

Background: Expression of breast tumor kinase (Brk) is linked to breast carcinoma.
Results: SOCS3 binds to Brk and inhibits its ability to tyrosine phosphorylate STAT3.
Conclusion: To date SOCS3 is the only negative modulator described for Brk, and it may play a significant role as a tumor suppressor.
Significance: Understanding the mechanism by which SOCS3 inhibits Brk provides knowledge to silence its action in tumor progression.
Keywords: Phosphotyrosine, Protein Kinases, Protein-Protein Interactions, Signal Transduction, STAT3, Brk, SOCS3
Abstract
Breast tumor kinase (Brk) was originally isolated from a human metastatic breast tumor, but also is found expressed in other epithelial tumors and in a subset of normal epithelia. Brk is a tyrosine kinase and its expression in breast carcinoma has been linked to tumor progression. The signal transducer and activator of transcription 3 (STAT3) is one of the substrate targets of Brk, and elevated tyrosine phosphorylation of STAT3 is known to contribute to oncogenesis. Conventional activation of STAT3 occurs in response to cytokine stimulation of Janus tyrosine kinases (JAK). One of the negative regulators discovered in cytokine signaling of the JAK-STAT pathway is the suppressor of cytokine signaling 3 (SOCS3). In this report we describe the finding that SOCS3 can also inhibit the unconventional target, Brk. Investigation of the mechanism by which SOCS3 inhibits Brk reveals the SOCS3 protein binds to Brk primarily via its SH2 domain, and its main inhibitory effect is mediated by the SOCS3 kinase inhibitory region (KIR). SOCS3 has only a modest effect on promoting Brk degradation, and this requires the C-terminal SOCS box domain. SOCS3 is the only known inhibitor of Brk, and knowledge of the mechanisms by which SOCS3 inhibits Brk may lead to methods that block Brk in cancer progression.
Introduction
Accumulating evidence points to a causal relationship between expression of breast tumor kinase (Brk)2 (also known as protein tyrosine kinase 6) and high-grade breast tumors (1–4). Although the function of Brk is not limited to the mammary gland, aberrant expression has been linked to breast cancer. Brk expression in breast carcinoma also correlates with overexpression of the human epidermal growth factor receptor (HER/EGFR/ErbB) family (5–8). The co-expression of ErbB and Brk has led to studies demonstrating physical association of Brk with ErbB, inhibition of ErbB internalization by Brk, Brk enhancement of the mitogenic and tumorigenic responses of ErbB with prolonged activation of MAPK pathways, and promotion of ErbB stimulated cell migration by Ras and Rac1 activation (3, 8–12).
Brk belongs to a family of tyrosine kinases distantly related to the Src family (2). Brk possesses both Src homology 2 (SH2) and SH3 domains that modulate its activity and play a role in substrate recognition (13, 14). However, it differs from the Src family in that it lacks myristoylation or palmitoylation signals and it is found in both cytoplasmic and nuclear compartments. In addition, the activity of Brk is correlated with its level of protein expression and is not dependent on an inducing ligand. To date more than ten substrates have been identified for Brk and these include two members of the signal transducer and activator of transcription (STAT) family, STAT3 and STAT5b (15, 16). The relevance of these substrates resides in the fact that constitutive activation of STAT3 and STAT5 by tyrosine phosphorylation has been causally linked to cancer development (17, 18). In addition, down-regulation of Brk in breast tumor cells leads to reduced tyrosine phosphorylation of STAT3 (19).
Cytokine activation of STAT3 is critical for a wide range of physiological responses (20). However, persistent cytokine signaling elicits deleterious effects, and for this reason negative regulation is critical to maintain an appropriate biological balance. STAT3 was first identified as a DNA-binding factor activated in response to interleukin-6 (IL-6) (21–23). Following IL-6 stimulation, Janus kinases (JAKs) that are associated with the receptor can tyrosine-phosphorylate both the receptor and recruited STAT3 (24). Phosphorylation of STAT3 promotes dimerization via reciprocal phosphotyrosine and SH2 domain interactions, and the dimers are able to bind specific DNA sequences. One of the genes induced in response to STAT3 activation is the suppressor of cytokine signaling 3 (SOCS3) (25). SOCS3 is a member of a family of SOCS proteins that negatively regulate cytokine signal transduction (26). SOCS3 binds to phosphotyrosine residues on cytokine receptors, and inhibits receptor-associated JAK activity (27). In addition, SOCS3 appears to target phosphorylated JAK signaling complexes for ubiquitin mediated proteosomal degradation (28). The down-modulation of JAK activity by SOCS3 serves a critical role in cytokine signaling homeostasis.
