

Abstract
Chromosomal inversions are usually portrayed as simple two-breakpoint rearrangements changing gene order but not gene number or structure. However, increasing evidence suggests that inversion breakpoints may often have a complex structure and entail gene duplications with potential functional consequences. Here, we used a combination of different techniques to investigate the breakpoint structure and the functional consequences of a complex rearrangement fixed in Drosophila buzzatii and comprising two tandemly arranged inversions sharing the middle breakpoint: 2m and 2n. By comparing the sequence in the breakpoint regions between D. buzzatii (inverted chromosome) and D. mojavensis (noninverted chromosome), we corroborate the breakpoint reuse at the molecular level and infer that inversion 2m was associated with a duplication of a ∼13 kb segment and likely generated by staggered breaks plus repair by nonhomologous end joining. The duplicated segment contained the gene CG4673, involved in nuclear transport, and its two nested genes CG5071 and CG5079. Interestingly, we found that other than the inversion and the associated duplication, both breakpoints suffered additional rearrangements, that is, the proximal breakpoint experienced a microinversion event associated at both ends with a 121-bp long duplication that contains a promoter. As a consequence of all these different rearrangements, CG5079 has been lost from the genome, CG5071 is now a single copy nonnested gene, and CG4673 has a transcript ∼9 kb shorter and seems to have acquired a more complex gene regulation. Our results illustrate the complex effects of chromosomal rearrangements and highlight the need of complementing genomic approaches with detailed sequence-level and functional analyses of breakpoint regions if we are to fully understand genome structure, function, and evolutionary dynamics.
Keywords: inversion, breakpoint, Drosophila, BAC, shotgun sequencing, transposable elements
Introduction
Changes in the structure of chromosomes have long been recognized as significant for the evolution of eukaryotes (Sturtevant 1917; Dobzhansky 1947; Sperlich and Pfriem 1986), and their role in speciation, adaptation, evolution of sex chromosomes, and also in human and animal disease is widely documented (Murphy et al. 2005; Lindsay et al. 2006; Darai-Ramqvist et al. 2008; Hoffmann and Rieseberg 2008; Chen et al. 2010; Kirkpatrick 2010). In recent years, a renewed interest in the study of chromosomal rearrangements has arisen as a consequence of the availability of genome sequences (Hoffmann and Rieseberg 2008; Hurles et al. 2008; Kirkpatrick 2010). The increased resolution of comparative genomic approaches has revealed that structural variants in general and chromosomal inversions in particular are much more common than previously thought (Coghlan et al. 2005; Feuk et al. 2005; Hoffmann and Rieseberg 2008; Kirkpatrick 2010). Genomic comparisons are also revealing that rather than being simple two-breakpoint rearrangements changing gene order, inversions may often entail gene duplications that can lead to genetic novelty and adaptation (Ranz et al. 2007; Kaessmann 2010; Furuta et al. 2011). Given the prevalence of chromosomal rearrangements and their associated functional consequences, understanding the mechanisms that generate them and their functional impact is necessary in order to understand the structure, function, and evolution of eukaryotic genomes.
The analyses of the chromosomal inversion breakpoints identified so far suggest that several mechanisms underlie their generation. For example, inversions can be generated by ectopic recombination between “homologous sequences” present at different chromosomal sites in opposite orientation (Petes and Hill 1988; Lim and Simmons 1994). The “homologous sequences” can be, for example, copies of transposable elements (TEs) that belong to the same TE family (Cáceres et al. 1999; Evans et al. 2007; Delprat et al. 2009), segmental duplications (Coulibaly et al. 2007), or tandemly arranged short repeats (Richards et al. 2005; Lobo et al. 2010). Inversions can also be generated by two simultaneous breaks in the same chromosome and repair by nonhomologous end joining (NHEJ), which sometimes leads to the duplication of sequences present at the breakpoints (Kehrer-Sawatzki et al. 2005; Matzkin et al. 2005; Sharakhov et al. 2006; Ranz et al. 2007). Because only a limited number of inversion breakpoints have been characterized so far, it is likely that additional analyses may elucidate novel molecular mechanisms generating them. Increasing our understanding of the mechanisms generating rearrangements should also improve our ability to design strategies to detect rearrangements, which is particularly interesting for rearrangements associated with human diseases (Chen et al. 2010).
The increased resolution of the techniques used to study chromosomal rearrangements also suggests that chromosome breakage is nonrandom. Breakpoint reuse, a term used as a shorthand for close clustering of breakpoints (Pevzner and Tesler 2003) appears to be a common phenomenon in eukaryotes (Pevzner and Tesler 2003; Bourque et al. 2004; Murphy et al. 2005; Gordon et al. 2007). However, there is some controversy about whether the identified cases of breakpoint reuse are real cases of reuse or a consequence of the limited resolution of the genomic studies since breakpoints are usually only assignable to regions spanning a few hundred kilobases (Kehrer-Sawatzki and Cooper 2008; Sankoff 2009). Here, we will use the terms “breakpoint clustering” and “breakpoint reuse” nonsynonymously to refer to the apparent reuse of breakpoints due to clustering and to the actual reuse at the molecular level, respectively. Therefore, a precise identification and characterization of sequences at inversion breakpoint sites and of adjacent sequences is needed in order to shed light on the nonrandom distribution of rearrangement breakpoints.
Detailed analyses of inversion breakpoint sequences are also needed in order to elucidate the functional consequences of chromosomal inversions. Several hypotheses have been proposed to explain the adaptive significance of inversions. First, inversions might have functional consequences due to the mutational impact of breakpoints (Sperlich and Pfriem 1986). Inversions might affect the expression, structure, and function of genes by removing or exchanging the regulatory regions of genes adjacent to the breakpoints, by disrupting the genes spanning the breakpoints, or by creating chimeric genes (Puig et al. 2004; Kehrer-Sawatzki and Cooper 2007; Hurles et al. 2008). The functional impact of inversions could also be due to their recombination-reduction effect (Sturtevant and Beadle 1936; Navarro and Ruiz 1997; Navarro et al. 1997). In heterozygotes, crossing-over and recombination are reduced inside the inverted segment and the alleles of the genes inside the inversion tend to be inherited as a single block. Accordingly, an inversion might be adaptive if it captures a favorable allele combination (Dobzhansky 1970; Charlesworth 1974) or a set of locally adapted genes (Kirkpatrick and Barton 2006). An inversion could also be favored if it captured a set of genes relatively free of recessive deleterious mutations (Nei et al. 1967). Alternatively, functional consequences of inversions might be due to their role in the formation and maintenance of selfish gene complexes that increase in frequency in populations through meiotic drive (Novitski 1967; Kirkpatrick and Barton 2006; Presgraves et al. 2009).
Drosophila is a good model organism to study the molecular mechanisms and the functional consequences of chromosomal inversions. Chromosomal inversions are particularly abundant in Drosophila both as intraspecific polymorphisms and as fixed differences between species (Krimbas and Powell 1992). Within the Drosophila genus, the species of the repleta group are particularly good models to study genome evolution because, besides the wealth of information on genome rearrangements and the availability of genomic resources, the species of this group have been used for studies of ecological adaptation and speciation for 70 years (Spencer 1941; Wharton 1942; Barker 1982; Manfrin and Sene 2006). Finally, the wealth of genetic tools and functional information available for Drosophila (Roy et al. 2010) facilitate the analysis of the potential functional consequences of inversion breakpoints in species of this genus.
Here, we used the Bacterial Artificial Chromosome (BAC) library and physical map of the Drosophila buzzatii genome (González et al. 2005) and the genome sequence of D. mojavensis (Clark et al. 2007), both species belonging to the repleta group, to isolate and characterize the breakpoints of a complex rearrangement of chromosome 2 fixed in all species of the buzzatii complex. This rearrangement comprises inversions 2m and 2n that were described by cytological studies as arranged in tandem and sharing the middle breakpoint (Ruiz and Wasserman 1993; González et al. 2007). The aims of this study are: 1) to determine the mechanisms that generated 2m and 2n fixed inversions, 2) to elucidate whether the described breakpoint clustering is in fact a breakpoint reuse at the sequence level, and 3) to assess potential functional effects of these inversions. To accomplish these aims we combined the classical in situ hybridization technique with shotgun sequencing of BAC clones, comparative genomics analyses and bioinformatic and experimental gene expression analyses. The combined results of these analyses allowed us to shed light not only into the mechanism that generated 2mn rearrangement but also on its functional consequences.
Materials and Methods
Drosophila Stocks
D. buzzatii stock st-1 (González et al. 2005), D. repleta stock 15084-1611.06 from Siboney, Cuba (University of California [UC] San Diego Drosophila Stock Center) and D. mojavensis stock 15081-1352.22 from Catalina Island, CA (UC San Diego Drosophila Stock Center) were used in this work. Additionally, the genomes of the 12 Drosophila species sequenced were used as reference in the comparative analyses (Clark et al. 2007).
D. buzzatii BAC Clones
BAC clones putatively encompassing inversion breakpoints were selected from the genomic library CHORI-225 (http://bacpac.chori.org), using the physical map of the D. buzzatii genome (González et al. 2005, 2007) available at the Genome Sciences Centre website (http://www.bcgsc.ca/ice). Clones containing breakpoints were identified because they produce a single signal when hybridized to D. buzzatii inverted chromosomes and two signals when hybridized to the noninverted chromosomes of D. repleta.
