in each case was exchanged into 50 mM sodium phosphate, 0.1 M NaCl, 10 mM ammonium sulfate, 2 mM dithiothreitol (DTT), and pH 7.5 (“crystallization buffer”). The purified A66C mutant protein contains a mixture of reduced and oxidized forms. To isolate the fully oxidized form of A66C, the purified protein was exchanged into crystallization buffer without DTT and subsequently air oxidized at room temperature for 3 weeks. An extinction coefficient of E280 nm (0.1%, 1 cm) =1.267, 45 was used to determine protein concentration for FGF-1 with the exception of C83T/C117V/L44F/F132W, SYM6ΔΔ/K12V/P134V, SYM10ΔΔ, and Symfoil-4P. Due to the variation in number of Trp and Tyr residues in these mutants, the extinction coefficient was determined by the method of Gill and von Hippel or densitometry analysis as reported previously.6, 10, 11 The resulting E280 nm (0.1%, 1 cm) values utilized were: C83T/C117V/L44F/F132W, 1.58; SYM6ΔΔ/K12V/P134V and SYM10ΔΔ, 1.31; Symfoil-4P, 0.32.
Heparin sodium salt, grade 1-A from porcine intestinal mucosa, was purchased from Sigma–Aldrich (St. Louis, MO). All other chemicals were purchased from Sigma–Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburg, PA).
Methods
Sample preparation
The purified FGF-1 proteins (WT and mutants) were dialyzed against 20 mM citrate-phosphate buffers at pH 3–8 and adjusted to an ionic strength of 0.15 M with NaCl, using a 3.5 kDa molecular-weight cutoff (Pierce, Rockford, IL) membrane. The dialysis step was carried out overnight at 4°C. A mixture of WT FGF-1 and heparin was prepared by adding heparin to the protein solution to achieve a 3:1 (w/w) ratio of heparin to protein.
CD spectroscopy
Far-UV CD spectra were recorded using either a Jasco J-810 (Tokyo, Japan) or Chirascan (Applied Photophysics) instruments, both equipped with a peltier type temperature controller. The protein concentration used was 0.2 mg/mL using a 0.1 cm path length quartz cuvette was used with a total volume of 0.2 mL. Full CD spectra were collected before and after temperature ramping at 10°C for wavelengths ranging from 260 to 200 nm with a resolution of 0.2 nm and a bandwidth of 1 nm over the pH range 3–8 at one unit intervals. The CD signal intensity changes at 228 nm were followed as the temperature was raised from 10 to 90°C at 2.5°C intervals with a scanning speed of 15°C/h and 5 min equilibration time at each temperature.
Intrinsic fluorescence spectroscopy and SLS
Intrinsic fluorescence measurements were recorded using a two-channel, four positions PTI Quantum Master fluorometer (Brunswick, New Jersey) equipped with a peltier temperature controlled cell holder. Spectra were obtained using excitation at 280 nm with emission spectra recorded between 290 and 390 nm at a resolution of 1 nm. Spectra were also obtained from 10 to 90°C at 2.5°C intervals in a 0.2 cm path length rectangular reduced volume cuvette containing 0.2 mL protein solution. Buffer spectra were collected and subtracted from the protein samples. Fluorescence emission peak positions and intensities were determined by a “dpoly” method using Origin 7.0 software. SLS intensity for the same sample was followed by using a right angle detector located 180° relative to the other detector, and the peak intensity change at 280 nm was followed as the temperature was raised.
Extrinsic fluorescence spectroscopy
Measurements were performed using ANS to detect exposure of apolar regions of the protein as the temperature was increased. A protein concentration of 0.08 mg/mL was used, and ANS was added to the protein solution to achieve an optimal ANS: protein molar ratio of 10:1. The resulting mixture was excited at 374 nm, and the emission spectra were recorded from 400 to 600 nm with a resolution of 1 nm.
Empirical phase diagrams
EPDs are designed to summarize and visualize physical characterization data as a colored diagram with two-dimensional axes of environmental stress conditions such as temperature and pH. The theory and calculation procedures to create EPDs are described in detail elsewhere.16, 17 The EPDs of FGF-1 and its mutants were constructed based on the following biophysical measurements: intrinsic fluorescence intensity ratio at two wavelengths (I305/I330 nm), intrinsic fluorescence peak intensity, CD at 228nm, SLS and ANS fluorescence intensity at 480nm. Complete data sets are presented in Supporting Information section. Data from the individual biophysical measurements (except for the I305/I330 nm fluorescence intensity ratio) were normalized as described previously.16, 17 All calculations were performed using MATLAB software (The Mathworks Inc., Natick, MA).