Continual activation of JAK/STAT pathways can promote cancer pathogenesis (18, 29, 30). The negative feedback of SOCS3 appears necessary to suppress inflammation and cellular proliferation. The promoter of the SOCS3 gene is often found silenced by methylation in cancers, and SOCS3 silencing allows unregulated cell growth (31). We have found that SOCS3 is not only a negative regulator of JAK cytokine signaling, it is also a negative regulator of Brk (15). To date this is the only negative modulator described for Brk, and it may play a significant role as a tumor suppressor. In this study we characterize how SOCS3 may play a role in negative feedback of Brk signaling and evaluate the mechanisms by which SOCS3 elicits this effect.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
HeLa, COS1, Hep3B, T47D, and MDA-MB-231 cells were cultured according to ATCC's guidelines. Cell proliferation was analyzed by trypan blue exclusion method and Countess automated cell counter system (Invitrogen). Doxycycline (Sigma) was used at 2 μg/ml, MG132 (Sigma) was used at 5 μm. DNA transfections were performed with TransIT-LT1 Reagent (Mirus).
Plasmids and Lentiviruses
The STAT3-GFP, Brk-V5, and Flag-SOCS3 plasmids have been described (15, 25, 32). The 3xFLAG-Brk dlSH3, dlSH2, and dlTK were previously described (13) or generated by cloning Brk-(1–190) a.a. into the p3xFLAG-CMV-7.1 vector (Sigma). The wild type or K219M mutant of Brk cDNA was PCR amplified and cloned into the pEGFP-C2 vector. Brk Y251F in GFP-Brk or Brk-V5 plasmid was generated by site-directed mutagenesis (Stratagene). SOCS3 deletion mutants were generated by PCR and cloned into the pEF-Flag-I vector, and SOCS3 point mutants were generated by site-directed mutagenesis. SOCS3 cDNA was cloned into the pGEX-KG vector in frame with GST for bacterial expression and purification. The tetracycline-inducible Brk (TO/Brk-myc) was generated by PCR and cloned into the pcDNA4/TO/myc-His vector (Invitrogen). The His-Ub plasmid has been described (33). Cells were infected with lentiviruses generated with pLenti6.3/V5-DEST Gateway Vector Kit (Invitrogen) to express SOCS3 or GFP. Cells were infected with a moi of 4 using 8 μg/ml of Polybrene (SIGMA). Four days post-infection cells were selected for lentiviral integration in 2.5–5 μg/ml blastidin for 2 weeks.
RT-PCR
RNA extraction was performed with SurePrep TrueTotal RNA Purification kit (Fisher) and cDNA was synthesized with M-MLV Reverse Transcriptase (Promega). PCR was performed with primers: SOCS3, forward 5′-CAGCTGGTGGTGAACGCAGTG and reverse 5′-GATGTAATAGGCTCTTCTGGG, and GAPDH, forward 5′-GGAGCCAAAAGGGTCATCATCTC and reverse 5′-AGTGGGTGTCGCTGTTGAGTC.
Bacterial Expression and Purification of GST-SOCS3
pGEX-KG plasmid expressing SOCS3 was transformed into BL21 codon plus bacteria and protein expression was induced with 0.2 mm IPTG in LB broth at 22 °C for 18 h. GST-SOCS3 was purified by batch affinity purification with glutathione-agarose beads (Sigma). Free glutathione was removed by dialysis after protein preparation. Concentration of the GST-SOCS3 protein was determined by Bio-Rad Protein Assay. The purified protein was stored at −80 °C until use.
In Vitro Binding Assay
Purified recombinant GST-SOCS3 protein or GST protein (15 μg each) was used in each binding reaction. Cell lysates containing GFP or GFP-Brk were prepared in RIPA buffer (50 mm Tris·Cl pH7.5, 150 mm NaCl, 5 mm EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS). Cellular protein (500 μg) was incubated with GST-SOCS3 protein bound to glutathione-agarose beads for 3 h, washed, and eluted. The eluted proteins were analyzed by Western blot. Images were captured with enhanced chemiluminescence system and x-ray film, and presented with Adobe Photoshop.
