

Abstract
Advanced oxidation protein products (AOPPs) are carried by oxidized plasma proteins, especially albumin and accumulate in subjects with renal disease and coronary artery disease. AOPPs represent an excellent novel marker of oxidative stress and their roles in the development of cardiovascular disease might be of great importance. Here, we show that in vitro–generated AOPP-albumin binds with high affinity to the high-density lipoprotein (HDL) receptor scavenger receptor class B type I (SR-BI). Already an equimolar concentration of AOPP-albumin to HDL blocked HDL association to SR-BI and effectively inhibited SR-BI–mediated cholesterol ester (CE) uptake. Interestingly, albumin extensively modified by advanced glycation end products (AGE-albumin), which is an established SR-BI ligand known to accumulate in renal disease, only weakly interfered with HDL binding to SR-BI. Furthermore, AOPP-albumin administration increased the plasma half-life of [3H]CE-HDL in control mice 1.6-fold (P=0.01) and 8-fold (P=0.0003) in mice infected with adenoviral vectors encoding human SR-BI. Moreover, albumin isolated from hemodialysis patients, but not albumin isolated from healthy controls, markedly inhibited SR-BI–mediated HDL-CE transfer in vitro dependent on the AOPP content of albumin. These results indicate that AOPP-albumin effectively blocks SR-BI in vitro and in vivo. Thus, depressed plasma clearance of HDL-cholesterol may contribute to the abnormal composition of HDL and the high cardiovascular risk observed in patients with chronic renal failure.
Keywords: AOPP, hemodialysis, myeloperoxidase, oxidative stress, HDL
Extracellular fluids contain only small amounts of antioxidant enzymes, and therefore plasma proteins are prone to oxidation by reactive oxidant species (ROS). In line with this observation, elevated levels of oxidized protein products, termed “advanced oxidation protein products” (AOPP), such as oxidized albumin, accumulate in the plasma of dialysis patients.1,2 It has been well documented that albumin is quite vulnerable to ROS3 and elevated levels of carbonyl groups of albumin have been reported in plasma of dialysis patients,4 liver cirrhosis and acute on chronic liver failure.5 One of the potential in vivo oxidants generated by the myeloperoxidase (MPO)-H2O2-halide system of activated phagocytes is hypochlorous acid (HOCl). MPO levels correlate with the AOPP plasma content in hemodialysis (HD) patients6,7 and up to 50% of AOPP are formed by MPO-dependent activities.7 Analyses of the molecular species and spectral properties of AOPP demonstrated that carbonyls and dityrosine containing cross-linked components of albumin result from MPO-derived HOCl.4,7 It has been recently shown that lipoproteins modified by HOCl bind with high affinity to the high-density lipoprotein (HDL) receptor scavenger receptor class B type I (SR-BI),8,9 in line with the finding that SR-BI recognizes a broad range of oxidized and modified proteins.10 Several studies have clearly revealed that SR-BI is protective against cardiovascular disease11 by acting as the primary pathway for disposal of HDL-borne cholesterol ester (CE) and triglycerides.10 It was shown that plasma clearance of HDL and the rate of apolipoprotein (apo)A-I biosynthesis, the main apoprotein of HDL, are decreased in patients with renal disease.12,13 Moreover, treatment of hypercholesterolemic rabbits with repeated intravenous injections of AOPP-albumin significantly increased plasma total cholesterol, which is mainly carried by HDL in rabbits.14 Chronic renal failure per se, without heavy proteinuria, does not significantly change SR-BI mRNA and protein abundance in the liver,15 indicating that AOPPs might interfere with plasma clearance of HDL.
Therefore, the goal of the present study was to test the hypothesis that AOPP-albumin interferes with HDL-CE clearance mediated by SR-BI by acting as a possible receptor antagonist.
Materials and Methods
Materials
NaOCl, organic solvents, potassium bromide, potassium iodide, chloramine-T, and fatty acid–free human serum albumin (HSA) were obtained from Sigma, isoflurane was from Pharmacia & Upjohn SA (Guyancourt, France). OxyBlot oxidized protein detection kit was from Millipore Corp (Molsheim, France). Ham’s-F12K medium was from Gibco (Life Technologies, Vienna, Austria), DMEM and FCS were obtained from Boehringer Ingelheim Bioproducts (Mannheim, Germany). HiTrap Blue HP, 1-mL columns were from (GE Healthcare, Vienna, Austria). Radiochemicals were purchased from PerkinElmer Life Sciences. All other reagents were obtained from Sigma (Vienna, Austria).
Methods
HDL and LDL Preparation
HDL (density range, 1.125 to 1.21 g/mL) and LDL (density, 1.063 g/mL) was prepared by discontinuous density ultracentrifugation of plasma obtained from HD patients or normolipidemic blood donors.8
Albumin Preparation
Albumin from HD patients (HD-albumin) and controls was separated from other plasma proteins by affinity chromatography using HiTrap Blue HP, 1 mL columns (GE Healthcare) according to the instructions of the manufacturer.
AOPP Assay
The AOPP assay included a sample preparation procedure to precipitate lipoproteins in the plasma to avoid assay interference (mostly by triglycerides, which are usually markedly elevated in chronic kidney failure) as recently described.16 MgCl2(5 μL of a 2 mol/L stock solution) and phosphotungstate (20 μL of a 4% stock solution in 0.19 mol/L NaOH) were mixed with 200 μL of EDTA plasma, centrifuged at 1000g for 20 minutes, on which the supernatant was carefully removed. AOPP were immediately measured in the supernatant at 340 nm under acidic conditions and expressed as chloramine-T equivalents.1
Modification of Albumin and LDL
AOPP-albumin was prepared in the absence of free amino acids/carbohydrates/lipids to exclude formation of advanced glycation end products (AGE)-like structures as described.17,18 Briefly, 10 mg of fatty acid–free HSA per milliliter of PBS was incubated with HOCl solution at 4°C for up to 1 hour at pH 7.4. The modified HSA preparations were passed over a PD10 column to remove unreacted HOCl and used immediately for experiments.