Two revisions of the data analysis methodologies are used in this work to improve the ability of EPDs to compare the conformational stability profiles of the FGF-1 mutants. First, the EPD data analysis technique was extended to better pursue direct color comparison of EPDs from multiple samples. Previously, regions of color transition, but not the color itself, have been compared between EPDs. Similar colors in different EPDs could not be interpreted, even if proteins had similar conformational states, because each EPD was generated separately using an arbitrary RGB color mapping scheme. In contrast, the colors of EPDs made with the revised methodology can be directly compared with each other, assuming the meaning of the results from the different biophysical measurements is consistent between different samples. In brief, experimental data sets are represented as n-dimensional vectors where n refers to the number of experiment techniques (five in this case). The condition space of the original EPD method consists of two environmental conditions (typically temperature and solution pH) whereas in this revised version the space is extended to incorporate multiple samples. Therefore, a sample condition space is defined with three components: sample, temperature, and solution pH. For a total of m conditions (=number of samples × number of temperature measurements × number of pH measurements), singular value decomposition of the m × n experimental data matrix is calculated to extract three orthogonal basis vectors associated with the three largest singular values, which are then mapped to RGB colors and visualized as EPDs.
The EPDs generated in this way are considered to display similar conformational behaviors across the different proteins based on an assumption that the collected experimental data represents similar physical behavior. If, however, the same data values represent different conformational states, these proteins are not processed together to generate EPDs for comparison purposes. For this reason, we divided WT FGF1 with and without heparin, as well as the 10 mutants into two groups (I and II) to generate EPDs (Table I), due to the significant differences in their intrinsic fluorescence behavior (see Supporting Information). This evaluation was done manually by visual assessment of the fluorescence data from each protein. Group I proteins include WT FGF-1 (A), WT FGF-1 with heparin (B), and the mutants L26D/H93G (C), C83T/C117V/K12V (D), P134V/C117V (E), K12V/C117V (F), A66C (G), and K12V/C117V/P134V (H). Group II proteins include C83T/C117V/L44F/F132W (I), SYM6ΔΔ/K12V/P134V (J), Symfoil-4P (K), and SYM10ΔΔ (L). Mutants in Group I contain a single Trp residue which is quenched in the protein's native state. Mutants in Group II contain either an unquenched Trp residue in the native state or do not contain a Trp residue. These differences lead to different fluorescence peak positions and intensities and the data sets cannot be directly compared. In summary for this work, the EPDs for FGF-1 proteins in each group (I and II) were created together as can be seen in Figures 5 and 6, respectively; therefore, the colors of EPDs should be compared directly within these two groups but not between the two groups.
The second revision implemented with the EPD methodology is the application of mathematical clustering analysis to computationally identify regions of structural transitions (e.g., color changes). Previously, structural transitions in the EPDs were estimated by visual inspection of the EPD with comparisons to the individual biophysical data sets. The subtle or gradual change in colors often presented in the EPD, however, makes the location of the transitions difficult to be determined visually. Therefore, clustering analysis that utilizes the same m × n experimental data matrix as the EPD can provide complementary information for structural transitions.
In this research, k-means clustering algorithm was chosen for the clustering analysis and implemented using a MATLAB toolbox downloaded from http://www.dcorney.com/ClusteringMatlab.html. The performance of various clustering algorithms will be the subject of future work.