Immunoprecipitation and Western Blot
For co-immunoprecipitation of endogenous Brk and SOCS3, T47D cells were lysed (400 mm NaCl, 50 mm Tris-pH 7.5, 5 mm EDTA, 0.5% Nonidet P-40, 10% glycerol), and 4 mg proteins were reacted overnight at 4 °C with either 2 μg of control rabbit IgG (Santa Cruz Biotechnology SC-2027) or rabbit anti-Brk antibody (Santa Cruz Biotechnology SC-1188). Immunocomplexes were collected on protein G agarose (Invitrogen), separated by SDS-PAGE and analyzed by Western blot with murine anti-SOCS3 antibody (Santa Cruz Biotechnology SC-51669) and murine anti-Brk (Santa Cruz Biotechnology SC-66003). Cells transfected with indicated plasmids were lysed (50 mm Tris·Cl pH8.0, 280 mm NaCl, 5 mm EDTA, and 0.5% Nonidet P-40) and 500 μg proteins were incubated with 1 μg of rabbit anti-SOCS3 antibody or control rabbit IgG (H103, Santa Cruz Biotechnology) at 4 °C for 3 h. Immunocomplexes were collected with protein G beads and analyzed by Western blot. Images were captured with enhanced chemiluminescence system and x-ray film, and presented with Adobe Photoshop. Primary antibodies used for Western blot included anti-phosphotyrosine STAT3 (B7, Santa Cruz Biotechnology), anti-STAT3 (H190, Santa Cruz Biotechnology), anti-Brk (C18, Santa Cruz Biotechnology), anti-c-Myc (9E10, Santa Cruz Biotechnology) anti-GFP (Roche), anti-V5 (Invitrogen), anti-Flag (M2, Sigma) and anti-α-tubulin (Sigma).
His-ubiquitination Assay
Cells were co-transfected with His-Ub, TO/Brk-myc, and pcDNA6/TR (Invitrogen) with or without Flag-SOCS3 or SOCS3 dlbox (1–185 a.a.) for 24 h and then treated with doxycycline with or without MG132 for 24 h. Cells were harvested and total histidine-tagged proteins were purified on nickel charged resins (Ni-NTA-agarose beads, Qiagen) (34). Myc-tagged Brk proteins were detected by Western blot with anti-c-Myc antibody. Images were captured with enhanced chemiluminescence system and x-ray film, and presented with Adobe Photoshop.
RESULTS
Association of Endogenous Brk and SOCS3
Previously we demonstrated the ability of ectopic expression of SOCS3 to inhibit Brk phosphorylation of STAT3 on tyrosine 705 (15). This result suggested SOCS3 could bind Brk and inhibit its tyrosine kinase activity. To evaluate protein interactions of endogenous proteins, we tested Brk and SOCS3 association by co-immunoprecipitation from T47D cells, a breast tumor cell line that expresses Brk. Brk was immunoprecipitated from cell lysates, and immunocomplexes were analyzed by Western blot with antibody to SOCS3 (Fig. 1A). Immunocomplexes with antibody to Brk clearly detected associated SOCS3 protein. This result indicates an association of endogenous SOCS3 with Brk in vivo.
FIGURE 1.

A, co-immunoprecipiation of endogenous SOCS3 and Brk. T47D breast cancer cell lysates were immunoprecipitated (IP) with control rabbit immunoglobulin (cIg) or rabbit anti-Brk. Western blots (WB) were performed with murine antibody to SOCS3 or Brk. Molecular mass markers are shown adjacent to 20% input. B, induction of endogenous SOCS3 mRNA with Brk expression. Hep3B cells were transfected with tetracycline-inducible Brk and tetracycline repressor for 24 h and treated with doxycycline for 4, 6, 8, 12, and 24 h. Total RNA was prepared and endogenous SOCS3 mRNA was detected by RT-PCR using specific primers. GAPDH mRNA was amplified as internal control. Brk expression was assayed from cell lysates by SDS-PAGE and Western blot (WB) with specific c-Myc (Brk tag) antibodies (bottom panel).
SOCS3 Gene Is Induced in Response to Brk
Biological systems are usually self-limiting. Our observation that SOCS3 could inhibit the action of Brk suggested the SOCS3 gene to be a downstream target and negative regulator of Brk. Enzymatic activity of Brk is constitutive and is coincident with expression of the protein. Since Brk is not induced or activated by a known ligand, we evaluated the effects of its expression with a tetracycline-inducible system. We co-expressed an inducible Brk gene and a gene encoding the tetracycline repressor engineered to be an activator in response to the tetracycline derivative doxycycline. The tetracycline-inducible Brk gene was introduced in cells that lack detectable expression of Brk, and its effects were evaluated on expression of the endogenous SOCS3 gene. Cells were treated with doxycycline, and SOCS3 mRNA levels were evaluated by reverse transcription polymerase chain reaction (RT-PCR) (Fig. 1B). Expression of the Brk gene was readily detected by 4 h following doxycycline, and induction of endogenous SOCS3 mRNA was apparent by 6 h. The data provide clear evidence that endogenous SOCS3 is induced in response to expression of Brk, as would be expected for a limiting negative regulator. The mRNA levels of SOCS3 decreased with time, similar to the transient response seen with IL-6 stimulation (35). The results support the reasoning that Brk activates STAT3 which in turn induces the expression of SOCS3 by binding to a target site in the promoter of the SOCS3 gene (36).