AGE-albumin was prepared as described previously.18 Briefly, 0.5 g of fatty acid–free HSA was dissolved with 3.0 g of d-glucose in 10 mL of 0.5 mol/L sodium phosphate buffer (pH 7.4) containing 0.05% NaN3. Each solution was deoxygenated with nitrogen, sterilized by ultrafiltration, and incubated for 90 days at 37°C in the dark. The samples were then passed over a PD 10 column and used for experiments.
Copper-induced oxidation of LDL was performed as described.9
Amino Acid Analysis
Aliquots of native, AOPP, and AGE-modified albumin (500 μg of protein) were lyophilized in 5-mL ampoules and purged with nitrogen before hydrolysis in constant boiling 6 N HCl (24 hour, 120°C). Amino acid analysis was performed on a Biotronics analyzer as described.18
Labeling Procedures
HDL and albumin labeling with 125I-Na iodination was performed as described8 using N-bromosuccinimide as the coupling agent.
HDL was labeled with [cholesteryl-1,2,6,7-3H]palmitate by CE transfer protein-catalyzed transfer from donor liposomes as described.8
Cell Culture
Chinese hamster ovary (CHO) cells: LdlA cells (LDL receptor-deficient CHO cell line) and stable transfectants expressing murine SR-BI (LdlA[SR-BI]) were cultured and maintained as described.8 Both cell lines were kindly provided by Dr Monty Krieger (Massachusetts Institute of Technology, Boston).
SDS-PAGE and Western Blotting
SDS-PAGE and subsequent Western blotting experiments of plasma and albumin preparations were performed with 3.75% to 20% polyacrylamide gradient gels as described.19
Detection of Carbonylated Proteins
Carbonylated albumin was detected using the chemical and immunologic reagents of the OxyBlot oxidized protein detection kit according to the instructions of the manufacturer.
Cell Association Studies
LdlA(SR-BI) and control LdlA7 cells were incubated for 2 hours at 37°C with 125I-labeled albumin/HDL (0 to 800 μg/mL) in DMEM. Specific cell association of 125I-labeled proteins to SR-BI was determined by subtracting association to control LdlA7 cells from LdlA(SR-BI) cells as described.8
Recombinant Adenovirus Preparation
Generation of adenoviral vectors encoding hSR-BI (Ad/hSR-BI) or lacZ cDNA (Ad/lacZ) have been described previously.20
125I-AOPP-Albumin and 3H-CE-HDL Turnover In Vivo
Three-month-old male BALB/c mice were anesthetized with isoflurane and infected via the tail vein with 2×109 virus particles of Ad/hSR-BI and Ad/lacZ.
125I-AOPP-Albumin Turnover
Four days after infection, 5 mice of Ad/hSR-BI and Ad/lacZ mice were injected via tail vein with 100 μg of 125I-AOPP-albumin (1×107 cpm) or control 125I-albumin in 100 μL of PBS. Blood samples were drawn from anesthetized mice starting at 30 seconds up to 60 minutes by retroorbital puncture.
3H-CE-HDL Turnover
Four days after infection, 5 mice of Ad/hSR-BI and Ad/lacZ mice were injected via tail vein with a mixture comprising 50 μg of [3H]CE-HDL (5×105 cpm) and either native albumin or AOPP-albumin (5 mg) in 100 μL of PBS. Blood samples were drawn from anesthetized mice at 30 seconds up to 60 minutes by retroorbital puncture. Plasma samples were analyzed by liquid scintillation counting.
Liver samples were harvested from mice killed by an overdose of isoflurane to measure uptake of 125I-albumin preparations or 3H-CE-HDL and 50 μg liver homogenates were analyzed by Western blotting to determine SR-BI expression. All animal experiments were approved by the Austrian Ministry of Education, Science and Culture according to the Regulations for Animal Experimentation.
Blood Collection and Plasma Isolation
Blood was taken from nondiabetic, HD patients and age matched control subjects at the time of routine laboratory investigations in agreement with the Ethical Committee of the Medical University of Graz. Blood (5 mL) was collected in standard sterile polystyrene vacuum tubes with 5 mmol/L EDTA. Following centrifugation (600g, 10 minutes), the plasma was stored in 500 μL aliquots at −70°C until use.
Statistical Analysis
Statistical differences between groups were tested for normal distribution and then analyzed by Student’s t and ANOVA test using Prism software (GraphPad). Significance was accepted at the following levels: P<0.05 (*), P<0.01 (**), P<0.001 (***).
Results
Preparation and Characterization of AOPP-Albumin and AGE-Albumin
AOPPs are formed by MPO-derived HOCl in vivo1,7; therefore, we prepared AOPP-albumin by treating albumin with HOCl: albumin molar ratios ranging from 25 to 100:1. These molar HOCl:albumin ratios were chosen according to a recent study showing that an HOCl:albumin ratio of ≈35:1 resembles AOPP-albumin isolated from HD patients.4 HOCl modification was performed in the absence of free amino acids/carbohydrates/lipids to avoid formation of AGE-like structures.14,18 AGE-albumin was prepared by incubating albumin with an excess glucose for 90 days. Treatment of albumin with HOCl induced protein aggregation or cross-linking that was evident by the presence of additional bands in the higher molecular range and a pronounced formation of albumin associated carbonyls, in line with a recent study4 (Figure 1). AGE-albumin showed strong cross-linking, whereas the carbonyl content of AGE-albumin was comparatively low. No carbonyls could be detected in unmodified albumin. To identify preferential amino acid targets of AOPP-albumin and AGE-albumin, an amino acid analysis was performed. It is evident that lysine, tyrosine, histidine, arginine, and cysteine were oxidized by HOCl dose-dependently up to 20% to 70% at the highest HOCl:albumin molar ratio, whereas 40% to 70% of lysine, arginine, and histidine residues were modified in AGE-albumin (Table 1).