BAC Walking
Primers were designed in the D. mojavensis genome preferentially in coding regions using PRIMER software (supplementary table S6 in additional file 3, Supplementary Material online). Amplification reactions were carried out in a total volume of 50 μl: 50–200 ng of DNA as template, 40 pmols of each primer, 200 μM of dNTPs, 1.5 mM Mg2Cl, and 1 U of Taq DNA Polymerase (Platinum Taq DNA Polymerase High Fidelity; Invitrogen; Carlsbad, CA). When polymerase chain reactions (PCRs) were carried out using DNA from a species different from that used to design the primers as template, the annealing temperature of the PCR was set 5 °C lower than the suggested temperature to account for putative nucleotide mismatches.
In Situ Hybridization
In situ hybridizations were carried out as in Montgomery et al. (1987). Probes were labeled with biotin-16-dUTP (Roche Diagnostics; Mannheim Germany) by random primed labeling and hybridized to D. buzzatii and D. repleta salivary gland chromosome squashes. Interspecific hybridizations were performed at 25 °C, while the intraspecific ones were performed at 37 °C. The localization of the hybridization signals was done using the cytological maps of D. buzzatii (Ruiz and Wasserman 1993; González et al. 2005, 2007). Two types of probes were used, BAC clones from the D. buzzatii CHORI-225 library (González et al. 2005) and PCR products generated in the chromosome walk (supplementary table S7 in additional file 3, Supplementary Material online).
DNA Sequencing
PCR products were purified using QIAquick gel extraction kit (Qiagen; Valencia, CA, USA) and directly sequenced with the same primers used to amplify them. When no optimal sequencing chromatograms were obtained, the PCR products were cloned into the pGEM-T easy vector (Vector Systems I of Promega; Madison, WI) and sequenced using M13 forward and reverse primers. These PCR products were sequenced at Macrogen sequence service (http://www.macrogen.com). The same primers used to amplify the breakpoints were used for sequencing.
BAC ends were sequenced using T7 and SP6 primers of the vector pTARBAC2.1 at GATC-biotech sequence service (http://www.gatc-biotech.com). Two BAC clones were fully sequenced: BAC 1N19 (AC breakpoint) and 20O19 (EB breakpoint). Sequences have been submitted to GenBank (accession numbers JQ611723 and JQ611724). Random shotgun sequencing was provided by Macrogen (http://www.macrogen.com). Details of the sequencing process are given in supplementary table S9 in additional file 3 (Supplementary Material online). Gap closure was carried out in our lab. Two gaps were closed in BAC clone 20O19 using primer pair A (5′ TGTCGACTATAGTTAAGCGT 3′ and 5′ GGCAGTAGTCGTCGCATTAT 3′) and primer pair B (5′ GTGAGGCAATGCGTAACATT 3′ and 5′ CTTCTTGCTACGCATAATCT 3′). One gap was closed for BAC clone 1N19 using primer pair (5′ CTACGCAGATAAGCAGGCTT 3′ and 5′ AACTGTCAGCAGCAACGTGT 3′).
Similarity Searches and Sequence Annotation
Similarity searches against the D. mojavensis genome (CAF1 assembly released in February 2006) (Clark et al. 2007) were done using the BAC-end sequences as queries and PCR products generated in the chromosome walk and in the amplifications of the breakpoint regions. These searches were done with BlastN implemented at DroSpeGe (Gilbert 2007) and bl2seq implemented at NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The two sequenced BAC clones were completely annotated. The annotation was done using the online software gene predictors GeneID (Parra et al. 2000) and GENESCAN (Tiwari et al. 1997). Gene predictions were compared with the GLEANR models of the D. mojavensis genome annotation available at DroSpeGe (Gilbert 2007) and to D. melanogaster annotations available at FlyBase (http://flybase.org) (Tweedie et al. 2009). Alignments were performed using MUSCLE (Edgar 2004) available at the EBI website (http://www.ebi.ac.uk) and modified as needed to adjust splicing, start and stop codons and frames.
For the annotation of TEs and repeat sequences in the two sequenced BAC clones, similarity searches were carried out against the TE database in the DPDB website (Casillas et al. 2005), Repeatmasker (Repbase library based) (Jurka et al. 2005), and DroSpeGe (ReAs library based) (Gilbert 2007). This limited our search to TEs that are conserved among species. We also perform similarity searches against the nonredundant nucleotide database at the NCBI website (Tatusova and Madden 1999) to identify any potential TE sequence not included in TE databases. After this general search, a more detailed analysis of the breakpoint regions was performed using bl2seq (Tatusova and Madden 1999). In each case, our sequence was used as query and the sequence of the TE producing the best hit in the previous search was used as subject. Only hits with a minimum size of 40 bp and E value ≤ 1 × 103 were considered significant.
RepeatMasker also allowed us to look for AT-rich regions and low complexity repeats such CCG repeats. Bl2seq was also used to search for palyndromic sequences. BlastN and bl2seq were used to search for Conserved Noncoding Sequences (CNSs). Overrepresented conserved Transcription Factor Binding Sites (TFBSs) were searched using Whole Genome rVISTA implemented in http://genome.lbl.gov/vista webpage.
Dating the Duplication
We aligned the CDS of Dmoj/CG4673 and Dbuz/CG4673 and estimated Ks (number of synonymous substitutions per synonymous site) using DnaSP (Librado and Rozas 2009). Divergence time between this two species is 11.3 myr (Russo et al. 1995; Tamura et al. 2004) (Oliveira DCSG, Almeida FC, O'Grady P, Etges WJ, Armella MA, DeSalle R, personal communication). The rate of neutral evolution (r) per site per million years (r = Ks/2T) is therefore 0.0187 × 10−6. We then estimated the number of changes between the two duplicated regions (taking into account both coding and noncoding sequences), and we used r to estimate T.
RNA Extraction and RT-PCR
Total RNA was isolated from different developmental stages (0–1.30, 0–2.15, 0–2.30\, and 0–22 h embryos, larvae, pupae, adult males and females) of D. buzzatii with RNeasy Kit (Quiagen; Valencia, CA) and treated with 1 unit of DNase I (Applied Biosystems/Ambion; Austin, TX) for 30 min at 37 °C to eliminate DNA contamination. cDNA was synthesized with First Strand cDNA Synthesis Kit (Roche Diagnostics; Mannheim, Germany) using ∼500 ng of the DNase I-treated RNA and Oligo-dT primer (Promega; Madison, WI). RT-PCRs were performed with volumes as previously described for PCR adding 1 μl of the retrotranscriptase reaction. As a control of the reverse transcription reaction, Gapdh (438 bp) was amplified with H1 and H2 primers (data not shown) (Puig et al. 2004). Reactions without retrotranscriptase (RT-) were carried out to control for DNA contamination (data not shown).
The name of the primers used for RT-PCR corresponds to the region where they were designed for example primer 5R was designed in exon 5 (supplementary table S3 in additional file 3, Supplementary Material online). Only one RT-PCR was performed for D. buzzatii CG4673 with primers designed in exons 5 and 6. Four different RT-PCRs were performed for D. buzzatii ΨCG4673 that attempted to amplify the regions that showed homology with exons 2, 3, 7, and to the 3′UTR region (supplementary table S3 in additional file 3, Supplementary Material online). Position 3′ of these primers were located in a mismatch to amplify specifically ΨCG4673 in AC breakpoint. We sequenced the five RT-PCR products, and we confirmed that they corresponded to exons 5–6 of CG4673 and exons 2, 3, 7, and to the 3′ UTR of ΨCG4673.
Rapid Amplification of Complementary DNA End
Rapid Amplification of Complementary DNA End (RACE) experiments were carried out with DNase I-treated total RNA from embryos and adults of D. buzzatii using specific adapters and primers from FirstChoice RLM-RACE (Applied Biosystems/Ambion; Austin, TX, USA). For CG4673, 5′RACE primers were designed in the first exon (1L and 1Lint; supplementary table S3 in additional file 3, Supplementary Material online) and primers for 3′RACE in the last exon (8R and 8Rint; supplementary table S3 in additional file 3, Supplementary Material online). For ΨCG4673, 5′RACE primers were designed in exon 2 (2L and 2Lint; supplementary table S3 in additional file 3, Supplementary Material online) and primers for 3′RACE in exon 7 (7R and 7Rint; supplementary table S3 in additional file 3, Supplementary Material online).
Northern Blot
Northern blot hybridization was performed as described in De et al. (1990). RNA from embryos, larvae, pupae, adult males and females was used. Two different membranes were hybridized one with 5–10 μl (∼1,300 ng/μl) of total RNA and the other one with 1–3 μl (∼1,300 ng/μl) of total RNA. Samples were loaded with 3V of Northern Formaldehyde Load Dye (Applied Biosystems/Ambion; Austin, TX). Running buffer 10× for formaldehyde gels (Applied Biosystems/Ambion; Austin, TX, USA) was used to run the gels in denaturalizing conditions. The probe was amplified with primers 5R and 7L (supplementary table S3 in additional file 3, Supplementary Material online). Hybridization was performed with 5 μl (15.3 ng/μl) of the [32P]UTP-labeled probe. Hybridization was carried out for 16 h at 65°C with Perfect Hyb Plus buffer (Sigma–Aldrich; St. Louis, MO). Two washes with 2× saline sodium citrate/ 0.1% sodium dodecyl sulfate were done before exposition.