The KIR Domain in SOCS3 Is Required for Inhibition of the Brk Kinase
The exact means by which SOCS3 inhibits IL-6 cytokine signaling appears to be multidimensional involving receptor binding, JAK kinase inhibition, and JAK degradation (37, 38). A recent study demonstrates SOCS3 inhibits JAK2 by a mechanism that does not involve competition for either ATP or substrate binding (27) The SOCS proteins were originally characterized to possess a conserved C-terminal module of about 40 amino acids termed the SOCS box (Fig. 2A). The SOCS box was later found to facilitate binding to an E3 ubiquitin ligase. The SOCS proteins also have a central SH2 domain that is involved in binding to tyrosine-phosphorylated substrates, and an extended region of the SH2 domain that contributes to substrate binding called the extended SH2 domain (ESS). SOCS3 has a PEST domain that signals its rapid protein degradation and may contribute to decreasing levels seen in the Brk inducible expression system (Fig. 1B) (39). The N terminus of the SOCS family is varied, and the SOCS3 protein contains a small kinase inhibitory domain (KIR).
FIGURE 2.

Requirement of SOCS3 KIR domain for Brk inhibition. A, linear diagram of SOCS3 deletion mutants. Numbers correspond to amino acids. B, COS1 cells were co-transfected with STAT3-GFP, Brk-V5 with or without Flag-SOCS3 full-length (FL) or SOCS3 deletion mutants, and serum-starved for 24 h. Cell lysates were subjected to SDS-PAGE and Western blot with specific anti-STAT3 phosphotyrosine, anti-GFP (STAT3 tag), anti-V5 (Brk tag) and anti-Flag (SOCS3 tag) antibodies. C, cells were co-transfected with STAT3-GFP, Brk-V5 with or without Flag-SOCS3 wide type (WT) or SOCS3 L22D, F25A, or double mutant LF22/25DA (LF). Lysates were prepared and analyzed by SDS-PAGE and Western blot with specific anti-STAT3 phosphotyrosine, anti-GFP, anti-V5, and anti-Flag antibodies.
To determine the mechanism by which SOCS3 inhibits Brk, we first evaluated, which domains are required for the inhibition of kinase activity (Fig. 2B). Several SOCS3 deletion mutants were generated and tested for their ability to inhibit Brk-induced STAT3 phosphorylation. Cells were co-transfected with genes encoding STAT3-GFP and Brk, with or without SOCS3 deletion mutants, and STAT3 phosphorylation was detected by Western blot with a specific STAT3 phosphotyrosine antibody. Full-length SOCS3 was clearly able to suppress phosphorylation of STAT3 by Brk in this system. However, the SOCS3 N-terminal deletions expressing amino acids 34–225 or 46–225 were not able to suppress STAT3 phosphorylation. Both of these deletion mutants lack the KIR domain, suggesting the KIR domain plays a critical role in the inhibition of Brk activity. Expression of a SOCS3 deletion mutant that lacks the SOCS box (amino acids 1–185) was fully competent to inhibit Brk-mediated STAT3 phosphorylation similar to full length SOCS3. These results indicate that the SOCS box is not required for the ability of SOCS3 to inhibit Brk, but suggest the KIR domain is necessary.
Closer examination of the role of the KIR domain was performed with site directed mutagenesis. A number of SOCS3 KIR point mutants, including L22D and F25A, have been reported to abrogate the ability of SOCS3 to inhibit cytokine signaling (40). For this reason we generated KIR single mutants L22D and F25A, and double mutant LF22/25DA and tested whether SOCS3 with these point mutations could inhibit Brk phosphorylation of STAT3. The results indicated that SOCS3 L22D and F25A partially blocked STAT3 phosphorylation by Brk, but the double mutant LF22/25DA abolished the ability of SOCS3 to inhibit Brk activity (Fig. 2C). These results confirm the requirement of the KIR domain for Brk inhibition by SOCS3.
SOCS3 Binding to Brk
SOCS3 inhibits IL-6 signaling by binding to a phosphorylated tyrosine site on the cytoplasmic domain of the gp130 receptor and by binding JAK2. SOCS3 inhibits JAK2 kinase activity, and appears to target the receptor for proteosomal degradation (27, 39, 41, 42). We demonstrated endogenous SOCS3 and Brk proteins interact (Fig. 1), but to evaluate their interaction in greater detail, we examined the physical interaction of recombinant proteins. Co-immunoprecipitation and Western blot studies with cells expressing GFP-tagged Brk and Flag-tagged SOCS3 demonstrated Brk was specifically detected in immune complexes with SOCS3, but was not detected with control antibody. (Fig. 3A). To determine direct physical association of Brk and SOCS3, we next evaluated whether bacterially expressed SOCS3 could bind Brk in vitro. Purified glutathione S-transferase (GST) or GST-SOCS3 fusion protein was immobilized on glutathione agarose beads and incubated with lysates from cells expressing Brk (Fig. 3B). Brk was detected by Western blot and clearly demonstrated binding to GST-SOCS3 protein but not to the GST control. The results indicate that SOCS3 binds directly to the Brk protein.