k-Means clustering is a mathematical procedure in which a number (m) of experimental observations are partitioned into k groups or clusters. Clusters are based on the proximity of observations to proximate means. Observations are defined as n-dimensional vectors (a1, a2, …, an). The method then partitions m observations into k-sets (k < n) P = [P1, P2, …Pk] to minimize the “within-cluster sum of squares”. More formally:
| (1) |
The number of clusters (k) is determined after several diagnostic runs with various integral numbers. Generally, k = 2–5 works well for protein characterization data because k should, to a first approximation, correspond to the number of conformational states portrayed in the EPD. When constructing an EPD, the clustering results are used to draw the boundary between different clusters. The clustering results identify regions of conformational changes in FGF-1 mutants (due to environmental stresses such as temperature and pH), and help to identify and compare regions displaying native state behaviors, and can be used to compare mutant FGF-1 proteins across the two groups, even though the two groups of EPDs display two different color sets.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Ioli V, Wiens B, Mellin T, Thomas K, Ellis R, Ryan J, White CJ. Effect of topically applied acidic fibroblast growth factor (FGF-1) on healing of chronic venous stasis and diabetic ulcers. Abstracts of the 5th Annual Symposium on Advanced Wound Care. 1992:150. [Google Scholar]
- 2.Schumacher B, Pecher P, von Specht BU, Stegmann T. Induction of neoangiogenesis in ischemic myocardium by human growth factors: first clinical results of a new treatment of coronary heart disease. Circulation. 1998;97:645–650. doi: 10.1161/01.cir.97.7.645. [DOI] [PubMed] [Google Scholar]
- 3.Comerota AJ, Throm RC, Miller KA, Henry T, Chronos N, Laird J, Sequeira R, Kent CK, Bacchetta M, Goldman C, Salenius JP, Schmieder FA, Pilsudski R. Naked plasmid DNA encoding fibroblast growth factor type 1 for the treatment of end-stage unreconstructible lower extremity ischemia: preliminary results of a phase I trial. J Vasc Surg. 2002;35:930–936. doi: 10.1067/mva.2002.123677. [DOI] [PubMed] [Google Scholar]
- 4.Wagoner L, Merrill W, Jacobs J, Conway G, Boehmer J, Thomas K, Stegmann T. Angiogenesis protein therapy with human fibroblast growth factor (FGF-1): results of a phase I open label, dose escalation study in subjects with CAD not eligible for PCI or CABG. Circulation. 2007;116:443. [Google Scholar]
- 5.Copeland RA, Ji H, Halfpenny AJ, Williams RW, Thompson KC, Herber WK, Thomas KA, Bruner MW, Ryan JA, Marquis-Omer D, Sanyal G, Sitrin RD, Yamazaki S, Middaugh CR. The structure of human acidic fibroblast growth factor and its interaction with heparin. Arch Biochem Biophys. 1991;289:53–61. doi: 10.1016/0003-9861(91)90441-k. [DOI] [PubMed] [Google Scholar]
- 6.Lee J, Blaber M. The interaction between thermostability and buried free cysteines in regulating the functional half-life of fibroblast growth factor-1. J Mol Biol. 2009;393:113–127. doi: 10.1016/j.jmb.2009.08.026. [DOI] [PubMed] [Google Scholar]
- 7.Tsai PK, Volkin DB, Dabora JM, Thompson KC, Bruner MW, Gress JO, Matuszewska B, Keogan M, Bondi JV, Middaugh CR. Formulation design of acidic fibroblast growth factor. Pharm Res. 1993;10:649–659. doi: 10.1023/a:1018939228201. [DOI] [PubMed] [Google Scholar]
- 8.Mach H, Volkin DB, Burke CJ, Middaugh CR, Linhardt RJ, Fromm JR, Loganathan D, Mattsson L. Nature of the interaction of heparin with acidic fibroblast growth factor. Biochemistry. 1993;32:5480–5489. doi: 10.1021/bi00071a026. [DOI] [PubMed] [Google Scholar]
- 9.Volkin DB, Tsai PK, Dabora JM, Gress JO, Burke CJ, Linhardt RJ, Middaugh CR. Physical stabilization of acidic fibroblast growth factor by polyanions. Arch Biochem Biophys. 1993;300:30–41. doi: 10.1006/abbi.1993.1005. [DOI] [PubMed] [Google Scholar]
- 10.Fan H, Li H, Zhang M, Middaugh CR. Effects of solutes on empirical phase diagrams of human fibroblast growth factor 1. J Pharm Sci. 2007;96:1490–1503. doi: 10.1002/jps.20796. [DOI] [PubMed] [Google Scholar]