Double-Stranded RNA Detection
Detection of double-stranded RNA (dsRNA) was carried out as in Aravin et al. (2007). RT-PCRs were performed with RNA of embryos and adults of st-1 line of D. buzzatii. Total RNA was treated with RNase ONE (Promega; Madison, WI) to degrade single-stranded RNA (D+). RNase-treated RNA was denatured and retrotranscribed as described for RT-PCR. Negative controls without retrotranscriptase (RT−), without RNase (D−), without the denaturalizing step (N) and using Oligo-dT primer (T) instead of random primers were performed. dsRNA detection were carried out with the primers 2R and 2L, 3R and 3L, 7R and 7L, and 3′ UTR-R, 3′UTR-L and Gapdh primers H1 and H2 as described for RT-PCR (supplementary table S3 in additional file 5, Supplementary Material online).
Results
High-Resolution Mapping and Isolation of Inversion Breakpoints
Following previous works (Wesley and Eanes 1994; Cáceres et al. 1999), the three breakpoint regions of inversions 2m and 2n were tentatively designated as AB, CD, and EF to refer to breakpoints in the noninverted chromosome, and AC, BE, and DF to describe breakpoints in the inverted chromosome (fig. 1). In both cases, A represents the most distally located region relative to the centromere, whereas F stands for the most proximal region.
Fig. 1.

Schematic representation of chromosome 2 arrangements in Primitive I (the “repleta” group ancestor), D. buzzatii and D. mojavensis. Three inversions took place between Primitive I and D. buzzatii. Inversions 2m and 2n share the middle breakpoint. Inversion 2z7 took place on a 2mn chromosome inverting BE breakpoint region. Seven inversions (not shown in this scheme) took place from Primitive I to D. mojavensis (2c, 2f, 2g, 2h, 2q, 2r, and 2s) and as a result, inversion breakpoint region CD is inverted in D. mojavensis.
To clone and sequence the 2m and 2n breakpoint regions, we used the BAC-based physical map of the D. buzzatii genome (González et al. 2005) and the genome sequence of D. mojavensis, the closest relative to D. buzzatii whose genome has been fully sequenced (Clark et al. 2007). We followed a procedure similar to that devised by Prazeres da Costa et al. (2009) comprising four experimental steps. 1) We used in situ hybridization to identify D. buzzatii BAC clones encompassing the breakpoints (table 1 and supplementary fig. S1 in additional file 1, Supplementary Material online). 2) We sequenced the ends of the identified BAC clones, and we located the sequences in the D. mojavensis genome, which allowed us to narrow down the position of the breakpoints to ∼100–150 kb regions (fig. 2). 3) We walked along the D. mojavensis genome by designing DNA probes and hybridizing them to the D. buzzatii chromosomes, which allowed us to further delimit the breakpoint regions in D. mojavensis (fig. 2): AB breakpoint region was located in ∼10 kb fragment between msi (A) and Ssadh (B); CD breakpoint was located in a ∼1.5 kb region between scrib (C), and Or98b (D); and EF breakpoint region was located in a ∼2.1 kb region between Wsck (E) and CG8147 (F). 4) We isolated the breakpoint regions in D. buzzatii by PCR using primers designed in D. mojavensis in the expected orientation according to figure 1 (Supplementary tables S8 in additional file 1, Supplementary Material online). Although the last step was attempted for the three breakpoints with different primers, it only worked out for DF breakpoint region. In D. buzzatii, the region between Or98b (D) and CG8147 (F), and containing DF breakpoint is 4.4 kb long (GenBank accession number JQ611725). Comparison of this 4.4 kb region with CD and EF regions of D. mojavensis further narrowed down the breakpoint to a ∼3.1 kb segment. For breakpoint regions AC and BE, we had to resort to an alternative step. 5) We sequenced the whole BAC clones containing the AC and BE inversion breakpoints (fig. 3).
Table 1.
D. buzzatii BAC Clones Containing the Breakpoints of Inversions 2m and 2n.
| Breakpoint Region | BAC | Size (base pairs)a | Cytological Coordinates in D. buzzatiib | Cytological Coordinates in D. repleta |
|---|---|---|---|---|
| AC | 1N19 | 138,197 | D3e/F2a | D3e and F2a |
| AC | 14B19c | 140,515 | D3e/F2a | D3e and F2a |
| AC | 15L19 | 133,077 | D3e/F2a | D3e and F2a |
| BE | 20O19c | 142,697 | D3e/F6h | D3e and F6h |
| BE | 22B03 | 128,945 | D3e/F6h | D3e and F6h |
| BE | 2N19 | 191,034 | D3e/F6h | D3e and F6h |
| DF | 16H04c | 124,185 | F6h/F2a | F6h and F2a |
| DF | 14E21 | 195,292 | F6h/F2a | F6h and F2a |
| DF | 8C14 | 156,485 | F6h/F2a | F6h and F2a |
a Estimates of BAC insert sizes taken from the D. buzzatii BAC-based physical map available at the Genome Sciences Centre (http://www.bcgsc.ca/ice).
b Cytological coordinates refer to the cytological map of D. repleta chromosome 2.
c These clones were identified by González et al. (2007).
Fig. 2.

Schematic view of the BAC walking through D. mojavensis genome to map the breakpoints of inversions 2m and 2n. Nucleotide coordinates above the gene structure map are from scaffold_6540 of D. mojavensis (Schaeffer et al. 2008). Black arrows show where the sequenced BAC ends were anchored. The discontinuous lines show the length of the BAC clones and therefore the maximum distance where the breakpoints can be located. Black boxes indicate successfully hybridized PCR probes, while gray boxes are probes that did not yield any hybridization signal. White arrows show the location of the different primers designed to attempt the amplification of the breakpoints in D. buzzatii (not drawn to scale).
Fig. 3.

Annotation of the sequenced D. buzzatii BACs 1N19 and 20O19 by comparison with the homologous regions in scaffold 6540 of D. mojavensis. Shaded rectangles include homologous regions between the two species. See supplementary additional data file 2 (Supplementary Material online) for further information.
BAC clone 1N19 is ∼134 kb long and contains complete coding sequence (CDS) of three genes and partial CDS for another two genes (fig. 3; GenBank accession number JQ611723). Clone 20O19 is ∼143 kb long and contains full CDS of 22 genes and partial CDS of another one (fig. 3; GenBank accession number JQ611724). Almost all genes (with the exception of CG4673; see below) have a similar exon–intron structure and identical orientation in D. buzzatii and D. mojavensis (fig. 3). By comparing these two BAC clones to D. mojavensis genome sequence, we confirmed that each clone contains a single synteny interruption (breakpoint). Instead of being located between msi (A) and scrib (C), as expected according to D. mojavensis chromosome annotation (fig. 2), in D. buzzatii AC breakpoint was located in a ∼15 kb region between CG12250 (A) and scrib (C). This is so because, in D. buzzatii, CG12250 and msi are overlapping instead of nested genes (fig. 3). Manual reannotation of CG12250 in D. mojavensis revealed that CG12250 and msi are also overlapping in this species (fig. 3). Comparison of this 15-kb region with AB and CD regions of D. mojavensis allowed us to further narrow down the breakpoint to a ∼700 bp segment. Finally, in D. buzzatii, BE breakpoint is located between genes Wsck (E) and Ssadh (B) in a ∼8 kb region (fig. 3). Comparison of this ∼8 kb region with AB and EF regions of D. mojavensis located the breakpoint in a ∼1.1 kb segment. A more detailed description of this experimental procedure is given in supplementary additional file 2 (Supplementary Material online).
Sequence Analysis of Breakpoint Regions in D. buzzatii and D. mojavensis
We performed a detailed analysis of the three breakpoint regions including annotation of 1) genes, 2) gene-associated sequences, 3) TE insertions, and 4) other structural features (fig. 4).
Fig. 4.

Annotation of breakpoint regions of inversions 2m and 2n for D. mojavensis (AB, CD, and EF) and D. buzzatii (AC, EB, and DF). CDS, TE fragments, duplicated and conserved sequences are shown. Lines under black arrows show the breakpoint junctions.
Gene Annotation
In D. mojavensis, the AB breakpoint region is located between CG12250 and Ssadh and contains three genes: CG5079 and CG5071 nested within intron 6 of CG4673 (fig. 4).
In D. buzzatii, the AC breakpoint region comprises the region between CG12250 and scrib, which is 15,343 bp long (fig. 4). Only one putatively functional ORF was detected in this region corresponding to CG5071, located 6,585 bp upstream of the CG12250 start codon. In D. buzzatii, CG5071 is a 2,004-bp long gene and encodes a 668 aa-long protein. Thus, in D. buzzatii, the coding region of this gene is considerably longer than in D. mojavensis (225 aa) and similar to that of D. melanogaster annotated CDS CG5071-RB (680 aa).
A small fragment with similarity to Dmoj\CG5079 was detected in D. buzzatii AC region (fig. 4). However, this fragment contains multiple stop codons and was therefore considered to be a pseudogene (ΨCG5079). Additionally, 16 small blocks (45–273 bp long) showing 65.2–88.9% identity to Dmoj/CG4673 were detected in this region (fig. 4 and supplementary table S1 in additional file 3, Supplementary Material online). The distance between the most remote CG4673 hits in the AC breakpoint region is 12,585 bp, which is similar to the distance between the orthologous hits in D. mojavensis (12,578 bp). However, we could not identify the start or stop codon of CG4673 in this region of the D. buzzatii genome and concluded that a degenerated, seemingly nonfunctional, copy of CG4673 (ΨCG4673) is present in this breakpoint region (fig. 4).