FIGURE 3.
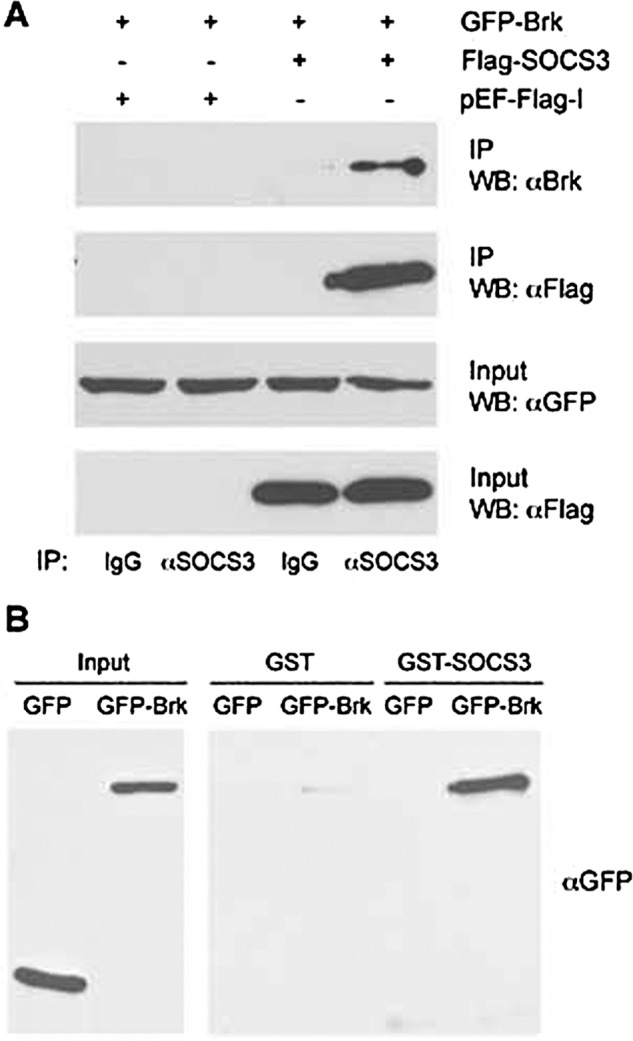
The interaction between SOCS3 and Brk. A, co-immunoprecipitation between SOCS3 and Brk. COS1 cells were co-transfected with GFP-Brk and Flag-SOCS3 or empty vector. Forty-eight hours after transfection, the cells were lysed, immunoprecipitated (IP) with anti-SOCS3 antibody or control rabbit IgG, and detected by Western blot (WB) with anti-Brk or anti-Flag antibody. An aliquot of each input was assayed with anti-GFP or anti-Flag antibody. B, in vitro binding assay of SOCS3 and Brk. GST-SOCS3 or GST proteins were immobilized on glutathione-agarose beads and incubated with lysates from cells expressing GFP-Brk or GFP. Bound Brk proteins were detected by Western blot with anti-GFP antibody.
Interaction between SOCS3 SH2 Domain and Brk
The SOCS3 protein binds to tyrosine-phosphorylated gp130 receptor by its SH2 domain (39, 41, 42). To examine whether the SH2 domain of SOCS3 mediates binding to Brk, we evaluated the behavior of several constructs containing regions of the SH2 domain. Three constructs were generated: SOCS3 amino acids 46–185 encoding the entire SH2 domain with the embedded 35 amino acid PEST motif; SOCS3 amino acids 130–225 containing the PEST motif, C-terminal portion of the SH2 domain, and the SOCS box; and SOCS3 amino acids 46–129 containing the N-terminal portion of the SH2 domain (39) (Fig. 4A). Cells expressing GFP-tagged Brk and either Flag-tagged full-length SOCS3 or the three SH2 domain constructs were tested for protein-protein association. The SOCS3 proteins were immunoprecipitated with anti-SOCS3 antibody and Brk was detected in immune complexes by Western blot. The results indicate that full-length SOCS3, the complete SOCS3 SH2 domain (46–185 a.a.), and the amino portion of the SOCS3 SH2 domain (46–129 a.a.) bind to Brk. However the SOCS3 fragment containing the carboxyl portion of the SH2 domain and the SOCS box (130–225 a.a.) was not able to bind to Brk. The results suggest that the binding between SOCS3 and Brk is mediated by the SOCS3 SH2 domain.
FIGURE 4.