- 11.Harris JM, Chess RB. Effect of PEGylation on pharmaceuticals. Nature Rev Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 12.Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev. 2002;54:531–545. doi: 10.1016/s0169-409x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- 13.Monfardini C, Schiavon O, Caliceti P, Morpurgo M, Harris JM, Veronese FM. A branched monomethoxypoly(ethylene glycol) for protein modification. Bioconjug Chem. 1995;6:62–69. doi: 10.1021/bc00031a006. [DOI] [PubMed] [Google Scholar]
- 14.Basu A, Yang K, Wang M, Liu S, Chintala R, Palm T, Zhao H, Peng P, Wu D, Zhang Z, Hua J, Hsieh MC, Zhou J, Petti G, Li X, Janjua A, Mendez M, Liu J, Longley C, Zhang Z, Mehlig M, Borowski V, Viswanathan M, Filpula D. Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug Chem. 2006;17:618–630. doi: 10.1021/bc050322y. [DOI] [PubMed] [Google Scholar]
- 15.Kurtzman AL, Govindarajan S, Vahle K, Jones JT, Heinrichs V, Patten PA. Advances in directed protein evolution by recursive genetic recombination: applications to therapeutic proteins. Curr Opin Biotechnol. 2001;12:4171–4197. doi: 10.1016/s0958-1669(00)00228-7. [DOI] [PubMed] [Google Scholar]
- 16.Kueltzo LA, Ersoy B, Ralston JP, Middaugh CR. Derivative absorbance spectroscopy and protein phase diagrams as tools for comprehensive protein characterization: a bGCSF case study. J Pharm Sci. 2003;92:1805–1820. doi: 10.1002/jps.10439. [DOI] [PubMed] [Google Scholar]
- 17.Maddux NR, Joshi SB, Volkin DB, Ralston JP, Middaugh CR. Multidimensional methods for the formulation of biopharmaceuticals and vaccines. J Pharm Sci. 2011;100:4171–4197. doi: 10.1002/jps.22618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22:1383–1391. doi: 10.1038/nbt1030. [DOI] [PubMed] [Google Scholar]
- 19.Lubiniecki AS, Marcia Federici M. Comparability is not just analytical equivalence. Biologicals. 2006;34:45–47. doi: 10.1016/j.biologicals.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 20.Lubiniecki A, Volkin DB, Federici M, Bond MD, Nedved ML, Hendricks L, Mehndiratta P, Bruner M, Burman S, Dalmonte P, Kline J, Ni A, Panek ME, Pikounis B, Powers G, Vafa O, Siegel R. Comparability assessments of process and product changes made during development of two different monoclonal antibodies. Biologicals. 2011;39:9–22. doi: 10.1016/j.biologicals.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Schenerman MA, Hope JN, Kletke C, Singh JK, Kimura R, Tsao EI, Folena-Wasserman G. Comparability testing of a humanized monoclonal antibody (Synagis) to support cell line stability, process validation, and scale-up for manufacturing. Biologicals. 1999;27:203–215. doi: 10.1006/biol.1999.0179. [DOI] [PubMed] [Google Scholar]
- 22.Brych SR, Dubey VK, Bienkiewicz E, Lee J, Logan TM, Blaber M. Symmetric primary and tertiary structure mutations within a symmetric superfold: a solution, not a constraint, to achieve a foldable polypeptide. J Mol Biol. 2004;344:769–780. doi: 10.1016/j.jmb.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 23.Culajay JF, Blaber SI, Khurana A, Blaber M. Thermodynamic characterization of mutants of human fibroblast growth factor 1 with an increased physiological half-life. Biochemistry. 2000;39:7153–7158. doi: 10.1021/bi9927742. [DOI] [PubMed] [Google Scholar]
- 24.Dubey VK, Lee J, Somasundaram T, Blaber S, Blaber M. Spackling the crack: stabilizing human fibroblast growth factor-1 by targeting the N and C terminus beta-strand interactions. J Mol Biol. 2007;371:256–268. doi: 10.1016/j.jmb.2007.05.065. [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Brych SR, Lee J, Logan TM, Blaber M. Identification of a key structural element for protein folding within beta-hairpin turns. J Mol Biol. 2003;328:951–961. doi: 10.1016/s0022-2836(03)00321-8. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Blaber M. Experimental support for the evolution of symmetric protein architecture from a simple peptide motif. Proc Natl Acad Sci U S A. 2011;108:126–130. doi: 10.1073/pnas.1015032108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J, Blaber M. Structural basis of conserved cysteine in the fibroblast growth factor family: evidence for a vestigial half-cystine. J Mol Biol. 2009;393:128–139. doi: 10.1016/j.jmb.