The D. buzzatii EB breakpoint region is located in the 8,242-bp long intergenic region between Wsck and Ssadh (fig. 4). A seemingly complete copy of gene CG4673 is present between Wsck and Ssadh (supplementary table S2 in additional file 3, Supplementary Material online). This gene putatively encodes three transcripts that are similar in exon number and exon size to those present in D. mojavensis and D. melanogaster (supplementary table S2 in additional file 3, Supplementary Material online). However, D. buzzatii gene is considerably shorter than D. mojavensis and D. melanogaster genes due to a 20-fold size reduction of intron 6 (table 2 and supplementary table S2 in additional file 3, Supplementary Material online). The CG4673 coding sequence is highly conserved (∼79% identity with D. melanogaster and ∼90% identity with D. mojavensis; table 2). Estimates of the ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site (ω) average 0.05 indicating that the three transcripts are subject to strong purifying selection (table 2).
Table 2.
Comparison of CG4673 Gene of D. buzzatii (EB breakpoint region) with D. mojavensis and D. melanogastera.
|
Dbuz/Dmoj |
Dbuz/Dmel |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Region | Dbuz (base pairs) | Dmoj (base pairs) | Dmel (base pairs) | Identity (%) | Ka/Ks | ω | Identity (%) | Ka/Ks | ω |
| RA transcript | 3,015 | 11,726 | 12,290 | — | — | — | — | — | — |
| RA CDSb | 1,968 | 1,962 | 1,959 | 90.6 | 0.0235/0.4218 | 0.0557 | 79.6 | 0.0723/1.4327 | 0.0505 |
| RB/RD transcriptc | 3,701 | 12,418 | 13,317 | — | — | — | — | — | — |
| RB/RD CDS | 1,881 | 1,875 | 1,875 | 90.0 | 0.0259/0.4363 | 0.0593 | 78.9 | 0.0728/1.4874 | 0.0489 |
a In D. melanogaster, CG4673 encodes three strongly supported transcripts, RA, RB, and RD. In D. mojavensis, only RA transcript is annotated; however, a detailed analysis of this region allowed us to annotate the other two transcripts.
b Not including UTRs.
c Transcripts RB and RD differ in the UTR regions.
The presence of CG4673 in EB region reveals that the region containing this gene was duplicated in the D. buzzatii lineage. However, no traces of the two genes (CG5079 and CG5071) nested within intron 6 of Dmoj\CG4673 were found within Dbuz\CG4673 intron 6 suggesting that these genes were deleted after the duplication. Two pieces of evidence suggest that the duplication of the region containing CG4673 gene occurred at the same time as the inversion. First, this duplication is present in other species of the buzzattii complex that also have the 2mn rearrangement (data not shown) but it is not present in the genome sequence of the other 12 Drosophila species that lack this rearrangement (Clark et al. 2007). Second, based on sequence analyses, we estimated that the duplication took place ∼8 Ma, which also agrees with the duplication taking place at the same time as the inversion, since the inversion must have happened prior to the divergence of the species of the buzzatti complex (∼8.4 Ma) and after the divergence between the buzzatii complex species from D. mojavensis (∼11.3 Ma) (Russo et al. 1995; Tamura et al. 2004) (Oliveira DCSG, Almeida FC, O'Grady P, Etges WJ, Armella, R. DeSalle MA, personal communication).
Furthermore, other than the duplication, a microinversion also happened in this genomic region. Taking into account the orientation of the genes in D. mojavensis, we would expect Ssadh and CG4673 to be in the same orientation in D. buzzatii. However, the orientation of CG4673 is reversed relative to what is expected. Note also that the duplicated region is flanked by a 121-bp long duplicated sequence with 72.7% identity (E value = 2 × 1016) (fig. 4). However, this duplicated sequence is found only once in D. mojavensis AB region and in D. buzzatii AC region, and in both cases, it is located upstream of CG4673 (fig. 4) suggesting that it was not duplicated before the microinversion.
Finally, DF breakpoint in D. buzzatii is located in a 4,400 bp segment between Or98b and CG8147 (fig. 4). When the DF sequence was compared with the CD region of D. mojavensis, two blocks with significant similarity were detected, one is 408 bp long with 87.7% identity (E value = 2 × 10147) and another one is 93 bp long with 70.9% identity (E value = 9 × 10). Although these conserved sequences do not correspond to the coding region of Or98b or any other known gene, they are located upstream of Or98b in D. mojavensis and might contain enhancers or other regulatory elements for this gene.
Gene-Associated Sequences
We looked for highly CNSs and TFBSs in the breakpoint regions to check whether the inversion disrupted or changed their location. Neither CNSs nor TFBSs were found in any of the three breakpoint regions.
TE Annotation
In D. mojavensis, six blocks of similarity with known Helitrons (Feschotte and Pritham 2007) were found in the CD region, while no similarities to any TE were found either in AB or EF regions (fig. 4). In the three breakpoint junctions of D. buzzatii, we found small blocks of similarity with BuT5 element (fig. 4 and table 3) (Casals et al. 2006). In the D. buzzatii AC breakpoint region, two other fragments similar to ISBu2, two similar to BS2, and finally, two more similar to Helitron1 were detected upstream of CG5071, between the most remote fragments of ΨCG4673 (fig. 4). ISBu elements are also Helitrons (Yang and Barbash 2008). BS2 is a LINE element (Capy 1998) and the only class I element found in these regions. Finally, another fragment similar to ISBu2 and two fragments with identity with transib3 (Kapitonov and Jurka 2003) of D. melanogaster were annotated in the DF breakpoint junction.
Table 3.
TEs detected at the breakpoint regions of inversions 2m and 2n in D. buzzatii.
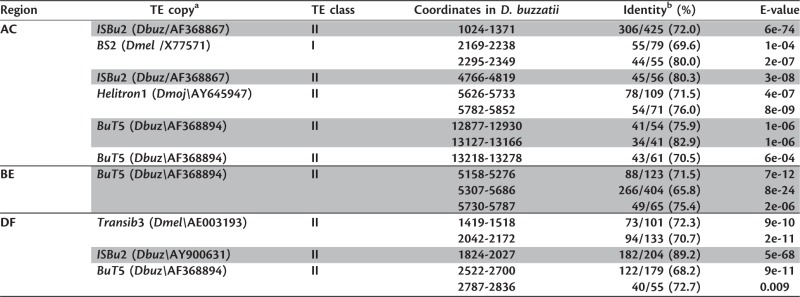 |
a The species and the accession number that allowed us to identify each TE copy is given in parentheses.
b Only hits with a minimum size of 40 bp and E-value ≤ 1e-03 are included in this list.
Annotation of Other Structural Features
Using several bioinformatic tools, we searched for structural features in the D. buzzatii breakpoint regions, such as AT-rich regions, palindromic sequences, or CCG repeats, that may contribute to chromosomal instability (Kurahashi et al. 2007; Zhang and Freudenreich 2007; Kolb et al. 2009). We did not find any sequence that suggests that the breakpoint regions are predisposed to breaks.
Analysis of the Breakpoint Regions in the 12 Drosophila Sequenced Genomes
The gene arrangements at the breakpoint regions of inversions 2m and 2n were also investigated in the 12 Drosophila sequenced genomes (supplementary fig. S2 in additional file 1, Supplementary Material online). The noninverted arrangement of D. mojavensis (AB, CD, and EF) was also found in D. pseudoobscura and D. persimilis (Sophophora subgenus) and D. virilis and D. grimshawi (Drosophila subgenus) suggesting that this is probably the ancestral gene arrangement of the genus. The order and orientation of the genes in the AB breakpoint region is conserved in all 12 sequenced species suggesting that this breakpoint region has been interrupted in D. buzzatii and its close relatives only. Region CD has been rearranged two other times: once in the lineage leading to the melanogaster group and once in the lineage leading to D. willistoni. Other than in D. buzzatii, the EF breakpoint region has also been rearranged in the lineage leading to the melanogaster group of species.
Expression Analysis of CG4673 and ΨCG4673 in D. buzzatii
As a first approximation to the study of the possible functional consequences of 2mn rearrangement, we analyzed the expression of CG4673 and ΨCG4673. Specifically, we used a combination of bioinformatic and experimental techniques in order to shed light on the expression, molecular structure, and function of D. buzzatii CG4673 gene and ΨCG4673 (see Materials and Methods).
Transcription Analyses
We first performed RT-PCR to check whether CG4673 and/or ΨCG4673 are transcribed (supplementary table S3 in additional file 3, Supplementary Material online). Specific primers for CG4673 and for ΨCG4673 were used (see Materials and Methods). RT-PCR reactions performed without retrotranscriptase yield no signal indicating the absence of DNA contamination (results not shown). Amplification products were obtained for both CG4673 and ΨCG4673 in embryos, larvae, pupae, and adults, revealing that both genes are transcribed in D. buzzatii (supplementary fig. S3 in additional file 1, Supplementary Material online).
A Northern Blot was also carried out to detect how many CG4673-related transcripts are present in D. buzzatii (supplementary fig. S4 in additional file 1, Supplementary Material online). Drosophila buzzatii total RNA extracted from different developmental stages was hybridized with a probe amplified with primers designed in exons 5 and 7 of CG4673. A clear and unique signal of approximately 2,100 bp, which corresponds to the expected size of CG4673 transcripts (∼2,000 bp; table 2), was observed in the hybridization with total mRNA of D. mojavensis. This result indicates that the probe did not hybridize nonspecifically with any other similar sequence, alternative transcript or unspliced mRNA.