SOCS3 SH2 domain binds to Brk. A, COS1 cells were co-transfected with GFP-Brk and Flag-SOCS3 full-length (FL) or SOCS3 deletion mutants expressing 46–185 a.a, 130–225 a.a., or 46–129 a.a.. Cells lysates were immunoprecipitated (IP) with anti-SOCS3 antibody and analyzed by Western blot (WB) with anti-GFP (Brk) antibody. An aliquot of each input was analyzed by Western blot with anti-GFP or anti-Flag antibody shown in lower panels. B, cells were co-transfected with GFP-Brk with or without Flag-SOCS3 SH2 46–129 a.a. or the 46–129 a.a. R71E point mutation. Cells lysates were prepared, immunoprecipitated with anti-SOCS3 antibody and analyzed by Western blot with anti-GFP antibody. An aliquot of each input was analyzed by Western blot with anti-GFP or anti-Flag antibody (lower panels).
Mutagenesis studies of SOCS3 have identified a critical arginine 71 within the SH2 domain that contributes to binding phosphotyrosine targets (40). To study the role of arginine 71 in the ability of the SOCS3 SH2 domain to interact with Brk, we mutated arginine 71 to glutamic acid (R71E) in the SOCS3 construct containing 46–129 amino acids. The SOCS3 mutation was expressed with Brk and evaluated for its ability to co-immunoprecipitate with Brk (Fig. 4B). The arginine mutation in SOCS3 46–129 R71E abolished its binding to Brk. These results indicate the SH2 domain is involved in binding to Brk.
SOCS3 Binds to the Tyrosine Kinase Domain of Brk
Brk possesses a tyrosine kinase domain located at its C terminus, a central SH2 domain, and a SH3 domain at the N terminus (2). To determine if a particular region of Brk is involved in association with SOCS3, we assessed the ability of various deletion mutants of Brk to bind SOCS3 (Fig. 5). The SOCS3 protein was produced as a bacterial GST fusion and bound to glutathione-agarose beads. As a source of Brk proteins, mammalian cells were transfected with genes encoding Flag-tagged Brk lacking the SH3 domain, or the SH2 domain, or the kinase domain. Cell lysates were incubated with the GST-SOCS3 bound to glutathione beads and bound proteins were eluted. The presence of Brk in the complexes was detected by Western blot. The results indicate that Brk deletions lacking the SH2 domain or the SH3 domain both bind efficiently to SOCS3 (Fig. 5A). However, deletion of the tyrosine kinase domain from Brk abrogates SOCS3 binding. These data suggest that the kinase domain of Brk is required for the interaction with SOCS3.
FIGURE 5.

The tyrosine kinase domain in Brk mediates interaction with SOCS3. A, binding assays of SOCS3 and Brk deletion mutants. Bacterially expressed GST or GST-SOCS3 proteins were immobilized on glutathione-agarose beads and incubated with lysates from cells expressing Flag-Brk or Flag-Brk deleted for the SH3 domain (dlSH3), the SH2 domain (dlSH3), or the tyrosine kinase domain (dlTK). Bound Brk proteins were detected by Western blot with anti-Flag antibody. Cell lysates were analyzed as input controls. B, co-immunoprecipitation between SOCS3 and Brk K219M. Cells were co-transfected with GFP-Brk wild type (WT) or Brk K219M with or without Flag-SOCS3. Cells lysates were immunoprecipitated (IP) with anti-SOCS3 antibody and Western blots (WB) were performed with anti-GFP or anti-Flag antibody. An aliquot of each input was analyzed with anti-GFP or anti-Flag antibody. C, binding of wild type GFP-Brk (WT) or GFP-Brk with a site mutation Y251F was evaluated. Protein lysates from cells expressing the Brk proteins were incubated with immobilized GST or GST-SOCS3 proteins. Bound Brk was evaluated by Western blot with Brk antibodies. Brk protein input is shown in bottom panel. D, Brk Y251F kinase activity is not impaired. Lysates from cells co-expressing STAT3-GFP with WT Brk or Brk Y251F were subjected to SDS-PAGE and Western blot with specific anti-STAT3 phosphotyrosine, anti-STAT3, or anti-Brk antibody. E, conceptual model of SOCS3 binding and inhibition of Brk.
The kinase activity of Brk has been shown to be dependent on the ATP binding site at lysine 219 (14). A mutation substituting methionine for lysine 219 (K219M) renders the Brk kinase inactive so that it does not display the usual autophosphorylation activity. To determine if the kinase activity of Brk was required for its recognition by SOCS3, binding to the kinase dead Brk mutant was tested. Brk K2191M showed impaired binding (estimate 4-fold decrease relative to input), indicating that the auto-phosphorylation site of Brk contributes to recognition by SOCS3 (Fig. 5B). However, it is possible that other phosphorylated tyrosines in the kinase domain participate.