Figure 1.
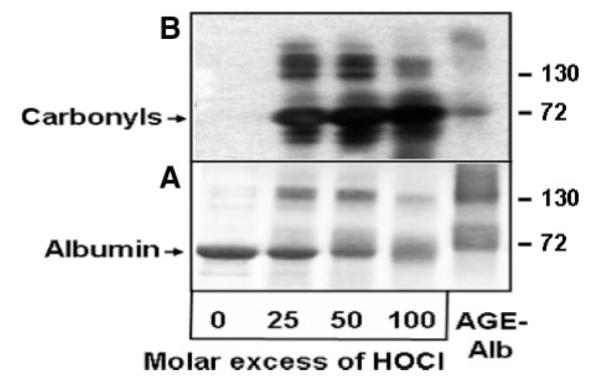
SDS-PAGE and Western blot showing the carbonyl content of modified albumin. Native albumin (Alb) was modified by indicated HOCl:albumin molar ratios, and AGE-albumin was prepared as described in Materials and Methods. A, Twenty micrograms of albumin preparations were subjected to SDS-PAGE (5% to 20%) under reducing conditions and subsequently visualized with Coomassie staining. B, The albumin preparations were derivatized with dinitrophenylhydrazine, subjected to SDS-PAGE, and transferred to poly(vinylidene difluoride) membranes. Albumin-associated carbonyls were detected with an anti-DNP antibody. The molecular mass (kDa) of the marker proteins is indicated at the right.
Table 1.
Amino Acid Characterization of Modified Albumin Preparations
| Alb | HOCl:Alb (25:1) | HOCl:Alb (100:1) | AGE-Alb | |
|---|---|---|---|---|
| Cysteine | 6.8 | 7.7 | 4.2 | 8.0 |
| Tyrosine | 40.9 | 35.3 | 8.8 | 34.4 |
| Lysine | 99.8 | 84.1 | 29.7 | 27.4 |
| Histidine | 31.4 | 31 | 23.3 | 18.8 |
| Arginine | 46.4 | 46.5 | 35.5 | 28.0 |
Amino acid analysis was performed as described in Materials and Methods. Results are expressed in micrograms of amino acid per milligram of albumin.
Cell Association Studies
To obtain specific binding data of AOPP-albumin preparations to SR-BI, SR-BI–overexpressing CHO cells (LdlA[SR-BI]) and control LdlA7 cells that show only minimal SR-BI expression (see insert, Figure 2A) were incubated with increasing concentrations of in vitro– modified 125I-labeled AOPP-albumin and native albumin. As shown in Figure 2A, SR-BI specifically mediated binding of 125I-AOPP-albumin, and binding curves exhibited a dose-dependent saturation pattern (Figure 2A). Nonlinear regression analysis revealed a marked increase in binding affinity (Kd values are listed in Table 2) for AOPP-albumin related to the increased AOPP content of albumin (Table 2). Next, we investigated whether AOPP-albumin interferes with HDL association to SR-BI and subsequently inhibits HDL-CE delivery. For this purpose, we performed competition experiments with a constant amount of 125I-HDL or 3H-CE–labeled HDL (10 μg/mL) in the presence of increasing concentrations of native and modified albumin. As shown in Figure 2B, AOPP-albumin effectively displaced 125I-HDL from LdlA(SR-BI) cells and attenuated SR-BI–mediated CE transfer (Figure 2C), whereas native albumin showed no effect. Thus, AOPP-albumin is a potent SR-BI antagonist. In contrast, the known SR-BI ligand AGE-albumin21 showed only weak inhibitory activity on SR-BI–mediated CE transfer (Figure 2C) compared to AOPP-albumin. The calculated IC50 values are listed in Table 2. Of note, AOPP-albumin more effectively blocked SR-BI–mediated HDL-CE-transfer than 125I-HDL binding. This is in line with the observation that iodination of HDL increases the affinity of HDL to SR-BI (G. Marsche, unpublished data, 2008).
Figure 2.

SR-BI–mediated cell association of 125I-labeled AOPP-Alb and competition of 125I-HDL and [3H]CE-HDL association to SR-BI. LdlA(SR-BI) and LdlA7 cells were incubated in DMEM for 2 hours at 37°C with increasing concentrations of 125I-albumin (■) and 125I-AOPP-albumin preparations containing an AOPP content of 3.9±0.4 μmol/L (▲), 12.9±0.8 μmol/L (▼), and 33.2±5.2 μmol/L (◆). The AOPP values for albumin preparations (1 mg/mL) are expressed as chloramine-T equivalents. Western blot analysis of SR-BI and β-actin (loading control) expression in LdlA(SR-BI) and LdlA7 cells is shown in the inset (A). LdlA(SR-BI) and LdlA7 cells were incubated in DMEM for 2 hours at 37°C with 10 μg of protein per milliliter of 125I-HDL (B) and 3H-CE-HDL (C) in the presence of increasing concentrations of albumin (■), AGE-albumin (○), and AOPP-albumin preparations containing an AOPP content of 3.9±0.4 μmol/L (▲), 12.9±0.8 μmol/L (▼), and 33.2±5.2 μmol/L (◆). Subsequently, the cells were washed and lysed to determine the amount of associated label and the cellular protein content. Specific association to SR-BI was calculated by subtracting values obtained with LdlA7 cells from those with LdlA(SR-BI) cells. Data shown are the means±SD of triplicate determinations from 1 experiment of 3 (A) or means±SD of 3 independent experiments performed in triplicates (B and C).