A strong hybridization signal similar in size (∼2,100 bp) to that obtained in D. mojavensis was observed in all the lanes containing D. buzzatii mRNA (supplementary fig. S4 in additional file 1, Supplementary Material online). A smaller hybridization signal (∼1,400 bp) appears to be partially hidden in the majority of lanes. Finally, another hybridization signal (∼3,600 bp) was observed in the lane containing mRNA from pupae. We performed a second Northern Blot hybridization with a lower concentration of mRNA in an attempt to get more defined hybridization bands (supplementary fig. S4 in additional file 1, Supplementary Material online). However, in this second Northern Blot, the ∼2,100 bp signal was observed only in eggs and pupae. The smaller hybridization signal (∼1,400 bp) was clearly observed in pupae, and the ∼3,600-bp hybridization signal was not present. Overall, three different transcripts related with CG4673 (3,600, 2,100, and 1,400 bp long) were observed in D. buzzatii.
We hypothesized that two of the three identified transcripts correspond to the transcripts detected with RT-PCR: the 2,100-bp long transcript corresponds to the functional copy of CG4673 gene and the 3,600-bp long transcript to an unspliced form of ΨCG4673 (note that the splice sites are not conserved in ΨCG4673). The third transcript detected in the Northern Blot hybridization (∼1,400 bp) could be transcribed from the putative promoter annotated in the duplicated sequence (see below) that is located downstream and oriented toward CG4673 in D. buzzatii BE breakpoint (fig. 3). This antisense transcript would therefore be complementary with both transcripts found in the RT-PCR experiments and the annealing of the sense and antisense transcripts would form dsRNA molecules.
We tested whether we could detect dsRNA by performing RT-PCR with mRNA of D. buzzatii previously digested with RNase. RNase only digests single-strand RNA molecules and therefore, RT-PCR would only amplify if dsRNA were present. RT-PCRs were attempted with primers that specifically amplify exons 2, 3, and 7 of ΨCG4673. While no amplification was observed for ΨCG4673 exon 2, both exons 3 and 7 did amplify when digested samples were used (supplementary fig. S5 in additional file 1, Supplementary Material online). Therefore, exons 3 and 7 of ΨCG4673 of D. buzzatii would be protected from digestion with RNase suggesting that dsRNA molecules are present in D. buzzatii.
Transcription Start and Termination Sites
To identify the transcription start sites (TSS) of CG4673, we used Neural Network Promoter Prediction software (Reese 2001). We detected two putative TSS and their corresponding TATA boxes located 30 bp upstream of them (fig. 5). These TSS and TATA boxes were not conserved in D. mojavensis, D. grimshawi, or D. virilis. We also identify putative TSS from sequence conservation with closely related species (D. mojavensis, D. grimshawi, and/or D. virilis). When the sequence upstream of CG4673 of D. buzzatii was aligned with that of these species, two different conserved regions that could also act as TSS were identified with their corresponding TATA box and BR elements (binding site for transcription factors located immediately upstream of the TATA box; fig. 5). 5′ RACE experiments confirmed the presence of several TSS. We obtained a single band product but sequencing of this band resulted in overlapping chromatograms ∼200 bp upstream of the start codon.
FIG. 5.

Structure of the gene CG4673 in D. buzzatii. Polyadenylation signal (Poly-A), NPL4 domain, putative promoters (a–f), and putative TSS are shown. TSS refers to putative start sites found with Neural Network Promoter Prediction software. TSS* refers to putative start sites inferred from conservation with close species (for details, see text).
Using 3′ RACE, we identified the transcription end site of CG4673 in D. buzzatii 196 bp downstream of the stop codon (fig. 5). We also attempted to determine the start and end of the putative transcript of ΨCG4673. However, neither RACE experiments nor software analysis detected the putatively start or end of ΨCG4673 transcript.
Promoter Detection
We used the default threshold of McPromoter software (Ohler 2006) to find putative promoters in the 8.2 kb region between Ssadh and Wsck genes were CG4673 is located in D. buzzatii BE region. Two promoters, promoter e (score = 0.0407) and promoter f (score = 0.0506), were identified upstream and in the same orientation as CG4673 (fig. 5). Promoter e is located in the 121-bp duplicated sequence. Another four putative promoters were found in reverse orientation (fig. 5). Promoter b (score = 0.0351) and promoter c (score = 0.0367) were annotated in exonic sequence and are likely artifacts (note that these are the two promoters with lower scores). Promoter d (score = 0.0445) was located upstream of CG4673 and oriented toward Wsck gene. The most significant promoter, promoter a (score = 0.0555) was located in the 121-bp duplicated region in the 3′ intergenic region. Therefore, both duplicated regions in the BE breakpoint contain potential promoters.
No putative promoters were detected with McPromoter software in direct or reverse orientation for ΨCG4673 (AC breakpoint region).
CG4673 Function
Dbuz/CG4673 has a 79% nucleotide sequence identity with Dmel/CG4673 (table 2). In D. melanogaster, CG4673 has a zinc finger and a NPL4 domain (supplementary Table S4 in additional file 3, Supplementary Material online), and it is involved in nuclear transport and in the ubiquitin-proteosome degradation pathway (Bays and Hampton 2002). We searched for conserved domains in the putative protein encoded by Dbuz/CG4673 (Marchler-Bauer et al. 2009). We found similarity with a COG5100 domain (exons 2–7; E value = 3 × 10103), which corresponds to the NPL4 protein (Bays et al. 2001; Botta et al. 2001). This domain is composed of two subdomains: an active NPL4 domain (pfam05021, E value = 9 × 1092) and a zinc finger of the NPL4 superfamily (pfam05020, E value = 1 × 1065). The conservation of the functional domains in D. melanogaster and D. buzzatii proteins, suggest that CG4673 is also involved in nuclear transport and in the ubiquitin-proteosome degradation pathway in D. buzzatii (supplementary table S4 in additional file 3, Supplementary Material online).
Discussion
Detailed analyses of inversion breakpoints can help to explain not only processes related to chromosome evolution, such as whether breakpoint use is random or nonrandom, but also provide insight into the functional consequences of inversions. In this work, we have sequenced the inversion breakpoints of the 2mn rearrangement fixed in all species of the buzzatii complex. Our results shed light on the mechanism generating this rearrangement, corroborate the breakpoint reuse at the molecular level, and start uncovering its functional consequences.
Generation of Inversions 2m and 2n
Chromosomal inversions may be generated by diverse mechanisms and the features observed in the inversion breakpoints often provide clues for the mechanisms involved, supporting some of them and rejecting others. In this work, we found that the 2m inversion is associated with a ∼13 kb duplicated segment present at both breakpoints and was likely generated by staggered breaks followed by repair by NHEJ as depicted in figure 6. The phylogenetic distribution of the duplication and the estimate of the age of the duplication (based on sequence divergence analyses) are consistent with the duplication co-occurring with the inversion.
Fig. 6.
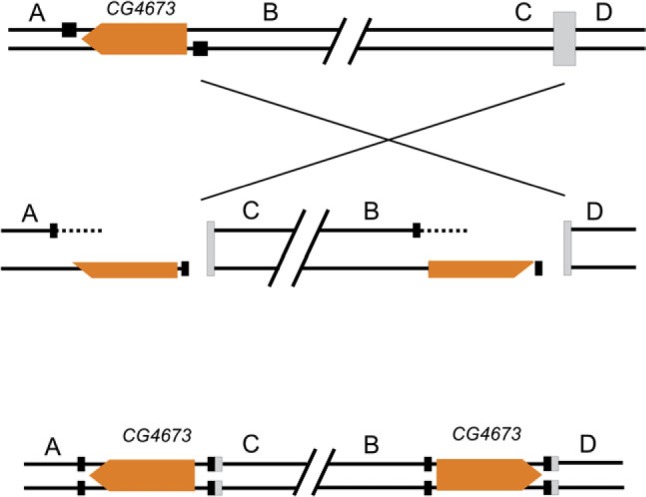
Schematic representation of the mechanism generating inversion 2m: single strand breaks (black squares) and double strand breaks (gray squares) followed by NHEJ repair.
The hybrid element insertion model (Gray et al. 1996; Gray 2000; Zhang and Peterson 2004) could explain inversion 2m and its associated duplication also as a single event if the participating BuT5 copies were originally inserted 5′ and 3′ of CG4673 in homologous chromosomes (supplementary fig. S6 in additional file 1, Supplementary Material online). However, this mechanism has only been described for a few transposon families and under particular conditions. Furthermore, BuT5 copies might have inserted in the 2m inversion breakpoint regions as secondary colonizers after the generation of the inversion as it has been reported previously (Cáceres et al. 2001; Casals et al. 2003; Delprat et al. 2009). Therefore, although an involvement of BuT5 in the generation of inversion 2m cannot be completely ruled out, we find the evidence unconvincing.
Alternative mechanisms, such as duplicative transposition of the ∼13 kb duplicated segment into the CD region followed by an event of ectopic recombination between the duplicated segments (inverting the region between the duplications) or duplication of the ∼13 kb region first and then the inversion breakpoint happening right in between of the two duplicated copies, are also possible. However, these two mechanisms are less parsimonious since they would require two independent events (the duplication and the inversion). Moreover, transposition is not as common as other types of rearrangements in Drosophila (Ranz et al. 2003; González et al. 2004; Bhutkar et al. 2007).