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Lee J, Dubey VK, Longo LM, Blaber M. A logical OR redundancy within the Asx-Pro-Asx-Gly type I beta-turn motif. J Mol Biol. 2008;377:1251–1264. doi: 10.1016/j.jmb.2008.01.055. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Blaber SI, Dubey VK, Blaber M. A polypeptide “building block” for the beta-trefoil fold identified by “top-down symmetric deconstruction”. J Mol Biol. 2011;407:744–763. doi: 10.1016/j.jmb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Srimathi T, Kumar TK, Kathir KM, Chi YH, Srisailam S, Lin WY, Chiu IM, Yu C. Structurally homologous all beta-barrel proteins adopt different mechanisms of folding. Biophys J. 2003;85:459–472. doi: 10.1016/S0006-3495(03)74491-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manning MC, Illangasekare M, Woody RW. Circular dichroism studies of distorted alpha-helices, twisted beta-sheets, and beta turns. Biophys Chem. 1988;31:77–86. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]
- 32.Khan MY, Villanueva G, Newman SA. On the origin of the positive band in the far-ultraviolet circular dichroic spectrum of fibronectin. J Biol Chem. 1989;264:2139–2142. [PubMed] [Google Scholar]
- 33.Berova N, Nakanishi K, Woody RW. Circular dichroism: principles and applications. 2nd ed. Wiley; 2000. [Google Scholar]
- 34.Mach H, Middaugh CR. Interaction of partially structured states of acidic fibroblast growth factor with phospholipid membranes. Biochemistry. 1995;34:9913–9920. doi: 10.1021/bi00031a013. [DOI] [PubMed] [Google Scholar]
- 35.Mach H, Ryan JA, Burke CJ, Volkin DB, Middaugh CR. Partially structured self-associating states of acidic fibroblast growth factor. Biochemistry. 1993;32:7703–7711. doi: 10.1021/bi00081a015. [DOI] [PubMed] [Google Scholar]
- 36.Pineda-Lucena A, Nunez De Castro I, Lozano RM, Munoz-Willery I, Zazo M, Gimenez-Gallego G. Effect of low pH and heparin on the structure of acidic fibroblast growth factor. Eur J Biochem. 1994;222:425–431. doi: 10.1111/j.1432-1033.1994.tb18881.x. [DOI] [PubMed] [Google Scholar]
- 37.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 38.Matulis D, Lovrien R. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys J. 1998;74:422–429. doi: 10.1016/S0006-3495(98)77799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joshi S, Bhambhani A, Zeng Y, Middaugh CR. Formulation and process development strategies for manufacturing biopharmaceuticals. Wiley & Sons Publication; 2010. [Google Scholar]
- 40.Linemeyer DL, Menke JG, Kelly LJ, DiSalvo J, Soderman D, Schaeffer MT, Ortega S, Gimenez-Gallego G, Thomas KA. Disulfide bonds are neither required, present, nor compatible with full activity of human recombinant acidic fibroblast growth factor. Growth Factors. 1990;3:287–298. doi: 10.3109/08977199009003671. [DOI] [PubMed] [Google Scholar]
- 41.Blaber M, DiSalvo J, Thomas KA. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry. 1996;35:2086–2094. doi: 10.1021/bi9521755. [DOI] [PubMed] [Google Scholar]
- 42.Gimenez-Gallego G, Conn G, Hatcher VB, Thomas KA. The complete amino acid sequence of human brain-derived acidic fibroblast growth factor. Biochem Biophys Res Commun. 1986;138:611–617. doi: 10.1016/s0006-291x(86)80540-x. [DOI] [PubMed] [Google Scholar]
- 43.Cuevas P, Carceller F, Ortega S, Zazo M, Nieto I, Gimenez-Gallego G. Hypotensive activity of fibroblast growth factor. Science. 1991;254:1208–1210. doi: 10.1126/science.1957172. [DOI] [PubMed] [Google Scholar]
- 44.Brych SR, Blaber SI, Logan TM, Blaber M. Structure and stability effects of mutations designed to increase the primary sequence symmetry within the core region of a beta-trefoil. Protein Sci. 2001;10:2587–2599. doi: 10.1110/ps.ps.34701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zazo M, Lozano RM, Ortega S, Varela J, Diaz-Orejas R, Ramirez JM, Gimenez-Gallego G. High-level synthesis in Escherichia coli of shortened and full-length human acidic fibroblast growth factor and purification in a form stable in aqueous solutions. Gene. 1992;113:231–238. doi: 10.1016/0378-1119(92)90400-j. [DOI] [PubMed] [Google Scholar]
- 46.Lee J, Blaber M. Increased functional half-life of fibroblast growth factor-1by recovering a vestigial disulfide bond. J Proteins Proteomics. 2010;1:37–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.