Table 2.
Binding Properties of Modified Albumin Preparations to SR-BI
| AOPP (μmol/L) | Kd (μg/mL) | Bmax (ng/mg cell protein) | IC50 | |
|---|---|---|---|---|
| Alb | 1.1±0.2 | ND | ND | ND |
| HOCl:Alb (25:1) | 3.9±0.4 | 99.3±14 | 181.6±14.6 | 33.8±2.9 |
| HOCl:Alb (50:1) | 12.9±0.8 | 45.3±2.4 | 210.0±4.7 | 8.5±2.6 |
| HOCl-Alb (100:1) | 33.2±5.2 | 10.5±1.9 | 183.3±9.1 | 4.1±1.3 |
| AGE-Alb | ND | ND | ND | 143.4±4.1 |
AOPP values for albumin preparations (1 mg/mL) are expressed as chloramine-T equivalents. Calculated Kd and Bmax values for binding of 125I-labelled albumin preparations to SR-BI and IC50 values for inhibition of [3H]CE-HDL (10 μg/ml) binding to SR-BI by albumin (Alb), AOPP-albumin (at indicated oxidant: protein molar ratio), and AGE-albumin are given. Calculations were performed by nonlinear regression analysis (GraphPad Prism). ND indicates not determined.
125I-AOPP-Albumin and 3H-CE-HDL Clearances In Vivo
Prompted by the high affinity binding of AOPP-albumin to SR-BI, we analyzed the fate of 125I-AOPP-albumin injected into mice. To test whether SR-BI is directly involved in AOPP-albumin clearance, we induced SR-BI expression by infecting mice with Ad/hSR-BI virus. Control mice were infected with Ad/lacZ. SR-BI expression in livers of Ad/hSR-BI and Ad/LacZ mice is shown in the insert of Figure 3A. hSR-BI expression in livers decreased plasma total cholesterol levels from 52±8 (Ad/LacZ) to 15±2.5 mg/dL. Plasma decay curves analyzed during a 1-hour period after injection (Figure 3A) showed that in control mice the calculated plasma half-life of 125I-AOPP-albumin is decreased (40.8±2.2 versus 123.3±1.7 minutes, P<0.0001) compared with 125I-albumin. Importantly, half-life of 125I-AOPP-albumin was further decreased in Ad/hSR-BI–treated mice (23.5±1.9 versus 40.8±2.2 minutes; P=0.0043). In contrast, hSR-BI expression did not exert any effect on 125I-albumin clearance. The decreased half-life of AOPP-albumin was reflected in its increased uptake by the liver as shown in Figure 3B.
Figure 3.

AOPP-albumin clearance and serum decay of [3H]CE-HDL in mice. One hundred micrograms (1×107 cpm) of 125I-albumin or 125I-AOPP-albumin (HOCl:Alb=50:1) were injected into control Ad/lacZ (n=10) and Ad/hSR-BI (n=10) mice. Subsequently, the serum decay of the tracer was monitored by counting of plasma samples collected by retroorbital puncturing over 1 hour. Liver homogenates were analyzed by Western blotting to determine SR-BI expression (inset) (A) and to estimate liver uptake of tracer (B). Ad/hSR-BI (n=10) and Ad/lacZ (n=10) mice were injected via tail vein with a mixture comprising 50 μg of [3H]CE-HDL (5×105 cpm) and either 5 mg of native albumin or 5 mg of AOPP-albumin (HOCl:Alb=50:1) in 100 μL of PBS. The AOPP content of AOPP-albumin (1 mg/mL)=12.9 μmol/L. Subsequently, the serum decay of the tracer was monitored by scintillation counting of plasma samples collected by retroorbital puncturing over 1 hour (C). Lipids were extracted from liver homogenates to estimate liver uptake of tracer by scintillation counting (D). Values represent the means±SEM of 5 mice.
To assess whether AOPP-albumin interferes with SR-BI mediated clearance of plasma HDL-CE in vivo, we examined the plasma clearance of 3H-CE-HDL coinjected with either native albumin or AOPP-albumin in Ad/hSR-BI and Ad/lacZ-infected mice. Plasma decay curves analyzed during a 1-hour period post injection showed, as expected, an hSR-BI–mediated decrease in the plasma half-life of 3H-CE-HDL coinjected with the native albumin, when compared with control Ad/lacZ mice (3.2±0.3 versus 48.0±2.3 minutes, P<0.0001) (Figure 3C). Most importantly, AOPP-albumin administration increased the plasma half-life of [3H]CE-HDL in control mice 1.6-fold (78.24±6.2 versus 48.0±2.3 minutes, P=0.01) and 8-fold (25.3±1.9 versus 3.2±0.3 minutes, P=0.0003) in mice infected with adenoviral vectors encoding human SR-BI. The increased plasma half-life of 3H-CE-HDL was accompanied by its decreased uptake by the liver as shown in Figure 3D. These in vivo data along with the cell culture experiments clearly demonstrate that AOPP-albumin effectively blocks SR-BI and interferes with SR-BI–mediated HDL-CE clearance in mice.