We have found few clues to explain the generation of inversion 2n. This inversion could have been generated by double strand breaks at both breakpoints and repair by NHEJ. As a matter of fact, as inversion 2m and 2n have never been found in isolation, we can not rule out the possibility that the two inversions were generated concurrently by the same mechanism (a three-point breakage followed by NHEJ repair). On the other hand, 2n might have resulted from ectopic recombination between BuT5 copies, as fragments of this transposon were found at both 2n breakpoints (BE and DF). However, the evidence is weak because these fragments are highly degraded and currently lack target site duplications.
The fixed inversions studied here are old (between ∼8.4 and 11.3 Ma) and some footprints of the events that generated them are not apparent anymore. This shortcoming could be overcome by studying younger polymorphic inversions. However, the study of fixed inversions should provide insights into what features make inversions successful whereas the evolutionary fate of polymorphic inversions is not known (as they might eventually be lost).
Breakpoint Reuse
Our molecular characterization of 2mn breakpoint regions has corroborated the breakpoint reuse previously suggested in this region (Ruiz and Wasserman 1993; González et al. 2007). If 2m and 2n were independent inversions but separated by a short distance, a small segment of the original CD region would be found in the middle breakpoint region of the derived chromosome. If 2m and 2n were overlapping by a short segment, this small segment would have been transferred between the distal and proximal breakpoints. None of these expectations hold true, that is, we could neither detect a small segment remaining in the middle breakpoint nor a small segment exchanged between distal and proximal breakpoints (fig. 4).
The detailed analysis of the 2mn breakpoint regions in D. buzzatii allowed us to identify an additional rearrangement, a microinversion, that took place in the EB breakpoint region. Furthermore, the comparison of the three 2mn breakpoint regions in the 12 Drosophila species sequenced (Clark et al. 2007) revealed that while AB region is conserved in the 12 species, CD and EF regions have been rearranged at least once during the evolution of the Drosophila genus (supplementary fig. S2 in additional file 1, Supplementary Material online) suggesting that these breakpoint regions have been repeatedly reused during evolution.
Although breakpoint clustering at the cytological level is common in Drosophila (González et al. 2007), only two other breakpoint reuse regions have been previously characterized at the sequence level (Richards et al. 2005; Ranz et al. 2007). Recurrent breakage might be revealing structural instability of these particular regions. However, the analyses of the breakpoint regions in D. buzzatii did not provide evidence for sequence features previously associated with fragile regions, such as AT-rich regions, CCG repeats, or palindromic sequences (Kurahashi et al. 2007; Zhang and Freudenreich 2007; Kolb et al. 2009). We neither found evidence for a high density of TE insertions in these regions. Alternatively, breakpoint reuse might be due not to an increase rate of generation of inversions in particular regions but to differential survival of inversions. Future analyses are necessary to unveil the actual causes of breakpoint reuse.
Functional Analyses of the Breakpoint Regions
The detailed analyses of the breakpoint regions also allowed us to shed light on the possible functional consequences of the 2mn rearrangement. We found that none of the breakpoints disrupt a coding region (fig. 4). Breakpoints could also affect the expression of nearby genes by disrupting, changing the location, or creating cis-regulatory elements. Although we did not find any evidence for CNSs or TFBSs in the breakpoint regions, the existence of species-specific regulatory regions cannot be ruled out since no polymorphism data is available for D. buzzatii.
Inversion 2m is associated with a 13 kb duplication of the region located between CG12250 and Ssadh and containing genes CG4673, CG5071, and CG5079 (fig. 4). CG5071 and CG5079 are nested within intron 6 of CG4673 and are flanked by the same two genes in all the other Drosophila genomes sequenced (Clark et al. 2007) indicating that this is the ancestral organization. However, the duplication of the region located between CG12250 and Ssadh and containing CG4673, CG5071, and CG5079 did not eventually lead to the duplication of any functional gene. In one of the duplicated fragments (AC breakpoint; fig. 4), CG4673 and CG5079 are now pseudogenes, while in the other duplicated fragment (BE breakpoint; fig. 4) CG5071 and CG5079 have been lost. Therefore, 2mn rearrangement eventually led to the net loss of one gene (CG5079). Although CG5079 is not associated with any gene ontology terms, RNAi analyses indicate that it might play a role in cell growth and viability (Boutros et al. 2004) suggesting that the loss of this gene might have functional consequences in D. buzzatii (supplementary table S4 in additional file 3, Supplementary Material online).
The ancestral copy of CG4673 has seemingly lost its function, while the derived copy is functional. This derived copy is much shorter than the ancestral one due to the loss of the two nested genes initially located in intron 6. Gene expression levels are negatively correlated with intron length, likely as a result of selection for transcriptional speed or economy (Castillo-Davis et al. 2002; Seoighe et al. 2005). As CG4673 is a highly expressed gene in D. melanogaster, we hypothesized that the loss of ∼9 kb in D. buzzatii increased the efficiency of transcription of this gene resulting in the conservation of the derived copy while the ancestral copy was allowed to degenerate.
Although the functional benefits of nested genes are not clear, gains of nested gene structures are more common than losses in Drosophila and other metazoans (Assis et al. 2008). It has been hypothesized that nested host gene structures present a mechanism for the coordinated regulation of functionally related gene pairs. However, many nested genes exhibit functions and expression patterns distinct from those of host genes suggesting that the gain of nested gene structures is neutral (Assis et al. 2008; Kumar 2009). This might also be the case for CG4673 and its two nested genes: CG4673 is involved in nuclear transport and in protein degradation while CG5071 has enzymatic activity and CG5079 might play a role in cell growth and viability (supplementary table S4 in additional file 3, Supplementary Material online). The level of expression of these three genes is not correlated (supplementary table S4 in additional file 3, Supplementary Material online). As hypothesized above, in D. buzzatii the loss of the nested structure in the derived copy might have contributed to the efficiency of expression of CG4673 leading to the degeneration of the ancestral copy of this gene.
CG4673 is flanked by a 121-bp long duplication that has promoter capability (fig. 5). We suggest that this 121 bp duplication was generated concurrently with the microinversion as a result of staggered single-strand breaks and NHEJ repair because the duplication is present only once in D. mojavensis AB and D. buzzatii AC regions (fig. 4). We further hypothesized that this 121-bp long sequence could be driving the transcription of both sense and antisense transcripts since the two copies are oriented toward CG4673, and we found that this was the case. Antisense transcripts can exert both positive and negative effects on gene regulation at different levels (Faghihi and Wahlestedt 2009; Werner and Swan 2010). For example, antisense transcription, but not the antisense RNA molecule itself, can modulate transcription of the sense RNA through transcriptional collision (Prescott and Proudfoot 2002). Antisense transcripts can also affect gene expression through DNA–RNA interactions or through the formation of RNA duplexes (Faghihi and Wahlestedt 2009). We indeed found evidence for the existence of CG4673 dsRNA in D. buzzatii. However, further analyses are necessary in order to elucidate whether dsRNA formation has an inhibitory influence on the sense transcript, for example, by inhibiting its translation, or on the other hand increases the expression of the gene, for example, by reducing mRNA decay (Faghihi and Wahlestedt 2009). In any case, our findings suggest that besides the 2mn rearrangement, the microinversion of CG4673 also had functional consequences. We also found evidence for transcription of ΨCG4673. Transcription of pseudogenes is not uncommon (Zheng and Gerstein 2007). Whether this transcription is spurious or has a biological function remains to be determined.
Finally, in D. buzzatii, CG5071 CDS is considerably longer than in D. mojavensis (668 aa and 225 aa long, respectively) but similar to D. melanogaster transcript RB (680 aa long). In D. melanogaster, this gene is involved in protein folding and shows moderately high expression in males (supplementary table S4 in additional file 3, Supplementary Material online). Sequence conservation between D. buzzatii and D. melanogaster proteins (74% identity) suggest that the function of this gene might be conserved in D. buzzatii.
In summary, the 2mn rearrangement fixed in all species of the buzzatii complex is currently associated with the loss of one gene (CG5079), the loss of a nested gene structure (CG4673 does not longer contain any other gene), and changes in gene structure and gene regulation (CG4673). Any of these changes or the combination of them might have been adaptive and therefore might have driven this rearrangement to fixation. Alternatively, this rearrangement could have increased in frequency through meiotic drive. CG4673 is involved in nuclear transport, and one of the best studied meiotic drive systems in Drosophila (SD system) comprises, as the main part of the driving chromosomes, a truncated duplicate of a nuclear transport protein, Sd-RanGAP, a short satellite region and often chromosomal rearrangements that restrict recombination between its components (Kusano et al. 2003; Presgraves et al. 2009).
Conclusion
Our detailed analyses of the breakpoints of the 2mn rearrangement present in all the species of the buzzatii complex have shown that other than the expected gene order change, this complex rearrangement was associated with changes in gene number, gene structure, and with the generation of an additional microinversion that has potentially functional consequences since it leads to the generation of an antisense transcript. These results indicate that 1) functional consequences of inversions are much more complex than those expected from a simple two-break rearrangement and that 2) in order to fully describe the genomic consequences of structural variants, genomic approaches should be complemented with detailed sequence-level and functional analyses of the breakpoint regions.
Supplementary Material
Supplementary Figures S1 to S6, Supplementary Data, and Supplementary Table S1_to_S9 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank Elena Casacuberta, Barbara Negre, and Natalia Petit for helpful comments on the manuscript. This research was supported by an FI-DGR Doctoral fellowship from Generalitat de Catalunya to O.C., by a “Ramón y Cajal” grant from the Spanish Ministry of Science and Innovation (RYC-2010-07306) to J.G., by the University of Texas at Arlington start-up funds and grant R01-GM071813 from National Institutes of Health to E.B., and by grant BFU2008-04988 from the Ministerio de Ciencia e Innovación (Spain) to A.R.