There are seventeen tyrosine residues in the Brk protein, and a specific tyrosine in the kinase domain matches a described consensus binding motif for SOCS3. Binding studies were performed with the SOCS3 SH2 domain and a library of phosphorylated tyrosine peptides. The studies identified selected sequences containing hydrophobic residues carboxyl to the phosphorylated tyrosine, especially with valine at the +3 position (43). This binding preference was also found in the crystal structure of SOCS3 in complex with the gp130 IL-6 receptor subunit (39). We identified a sequence in Brk at amino acids 249–256 (ALYAVVSV) that matched the binding site consensus, and evaluated its contribution to SOCS3 binding. A mutation was introduced in Brk substituting the tyrosine 251 a.a. with phenylalanine (Y251F), and binding to GST-SOCS3 was evaluated (Fig. 5C). The Brk Y251F mutation greatly impaired the ability of SOCS3 to bind. To ensure that Y251F did not reduce the kinase activity of Brk, phosphorylation of STAT3 was evaluated (Fig. 5D). The Brk Y251F site mutation did not reduce the ability of Brk to tyrosine phosphorylate STAT3. The results indicate that phosphorylated tyrosine 251 is a target for SOCS3 recognition.
Our analyses of the ability of SOCS3 to bind and inhibit Brk are consistent with a model in which the SH2 domain of SOCS3 binds to phosphotyrosine 251 in the Brk kinase domain. The binding allows the correct positioning of the KIR domain in SOCS3 to inhibit the tyrosine kinase of Brk (Fig. 5E). This physical interaction between SOCS3 and Brk phosphotyrosine 251 appears to be a primary but not exclusive interaction, as other tyrosine residues or Brk domains may also contribute to SOCS3 binding.
SOCS3 Has only Modest Effects on Brk Ubiquitination and Protein Levels
Evidence suggests that the SOCS box of SOCS proteins participates in the formation of E3 ligase complexes that can target substrates for ubiquitin-mediated proteasomal degradation. The SOCS box can interact with elongin C and B and additional recruitment of cullin5 and Rbx1 completes the formation of an E3 ligase complex (38). SOCS3 has been reported to target CD33-related receptors for proteasomal degradation, and to promote destruction of insulin receptor substrates and focal adhesion kinase (44–46). However, the major inhibitory action of SOCS3 on JAK2 does not appear to be dependent on degradation (27). For these reasons we determined whether SOCS3 could promote proteasomal degradation of Brk. To enhance detection of ubiquitination, a histidine-tagged ubiquitin gene was expressed in cells with the tetracycline inducible Brk system. Histidine-ubiquitin and ligated complexes were captured on nickel charged resins and Brk was detected by Western blot (Fig. 6A). Results indicated Brk was extensively ubiquitinated, with monoubiquitin and multiubiquitin or polyubiquitin chains independent of SOCS3 expression. Expression of SOCS3 modestly reduced the total amount of Brk protein in whole cell lysates less than 2-fold (WCL:Brk), and this was consistent with a partial decrease in the apparent ubiquitination of Brk. A proteasome inhibitor MG132 greatly increased ubiquitination of Brk and restored the Brk protein levels. To investigate whether the decrease in Brk levels promoted by SOCS3 was dependent on the SOCS box domain, we evaluated the effect of SOCS3 deleted for the SOCS box (1–185 a.a.). Expression of SOCS3 deleted for the SOCS box had no effect on Brk protein levels (Fig. 6B). Together the results indicate that SOCS3 can modestly reduce the protein level of Brk, and this affect requires the SOCS box.
FIGURE 6.

SOCS3 targets Brk for proteasomal degradation via the SOCS box. A, Brk levels decrease with SOCS3. Cells were transfected with tetracycline-inducible Brk, tetracycline repressor and His-Ub with or without Flag-SOCS3 for 24 h and treated with doxycycline with or without MG132 for 24 h. Histidine-tagged proteins were purified using Ni-NTA-agarose beads and Myc-tagged Brk proteins were detected by Western blot with anti-c-Myc antibody. An aliquot of each whole cell lysate (WCL) was analyzed by Western blot with anti-c-Myc, anti-Flag, or anti-tubulin antibody. B, SOCS box is required for Brk degradation. Cells were transfected with tetracycline-inducible Brk, tetracycline repressor, and His-Ub with or without Flag-SOCS3 WT or SOCS3 with a deletion in the SOCS box (dlbox) for 24 h and treated with doxycycline for 24 h. Protein level and ubiquitination of Brk were detected as described in A.