Effect of Albumin Isolated From HD Patients on SR-BI–Mediated HDL-CE Uptake
Elevated levels of AOPP-albumin accumulate in the plasma of HD patients. To test whether albumin from uremic patients binds to SR-BI and interferes with SR-BI–mediated HDL-CE clearance, we isolated albumin from HD patients with high plasma AOPP values and healthy age matched control subjects. The plasma content of AOPPs found in delipidated uremic plasma was significantly higher compared to controls (168.3±7.3 versus 41.0±1.5 μmol/L, mean±SEM, P<0.0001), respectively. To examine whether HD-albumin is able to interfere with SR-BI mediated CE uptake, LdlA(SR-BI) and control LdlA7 cells were incubated with 3H-CE–labeled HDL in the presence of albumin isolated from HD patients and controls, respectively. The calculated molar excess of albumin over HDL in plasma is ≈50 to 125 fold; therefore, we used a 70-fold molar excess of HD-albumin (1 mg/mL) over 3H-CE-HDL (25 μg/mL). As shown in Figure 4A, albumin isolated from HD patients contained a high carbonyl content (insert Figure 4A) and profoundly decreased 3H-CE-HDL association up to 50%. In contrast, albumin isolated from controls did not alter 3H-CE-HDL association. The inhibitory activity of uremic albumin was dose dependent and also operative at higher, receptor saturating concentrations of both 3H-CE-HDL (125 μg/mL) and HD-albumin (5 mg/mL) (data not shown). Next, we examined the relationship between the AOPP content of HD-albumin and its SR-BI inhibitory activity. For that purpose, albumin was isolated from HD patients with low and high plasma AOPP content. The AOPP content of 1 mg/mL isolated HD-albumin ranged from 2 to 10 μmol/L and was similar to the AOPP content of in vitro– generated AOPP-albumin (Table 2). As shown in Figure 4B, the SR-BI inhibitory activity of HD-albumin measured as the efficacy to attenuate 3H-CE-HDL association with LdlA7(SR-BI) cells correlated highly significantly with the AOPP content of HD-albumin (r=0.91, P<0.001). Increased oxidative stress in HD patients might also lead to formation of oxidized HDL, which binds with increased affinity to SR-BI.8 Therefore, we tested whether HD-albumin is able to block SR-BI association of HDL isolated from HD patients (HD-HDL). Indeed, HD-albumin (1 mg/mL; AOPP content, 7.6 μmol/L) displaced HD-HDL isolated from two patients (35% to 45%) to approximately the same extent as found for HDL isolated from controls (40%) (Figure 4C). This finding clearly indicates that the affinity of HD-HDL to SR-BI is similar to that of control HDL.
Figure 4.

HD-albumin–mediated competition of [3H]CE-HDL is dependent on the AOPP content. A, LdlA(SR-BI) cells and control LdlA7 cells were incubated in DMEM for 2 hours at 37°C with [3H]CE-HDL (25 μg of protein/mL) in the presence of 1 mg/mL albumin isolated from plasma of HD patients (n=8) and control subjects (n=5), respectively. Western blot of the carbonyl content and Ponceau red stain as a loading control of albumin of control subjects (n=5) and HD patients (n=8) are shown (inset in A). Values measured with LdlA7 cells were subtracted from LdlA(SR-BI) cells to obtain SR-BI–specific binding. B, Regression analysis between AOPP content of HD-albumin and SR-BI inhibitory activity. LdlA(SR-BI) cells were incubated in DMEM for 2 hours at 37°C with 25 μg/mL 3H-CE-HDL in the presence of 1 mg/mL albumin isolated from 17 HD patients with indicated AOPP content, and cell association of 3H-CE was measured. C, LdlA(SR-BI) cells were incubated in DMEM for 2 hours at 37°C with 20 μg/mL 125I-HDL isolated from 1 control and 2 HD patients in the presence of 1 mg/mL control albumin or HD-albumin isolated from HD patient 1 (AOPP content of 1 mg/mL=7.2 μmol/L).
Binding Properties of HD-Albumin and AOPP-Albumin to SR-BI
To determine the binding properties of HD-albumin to SR-BI, binding studies with LdlA(SR-BI) and control LdlA7 cells were performed (Figure 5A). Binding capacity of 125I-HD-albumin to LdlA(SR-BI) cells was higher compared to LdlA7 cells, indicating that SR-BI mediates binding of HD-albumin. Nonlinear regression analysis revealed a Kd value of 336±48 μg/mL and a Bmax value 324±35 ng/mg cell protein. To further demonstrate that HD-albumin and in vitro– generated AOPP-albumin bind to the same binding site(s) of SR-BI, competition experiments with known SR-BI ligands were performed. For that purpose, LdlA(SR-BI) and LdlA7 cells were incubated with 20 μg/mL of 125I-HD-albumin or in vitro– generated 125I-AOPP-albumin in the presence of 200 μg/mL competitors. As shown in Figure 5B, HOCl-albumin, HDL, and oxidized LDL effectively displaced 125I-HD-albumin, as well as 125I-AOPP-albumin. Importantly, AGE-albumin did not affect 125I-AOPP-albumin binding and only weakly interfered with 125I-HD-albumin binding, indicating that HD-albumin and in vitro– generated AOPP-albumin, but not AGE-albumin, share the same binding site on SR-BI. Only minimal competition was observed in LdlA7 cells.
Figure 5.