References
- Aravin AA, Hannon GJ, Brennecke J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science. 2007;318:761–764. doi: 10.1126/science.1146484. [DOI] [PubMed] [Google Scholar]
- Assis R, Kondrashov AS, Koonin EV, Kondrashov FA. Nested genes and increasing organizational complexity of metazoan genomes. Trends Genet. 2008;24:475–478. doi: 10.1016/j.tig.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JSF. Population genetics of Opuntia breeding Drosophila in Australia. Sydney (Australia): Academic Press; 1982. [Google Scholar]
- Bays NW, Hampton RY. Cdc48-Ufd1-Npl4: stuck in the middle with Ub. Curr Biol. 2002;12:R366–R371. doi: 10.1016/s0960-9822(02)00862-x. [DOI] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY. HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell. 2001;12:4114–4128. doi: 10.1091/mbc.12.12.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutkar A, Russo SM, Smith TF, Gelbart WM. Genome-scale analysis of positionally relocated genes. Genome Res. 2007;17:1880–1887. doi: 10.1101/gr.7062307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta A, Tandoi C, Fini G, Calabrese G, Dallapiccola B, Novelli G. Cloning and characterization of the gene encoding human NPL4, a protein interacting with the ubiquitin fusion-degradation protein (UFD1L) Gene. 2001;275:39–46. doi: 10.1016/s0378-1119(01)00649-7. [DOI] [PubMed] [Google Scholar]
- Bourque G, Pevzner PA, Tesler G. Reconstructing the genomic architecture of ancestral mammals: lessons from human, mouse, and rat genomes. Genome Res. 2004;14:507–516. doi: 10.1101/gr.1975204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N. Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science. 2004;303:832–835. doi: 10.1126/science.1091266. [DOI] [PubMed] [Google Scholar]
- Cáceres M, Puig M, Ruiz A. Molecular characterization of two natural hotspots in the Drosophila buzzatii genome induced by transposon insertions. Genome Res. 2001;11:1353–1364. doi: 10.1101/gr.174001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cáceres M, Ranz JM, Barbadilla A, Long M, Ruiz A. Generation of a widespread Drosophila inversion by a transposable element. Science. 1999;285:415–418. doi: 10.1126/science.285.5426.415. [DOI] [PubMed] [Google Scholar]
- Capy P. Evolutionary biology. A plastic genome. Nature. 1998;396:522–523. doi: 10.1038/25007. [DOI] [PubMed] [Google Scholar]
- Casals F, Caceres M, Ruiz A. The foldback-like transposon Galileo is involved in the generation of two different natural chromosomal inversions of Drosophila buzzatii. Mol Biol Evol. 2003;20:674–685. doi: 10.1093/molbev/msg070. [DOI] [PubMed] [Google Scholar]
- Casals F, González J, Ruiz A. Abundance and chromosomal distribution of six Drosophila buzzatii transposons: BuT1, BuT2, BuT3, BuT4, BuT5, and BuT6. Chromosoma. 2006;115:403–412. doi: 10.1007/s00412-006-0071-7. [DOI] [PubMed] [Google Scholar]
- Casillas S, Petit N, Barbadilla A. DPDB: a database for the storage, representation and analysis of polymorphism in the Drosophila genus. Bioinformatics. 2005;21(Suppl 2):ii26–ii30. doi: 10.1093/bioinformatics/bti1103. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for short introns in highly expressed genes. Nat Genet. 2002;31:415–418. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. Inversion polymorphism in a two-locus genetic system. Genet Res. 1974;23:259–280. doi: 10.1017/s0016672300014919. [DOI] [PubMed] [Google Scholar]
- Chen JM, Cooper DN, Ferec C, Kehrer-Sawatzki H, Patrinos GP. Genomic rearrangements in inherited disease and cancer. Semin Cancer Biol. 2010;20:222–233. doi: 10.1016/j.semcancer.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Clark AG, Eisen MB, Smith DR, et al. (244 co-authors) Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Coghlan A, Eichler EE, Oliver SG, Paterson AH, Stein L. Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet. 2005;21:673–682. doi: 10.1016/j.tig.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Coulibaly MB, Lobo NF, Fitzpatrick MC, Kern M, Grushko O, Thaner DV, Traore SF, Collins FH, Besansky NJ. Segmental duplication implicated in the genesis of inversion 2Rj of Anopheles gambiae. PLoS One. 2007;2:e849. doi: 10.1371/journal.pone.0000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darai-Ramqvist E, Sandlund A, Muller S, Klein G, Imreh S, Kost-Alimova M. Segmental duplications and evolutionary plasticity at tumor chromosome break-prone regions. Genome Res. 2008;18:370–379. doi: 10.1101/gr.7010208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De SK, McMaster MT, Andrews GK. Endotoxin induction of murine metallothionein gene expression. J Biol Chem. 1990;265:15267–15274. [PubMed] [Google Scholar]
- Delprat A, Negre B, Puig M, Ruiz A. The transposon Galileo generates natural chromosomal inversions in Drosophila by ectopic recombination. PLoS One. 2009;4:e7883. doi: 10.1371/journal.pone.0007883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. Genetics of natural populations; a response of certain gene arrangements in the third chromosome of Drosophila pseudoobscura to natural selection. Genetics. 1947;32:142–160. doi: 10.1093/genetics/32.2.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobzhansky T. Genetics of the evolutionary process. New York: Columbia University Press; 1970. [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AL, Mena PA, McAllister BF. Positive selection near an inversion breakpoint on the neo-X chromosome of Drosophila americana. Genetics. 2007;177:1303–1319. doi: 10.1534/genetics.107.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637–643. doi: 10.1038/nrm2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuk L, MacDonald JR, Tang T, Carson AR, Li M, Rao G, Khaja R, Scherer SW. Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet. 2005;1:e56. doi: 10.1371/journal.pgen.0010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta Y, Kawai M, Yahara K, et al. (12 co-authors) Birth and death of genes linked to chromosomal inversion. Proc Natl Acad Sci U S A. 2011;108:1501–1506. doi: 10.1073/pnas.1012579108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DG. DroSpeGe: rapid access database for new Drosophila species genomes. Nucleic Acids Res. 2007;35:D480–D485. doi: 10.1093/nar/gkl997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J, Casals F, Ruiz A. Duplicative and conservative transpositions of larval serum protein 1 genes in the genus Drosophila. Genetics. 2004;168:253–264. doi: 10.1534/genetics.103.025916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J, Casals F, Ruiz A. Testing chromosomal phylogenies and inversion breakpoint reuse in Drosophila. Genetics. 2007;175:167–177. doi: 10.1534/genetics.106.062612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J, Nefedov M, Bosdet I, et al. (14 co-authors) A BAC-based physical map of the Drosophila buzzatii genome. Genome Res. 2005;15:885–892. doi: 10.1101/gr.3263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon L, Yang S, Tran-Gyamfi M, et al. (11 co-authors) Comparative analysis of chicken chromosome 28 provides new clues to the evolutionary fragility of gene-rich vertebrate regions. Genome Res. 2007;17:1603–1613. doi: 10.1101/gr.6775107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray YH. It takes two transposons to tango: transposable-element-mediated chromosomal rearrangements. Trends Genet. 2000;16:461–468. doi: 10.1016/s0168-9525(00)02104-1. [DOI] [PubMed] [Google Scholar]
- Gray YH, Tanaka MM, Sved JA. P-element-induced recombination in Drosophila melanogaster: hybrid element insertion. Genetics. 1996;144:1601–1610. doi: 10.1093/genetics/144.4.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann AA, Rieseberg LH. Revisiting the impact of inversions in evolution: from population genetic markers to drivers of adaptive shifts and speciation? Annu Rev Ecol Evol Syst. 2008;39:21–42. doi: 10.1146/annurev.ecolsys.39.110707.173532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurles ME, Dermitzakis ET, Tyler-Smith C. The functional impact of structural variation in humans. Trends Genet. 2008;24:238–245. doi: 10.1016/j.tig.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20:1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci U S A. 2003;100:6569–6574. doi: 10.1073/pnas.0732024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN. Understanding the recent evolution of the human genome: insights from human-chimpanzee genome comparisons. Hum Mutat. 2007;28:99–130. doi: 10.1002/humu.20420. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN. Molecular mechanisms of chromosomal rearrangement during primate evolution. Chromosome Res. 2008;16:41–56. doi: 10.1007/s10577-007-1207-1. [DOI] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Sandig C, Chuzhanova N, Goidts V, Szamalek JM, Tanzer S, Muller S, Platzer M, Cooper DN, Hameister H. Breakpoint analysis of the pericentric inversion distinguishing human chromosome 4 from the homologous chromosome in the chimpanzee (Pan troglodytes) Hum Mutat. 2005;25:45–55. doi: 10.1002/humu.20116. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M. How and why chromosome inversions evolve. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000501. e1000501. doi:10.1371/journal.pbio.1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173:419–434. doi: 10.1534/genetics.105.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb J, Chuzhanova NA, Hogel J, Vasquez KM, Cooper DN, Bacolla A, Kehrer-Sawatzki H. Cruciform-forming inverted repeats appear to have mediated many of the microinversions that distinguish the human and chimpanzee genomes. Chromosome Res. 2009;17:469–483. doi: 10.1007/s10577-009-9039-9. [DOI] [PubMed] [Google Scholar]
- Krimbas CB, Powell J. Drosophila inversion polymorphism. Boca Raton (FL): CRC; 1992. [Google Scholar]