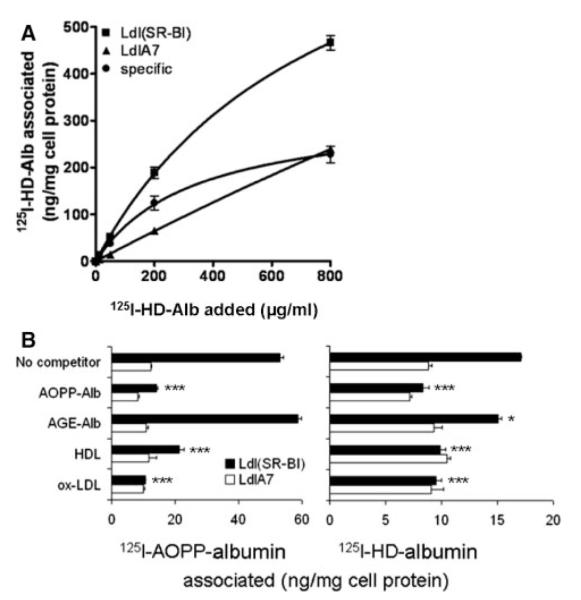
Specificity of HD-albumin binding to SR-BI. A, LdlA(SR-BI) and LdlA7 cells were incubated in DMEM for 2 hours at 37°C with increasing concentrations of 125I-HD-albumin (AOPP content of 1 mg/mL=7.2 μmol/L). Values measured with LdlA7 cells were subtracted from LdlA(SR-BI) cells to obtain SR-BI–specific binding. B, LdlA(SR-BI) and LdlA7 cells were incubated in DMEM for 2 hours at 37°C with 20 μg/mL in vitro– generated 125I-AOPP-albumin (HOCl:Alb=50:1; AOPP content of 1 mg/mL=12.9 μmol/L) and 125I-HD-albumin (AOPP content of 1 mg/mL=7.2 μmol/L) in the presence of 200 μg/mL indicated competitors. Data shown are the means±SD of triplicate determinations from 2 independent experiments.
Discussion
In the present study, we demonstrate that albumin isolated from nondiabetic HD patients effectively blocks SR-BI–mediated HDL-CE transfer. We provide evidence that the inhibitory activity of albumin from uremic patients is mainly mediated by an oxidized fraction of albumin. First, association studies performed with in vitro– generated AOPP-albumin, and albumin isolated from HD patients revealed that binding affinity to SR-BI is dependent on the AOPP content of albumin. Second, we demonstrate that AOPP-albumin effectively reduces SR-BI–mediated lipid tracer uptake in vitro and in vivo and that SR-BI is involved in the in vivo clearance of AOPP-albumin. Third, competition studies clearly showed that the SR-BI binding domain(s) of in vitro– generated AOPP-albumin and HD-albumin, but not AGE-albumin, are identical and overlap with binding domains for native HDL. This clearly indicates that the SR-BI inhibitory activity is not mediated by AGE-like structures.
Our results are in line with a recent study showing that AGE-albumin had no effect on HDL binding to SR-BI,21 indicating that the binding domain(s) of AGE-albumin and HDL are not identical. Furthermore, it has been recently shown that the affinity of AGE-proteins to SR-BI strictly depends on the extent of modification by AGEs. Only highly modified AGE-albumin is significantly recognized by SR-BI, whereas mildly, more physiological modified AGE-albumin shows no ligand activity.22
The inhibitory activity of in vitro– generated AOPP-albumin is stronger compared to albumin isolated from HD patients, despite similar AOPP content of albumin. Our binding data show that in vitro– generated AOPP-albumin with an AOPP content of 3.9 μmol/L binds with higher affinity (Kd value of ≈100 μg/mL) (Table 2) to SR-BI compared to isolated HD-albumin (AOPP content of 7.2 μmol/L; Kd value of ≈340 μg/mL) (Figure 5A). The reason for this difference is probably because ≈50% of the AOPP generation in HD patients results from MPO-independent modifications.7 Therefore, it is likely that non MPO-derived modifications, like the AGE-product pentosidine,1 with weak or no SR-BI inhibitory activity, may contribute to the AOPP content of HD-albumin. However, the observed inhibitory activity of HD-albumin is of clear physiological relevance. The calculated molar excess of albumin over HDL in plasma is ≈50- to 125-fold. Therefore, our data indicate that a physiological molar excess of HD-albumin over HDL (≈70 fold) may block up to 50% of HDL-CE delivery to SR-BI.
Our findings do not clarify the molecular mechanism responsible for AOPP-albumin binding to SR-BI. All of the identified modified proteins recognized by the multiligand receptor SR-BI hold in common a negative charge, suggesting that SR-BI recognizes the negative charge of proteins. In line with this finding, modification of albumin-lysine residues by the MPO product HOCl (leading to a decrease of positive charge) markedly affects albumin interaction with SR-BI (Tables 1 and 2). This observation is also supported by the finding that HOCl-modification of HDL and LDL also increases the affinity to SR-BI up to 10-fold.8,9 Thus, it can be speculated that under proinflammatory conditions, AOPP-albumin and oxidized lipoproteins may markedly impair SR-BI–mediated reverse cholesterol transfer.
It has been recently shown that a decreased clearance of HDL, as shown in SR-BI–deficient mice, renders HDL dysfunctional and proinflammatory.23 Another study showed that the HDL inflammatory index correlates with poor outcome in HD patients.24 This is of particular interest because besides transferring excess cholesterol to the liver, HDL has antioxidant, antiinflammatory, vasodilating, and antithrombotic properties.25 Moreover, hepatic expression of SR-BI has been shown to be a positive regulator of cholesterol efflux from macrophages26; hence, blockade of SR-BI by AOPP-albumin might directly contribute to foam cell formation in the arterial wall.
The risk for cardiovascular disease in a 30-year-old end-stage renal disease patient is similar to the calculated risk of a 70- to 80-year-old subject from the nonrenal population.27,28 Most importantly, AOPPs are risk factors for cardiovascular events in nondiabetic patients with renal disease.29 Chronic renal failure in humans results in profound lipid disorders, which arise largely from dysregulation of HDL and triglyceride-rich lipoprotein metabolism.13,15 Accordingly, an AOPP-albumin–induced decrease in SR-BI–mediated HDL-CE clearance, as observed in the present study, could promote the shift/redistribution of CE from HDL to the proatherogenic apoB containing lipoprotein pool.
Besides acting as a high-affinity ligand for SR-BI, as observed in the present study, AOPP-albumin is also recognized by CD3630 and the receptor for advanced glycation end products (RAGE).18,31,32 Furthermore, AOPP-albumin stimulates the oxidative burst and the synthesis of proinflammatory cytokines in neutrophils and monocytes,6 indicating that oxidized albumin may be a potent inflammatory mediator in vivo. Together these observed effects of in vitro– generated AOPP-albumin were not mediated by AGE-like structures, because the content of AGEs does not increase when albumin is modified by HOCl.14,18,32
In summary, we provide strong in vivo and in vitro evidence that AOPPs, which arise from the reaction between chlorinated oxidants and plasma proteins, are proinflammatory mediators that directly impair HDL metabolism and might therefore be potential key players in the development of cardiovascular disease.
Acknowledgments
Sources of Funding This work was supported by Austrian Science Foundation grants P21004-B02, P19424-B05, and P19473-B05 (to G.M., A. Heinemann, and S.F.); the Jubilee Fund; Austrian National Bank grants 11165 (to C.W.), 12778 (to S.F.), and 11967 (to A. Heinemann); the Lore Saldow Research Fund (to A. Hrzenjak); Lanyar Stiftung grants 314, 328, and 329 (to K.O., S.F., and G.M.); and by the PhD program “Molecular Medicine” of the Medical University Graz (to G.M. and M.H.)
Footnotes
Disclosures None.
References
- 1.Witko-Sarsat V, Friedlander M, Capeillére-Blandin C, Nguyen-Khoa T, Nguyen AT, Zingraff J, Jungers P, Descamps-Latscha B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996;49:1304–1313. doi: 10.1038/ki.1996.186. [DOI] [PubMed] [Google Scholar]
- 2.Mayer B, Zitta S, Greilberger J, Holzer H, Reibnegger G, Hermetter A, Oettl K. Effect of hemodialysis on the antioxidative properties of serum. Biochim Biophys Acta. 2003;1638:267–272. doi: 10.1016/s0925-4439(03)00093-0. [DOI] [PubMed] [Google Scholar]
- 3.Himmelfarb J, McMonagle E. Albumin is the major plasma protein target of oxidant stress in uremia. Kidney Int. 2001;60:358–363. doi: 10.1046/j.1523-1755.2001.00807.x. [DOI] [PubMed] [Google Scholar]
- 4.Capeillere-Blandin C, Gausson V, Descamps-Latscha B, Witko-Sarsat V. Biochemical and spectrophotometric significance of advanced oxidized protein products. Biochim Biophys Acta. 2004;1689:91–102. doi: 10.1016/j.bbadis.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Oettl K, Stadlbauer V, Petter F, Greilberger J, Putz-Bankuti C, Hallström S, Lackner C, Stauber RE. Oxidative damage of albumin in advanced liver disease. Biochim Biophys Acta. 2008;1782:469–473. doi: 10.1016/j.bbadis.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Witko-Sarsat V, Gausson V, Nguyen AT, Touam M, Drueke T, Santangelo F, Descamps-Latscha B. AOPP-induced activation of human neutrophil and monocyte oxidative metabolism: a potential target for N-acetylcysteine treatment in dialysis patients. Kidney Int. 2003;64:82–91. doi: 10.1046/j.1523-1755.2003.00044.x. [DOI] [PubMed] [Google Scholar]
- 7.Capeillere-Blandin C, Gausson V, Nguyen AT, Descamps-Latscha B, Drueke T, Witko-Sarsat V. Respective role of uremic toxins and myeloperoxidase in the uremic state. Nephrol Dial Transplant. 2006;21:1555–1563. doi: 10.1093/ndt/gfl007. [DOI] [PubMed] [Google Scholar]
- 8.Marsche G, Hammer A, Oskolkova O, Kozarsky KF, Sattler W, Malle E. Hypochlorite-modified high density lipoprotein, a high affinity ligand to scavenger receptor class B, type I, impairs high density lipoprotein-dependent selective lipid uptake and reverse cholesterol transport. J Biol Chem. 2002;277:32172–32179. doi: 10.1074/jbc.M200503200. [DOI] [PubMed] [Google Scholar]
- 9.Marsche G, Zimmermann R, Horiuchi S, Tandon NN, Sattler W, Malle E. Class B scavenger receptors CD36 and SR-BI are receptors for hypochlorite-modified low density lipoprotein. J Biol Chem. 2003;278:47562–47570. doi: 10.1074/jbc.M308428200. [DOI] [PubMed] [Google Scholar]
- 10.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 11.Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 12.Fuh MM, Lee CM, Jeng CY, Shen DC, Shieh SM, Reaven GM, Chen YD. Effect of chronic renal failure on high-density lipoprotein kinetics. Kidney Int. 1990;37:1295–1300. doi: 10.1038/ki.1990.114. [DOI] [PubMed] [Google Scholar]