- Kumar A. An overview of nested genes in eukaryotic genomes. Eukaryot Cell. 2009;8:1321–1329. doi: 10.1128/EC.00143-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Inagaki H, Hosoba E, Kato T, Ohye T, Kogo H, Emanuel BS. Molecular cloning of a translocation breakpoint hotspot in 22q11. Genome Res. 2007;17:461–469. doi: 10.1101/gr.5769507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano A, Staber C, Chan HY, Ganetzky B. Closing the (Ran)GAP on segregation distortion in Drosophila. Bioessays. 2003;25:108–115. doi: 10.1002/bies.10222. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lim JK, Simmons MJ. Gross chromosome rearrangements mediated by transposable elements in Drosophila melanogaster. Bioessays. 1994;16:269–275. doi: 10.1002/bies.950160410. [DOI] [PubMed] [Google Scholar]
- Lindsay SJ, Khajavi M, Lupski JR, Hurles ME. A chromosomal rearrangement hotspot can be identified from population genetic variation and is coincident with a hotspot for allelic recombination. Am J Hum Genet. 2006;79:890–902. doi: 10.1086/508709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo NF, Sangare DM, Regier AA, et al. (11 co-authors) Breakpoint structure of the Anopheles gambiae 2Rb chromosomal inversion. Malaria J. 2010;9:293. doi: 10.1186/1475-2875-9-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfrin MH, Sene FM. Cactophilic Drosophila in South America: a model for evolutionary studies. Genetica. 2006;126:57–75. doi: 10.1007/s10709-005-1432-5. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, Chitsaz F, et al. (28 co-authors) CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009;37:D205–D210. doi: 10.1093/nar/gkn845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzkin LM, Merritt TJ, Zhu CT, Eanes WF. The structure and population genetics of the breakpoints associated with the cosmopolitan chromosomal inversion In(3R)Payne in Drosophila melanogaster. Genetics. 2005;170:1143–1152. doi: 10.1534/genetics.104.038810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery E, Charlesworth B, Langley CH. A test for the role of natural selection in the stabilization of transposable element copy number in a population of Drosophila melanogaster. Genet Res. 1987;49:31–41. doi: 10.1017/s0016672300026707. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Larkin DM, Everts-van der Wind A, et al. (25 co-authors) Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;309:613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- Navarro A, Betran E, Barbadilla A, Ruiz A. Recombination and gene flux caused by gene conversion and crossing over in inversion heterokaryotypes. Genetics. 1997;146:695–709. doi: 10.1093/genetics/146.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Ruiz A. On the fertility effects of pericentric inversions. Genetics. 1997;147:931–933. doi: 10.1093/genetics/147.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Kojima KI, Schaffer HE. Frequency changes of new inversions in populations under mutation-selection equilibria. Genetics. 1967;57:741–750. doi: 10.1093/genetics/57.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitski E. Nonrandom disjunction in Drosophila. Annu Rev Genet. 1967;1:71–86. [Google Scholar]
- Ohler U. Identification of core promoter modules in Drosophila and their application in accurate transcription start site prediction. Nucleic Acids Res. 2006;34:5943–5950. doi: 10.1093/nar/gkl608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Blanco E, Guigo R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petes TD, Hill CW. Recombination between repeated genes in microorganisms. Annu Rev Genet. 1988;22:147–168. doi: 10.1146/annurev.ge.22.120188.001051. [DOI] [PubMed] [Google Scholar]
- Pevzner P, Tesler G. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc Natl Acad Sci U S A. 2003;100:7672–7677. doi: 10.1073/pnas.1330369100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prazeres da Costa O, Gonzalez J, Ruiz A. Cloning and sequencing of the breakpoint regions of inversion 5g fixed in Drosophila buzzatii. Chromosoma. 2009;118:349–360. doi: 10.1007/s00412-008-0201-5. [DOI] [PubMed] [Google Scholar]
- Prescott EM, Proudfoot NJ. Transcriptional collision between convergent genes in budding yeast. Proc Natl Acad Sci U S A. 2002;99:8796–8801. doi: 10.1073/pnas.132270899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC, Gerard PR, Cherukuri A, Lyttle TW. Large-scale selective sweep among segregation distorter chromosomes in African populations of Drosophila melanogaster. PLoS Genet. 2009;5:e1000463. doi: 10.1371/journal.pgen.1000463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig M, Cáceres M, Ruiz A. Silencing of a gene adjacent to the breakpoint of a widespread Drosophila inversion by a transposon-induced antisense RNA. Proc Natl Acad Sci U S A. 2004;101:9013–9018. doi: 10.1073/pnas.0403090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, González J, Casals F, Ruiz A. Low occurrence of gene transposition events during the evolution of the genus Drosophila. Evolution. 2003;57:1325–1335. doi: 10.1111/j.0014-3820.2003.tb00340.x. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Maurin D, Chan YS, von Grotthuss M, Hillier LW, Roote J, Ashburner M, Bergman CM. Principles of genome evolution in the Drosophila melanogaster species group. PLoS Biol. 2007;5:e152. doi: 10.1371/journal.pbio.0050152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem. 2001;26:51–56. doi: 10.1016/s0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- Richards S, Liu Y, Bettencourt BR, et al. (52 co-authors) Comparative genome sequencing of Drosophila pseudoobscura: chromosomal, gene, and cis-element evolution. Genome Res. 2005;15:1–18. doi: 10.1101/gr.3059305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, et al. (96 co-authors) Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010;330:1787–1797. doi: 10.1126/science.1198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Wasserman M. Evolutionary cytogenetics of the Drosophila buzzatii species complex. Heredity. 1993;70(Pt 6):582–596. doi: 10.1038/hdy.1993.85. [DOI] [PubMed] [Google Scholar]
- Russo CA, Takezaki N, Nei M. Molecular phylogeny and divergence times of drosophilid species. Mol Biol Evol. 1995;12:391–404. doi: 10.1093/oxfordjournals.molbev.a040214. [DOI] [PubMed] [Google Scholar]
- Sankoff D. The where and wherefore of evolutionary breakpoints. J Biol. 2009;8:66. doi: 10.1186/jbiol162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer SW, Bhutkar A, McAllister BF, et al. (38 co-authors) Polytene chromosomal maps of 11 Drosophila species: the order of genomic scaffolds inferred from genetic and physical maps. Genetics. 2008;179:1601–1655. doi: 10.1534/genetics.107.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoighe C, Gehring C, Hurst LD. Gametophytic selection in Arabidopsis thaliana supports the selective model of intron length reduction. PLoS Genet. 2005;1:e13. doi: 10.1371/journal.pgen.0010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharakhov IV, White BJ, Sharakhova MV, Kayondo J, Lobo NF, Santolamazza F, Della Torre A, Simard F, Collins FH, Besansky NJ. Breakpoint structure reveals the unique origin of an interspecific chromosomal inversion (2La) in the Anopheles gambiae complex. Proc Natl Acad Sci U S A. 2006;103:6258–6262. doi: 10.1073/pnas.0509683103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer EL. Inhibition of increase and activity of tobacco-mosaic virus under nitrogen-deficient conditions. Plant Physiol. 1941;16:227–239. doi: 10.1104/pp.16.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperlich D, Pfriem P. Chromosomal polymorphism in natural and experimental populations. London: Academic Press; 1986. [Google Scholar]
- Sturtevant AH. Genetic factors affecting the strength of linkage in Drosophila. Proc Natl Acad Sci U S A. 1917;3:555–558. doi: 10.1073/pnas.3.9.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtevant AH, Beadle GW. The relations of inversions in the X chromosome of Drosophila Melanogaster to crossing over and disjunction. Genetics. 1936;21:554–604. doi: 10.1093/genetics/21.5.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol. 2004;21:36–44. doi: 10.1093/molbev/msg236. [DOI] [PubMed] [Google Scholar]
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiology Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- Tiwari S, Ramachandran S, Bhattacharya A, Bhattacharya S, Ramaswamy R. Prediction of probable genes by Fourier analysis of genomic sequences. Comput Appl Biosci. 1997;13:263–270. doi: 10.1093/bioinformatics/13.3.263. [DOI] [PubMed] [Google Scholar]
- Tweedie S, Ashburner M, Falls K, et al. (11 co-authors) FlyBase: enhancing Drosophila gene ontology annotations. Nucleic Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Swan D. What are natural antisense transcripts good for? Biochem Soc Trans. 2010;38:1144–1149. doi: 10.1042/BST0381144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesley CS, Eanes WF. Isolation and analysis of the breakpoint sequences of chromosome inversion In(3L)Payne in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1994;91:3132–3136. doi: 10.1073/pnas.91.8.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton LT. Analysis of the repleta group of Drosophila. Univ Texas Pub. 1942;4228:23–52. [Google Scholar]
- Yang HP, Barbash DA. Abundant and species-specific DINE-1 transposable elements in 12 Drosophila genomes. Genome Biol. 2008;9:R39. doi: 10.1186/gb-2008-9-2-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Freudenreich CH. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol Cell. 2007;27:367–379. doi: 10.1016/j.molcel.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Peterson T. Transposition of reversed AC element ends generates chromosome rearrangements in maize. Genetics. 2004;167:1929–1937. doi: 10.1534/genetics.103.026229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Gerstein MB. The ambiguous boundary between genes and pseudogenes: the dead rise up, or do they? Trends Genet. 2007;23:219–224. doi: 10.1016/j.tig.2007.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.