- 13.Shoji T, Nishizawa Y, Nishitani H, Billheimer JT, Sturley SL. Impaired metabolism of high density lipoprotein in uremic patients. Kidney Int. 1992;41:1653–1661. doi: 10.1038/ki.1992.238. [DOI] [PubMed] [Google Scholar]
- 14.Liu SX, Hou FF, Guo ZJ, Nagai R, Zhang WR, Liu ZQ, Zhou ZM, Zhou M, Xie D, Wang GB, Zhang X. Advanced oxidation protein products accelerate atherosclerosis through promoting oxidative stress and inflammation. Arterioscler Thromb Vasc Biol. 2006;26:1156–1162. doi: 10.1161/01.ATV.0000214960.85469.68. [DOI] [PubMed] [Google Scholar]
- 15.Vaziri ND. Dyslipidemia of chronic renal failure: the nature, mechanisms, and potential consequences. Am J Physiol Renal Physiol. 2006;290:262–272. doi: 10.1152/ajprenal.00099.2005. [DOI] [PubMed] [Google Scholar]
- 16.Anderstam B, Ann-Christin BH, Valli A, Stenvinkel P, Lindholm B, Suliman ME. Modification of the oxidative stress biomarker AOPP assay: application in uremic samples. Clin Chim Acta. 2008;393:114–118. doi: 10.1016/j.cca.2008.03.029. [DOI] [PubMed] [Google Scholar]
- 17.Marsche G, Weigle B, Sattler W, Malle E. Soluble RAGE blocks scavenger receptor CD36-mediated uptake of hypochlorite-modified low-density lipoprotein. FASEB J. 2007;21:3075–3082. doi: 10.1096/fj.07-8316com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsche G, Semlitsch M, Hammer A, Frank S, Weigle B, Demling N, Schmidt K, Windischhofer W, Waeg G, Sattler W, Malle E. Hypochlorite-modified albumin colocalizes with RAGE in the artery wall and promotes MCP-1 expression via the RAGE-Erk1/2 MAP-kinase pathway. FASEB J. 2007;21:1145–1152. doi: 10.1096/fj.06-7439com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marsche G, Heller R, Fauler G, Kovacevic A, Nuszkowski A, Graier W, Sattler W, Malle E. 2-chlorohexadecanal derived from hypochlorite-modified high-density lipoprotein-associated plasmalogen is a natural inhibitor of endothelial nitric oxide biosynthesis. Arterioscler Thromb Vasc Biol. 2004;24:2302–2306. doi: 10.1161/01.ATV.0000148703.43429.25. [DOI] [PubMed] [Google Scholar]
- 20.Marsche G, Frank S, Raynes JG, Kozarsky KF, Sattler W, Malle E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem J. 2007;402:117–124. doi: 10.1042/BJ20061406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohgami N, Nagai R, Miyazaki A, Ikemoto M, Arai H, Horiuchi S, Nakayama H. Scavenger receptor class B type I-mediated reverse cholesterol transport is inhibited by advanced glycation end products. J Biol Chem. 2001;276:13348–13355. doi: 10.1074/jbc.M011613200. [DOI] [PubMed] [Google Scholar]
- 22.Nagai R, Mera K, Nakajou K, Fujiwara Y, Iwao Y, Imai H, Murata T, Otagiri M. The ligand activity of AGE-proteins to scavenger receptors is dependent on their rate of modification by AGEs. Biochim Biophys Acta. 2007;1772:1192–1198. doi: 10.1016/j.bbadis.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Van Eck M, Hoekstra M, Hildebrand RB, Yaong Y, Stengel D, Kruijt JK, Sattler W, Tietge UJ, Ninio E, Van Berkel TJ, Praticò D. Increased oxidative stress in scavenger receptor BI knockout mice with dysfunctional HDL. Arterioscler Thromb Vasc Biol. 2007;27:2413–2419. doi: 10.1161/ATVBAHA.107.145474. [DOI] [PubMed] [Google Scholar]
- 24.Kalantar-Zadeh K, Kopple JD, Kamranpour N, Fogelman AM, Navab M. HDL-inflammatory index correlates with poor outcome in hemodialysis patients. Kidney Int. 2007;72:1149–1156. doi: 10.1038/sj.ki.5002491. [DOI] [PubMed] [Google Scholar]
- 25.Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindner A, Charra B, Sherrard DJ, Schribner BH. Accelerated atherosclerosis in prolonged maintenance haemodialysis. N Engl J Med. 1974;290:697–701. doi: 10.1056/NEJM197403282901301. [DOI] [PubMed] [Google Scholar]
- 28.London GM, Drueke TB. Atherosclerosis and arteriosclerosis in chronic renal failure. Kidney Int. 1997;51:1678–1695. doi: 10.1038/ki.1997.233. [DOI] [PubMed] [Google Scholar]
- 29.Descamps-Latscha B, Witko-Sarsat V, Nguyen-Khoa T, Nguyen AT, Gausson V, Mothu N, London GM, Jungers P. Advanced oxidation protein products as risk factors for atherosclerotic cardiovascular events in nondiabetic predialysis patients. Am J Kidney Dis. 2005;45:39–47. doi: 10.1053/j.ajkd.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Iwao Y, Nakajou K, Nagai R, Kitamura K, Anraku M, Maruyama T, Otagiri M. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am J Physiol Renal Physiol. 2008;295:1871–1880. doi: 10.1152/ajprenal.00013.2008. [DOI] [PubMed] [Google Scholar]
- 31.Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE) J Biol Chem. 2008;283:27255–27269. doi: 10.1074/jbc.M801622200. [DOI] [PubMed] [Google Scholar]
- 32.Guo ZJ, Niu HX, Hou FF, Zhang L, Fu N, Nagai R, Lu X, Chen BH, Shan YX, Tian JW, Nagaraj RH, Xie D, Zhang X. Advanced oxidation protein products activate vascular endothelial cells via a RAGE-mediated signaling pathway. Antioxid Redox Signal. 2008;10:1699–1712. doi: 10.1089/ars.2007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]