Increased SOCS3 Expression Inhibits Breast Tumor Cell Proliferation
We have shown previously that breast tumor cell lines such as MDA-MB-231 express Brk concomitant with tyrosine-phosphorylated STAT3 (15). To evaluate the biological effects of SOCS3 on proliferation of breast tumor cells, we increased expression of SOCS3 by lentiviral gene transduction. MDA-MB-231 cells were infected with lentivirus expressing GFP as a control or the SOCS3 gene and selected for stable expression. Cells were plated at similar densities and evaluated for proliferation by trypan blue exclusion over the course of several days. Results of multiple independent experiments indicated that expression of SOCS3 inhibits the proliferation of these breast tumor cells (Fig. 7A). Western blots demonstrated the increased expression correlated with reduced STAT3 tyrosine phosphorylation (Fig. 7B). Similar results were obtained with another Brk positive breast tumor line, T47D (data not shown). Increased expression of SOCS3 has a negative impact on the proliferation of these breast tumor cells.
FIGURE 7.
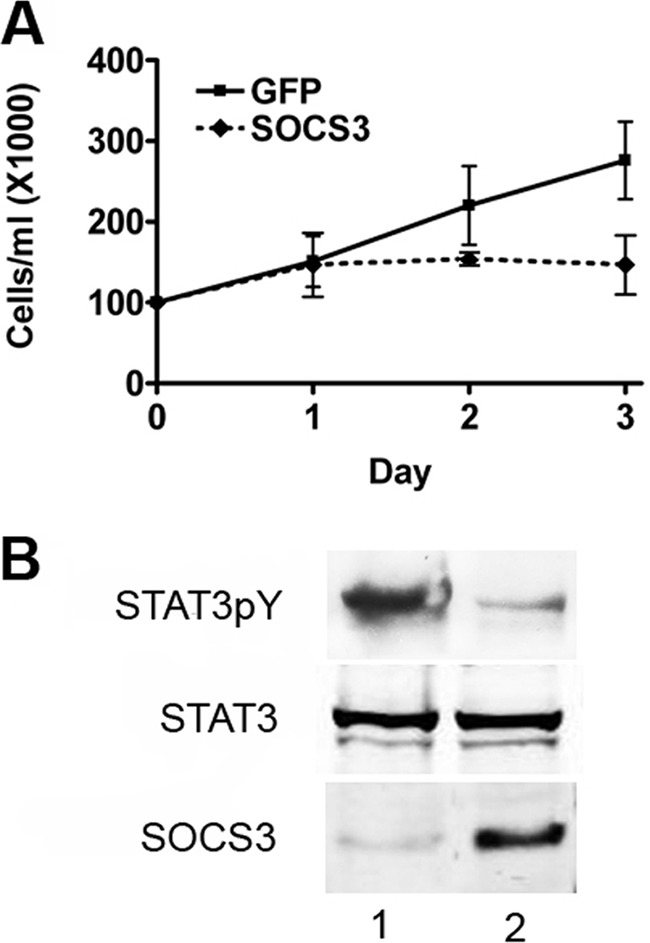
SOCS3 inhibits growth of breast cancer cell line MDA-MB-231. A, lentivirus infection was used to stably express SOCS3 or GFP in the MDA-MB-231 cell line. Similar cell numbers were seeded and proliferation was measured by trypan blue exclusion. Results are graphed showing the standard deviation of three independent experiments. B, Western blot analysis of MDA-MB-231 cells used in A expressing GFP (lane 1) or SOCS3 (lane 2) performed with antibodies to tyrosine phosphorylated STAT3 (STAT3pY), STAT3, or SOCS3.
DISCUSSION
The precise functions of Brk in normal tissues and cancer are only beginning to be understood. Enhanced expression of Brk correlates with tumorigenesis, whereas disruption of the murine Brk gene leads to increased growth of the small intestine (2, 48). The effects of Brk may be context dependent and influenced by positive or negative regulators. The ability to engage or disengage Brk can have a profound impact on a single cell and as a consequence on the entire organism. Therefore it is critical to understand the molecular mechanisms that regulate Brk to block its ability to promote cancer.
We identified the STAT3 transcription factor as one of the substrates of Brk (15). Persistent tyrosine phosphorylation of STAT3 induces the expression of genes that positively control cell survival, proliferation, and tumor cell invasion (18, 49–51). The SOCS3 gene is induced in response to activated STAT3, and this led to the finding that SOCS3 can inhibit the ability of Brk to phosphorylate STAT3 (15). In this report we demonstrate that expression of Brk can induce the endogenous SOCS3 gene, suggesting this is a negative feedback mechanism for Brk activity. SOCS3 is the only negative regulator of Brk as yet described. In breast cancer, loss of SOCS3 expression is associated with poor clinical outcome (52).
Our studies demonstrate a physical interaction between SOCS3 and Brk. Analyses of various mutations revealed the association appears to be mediated primarily by the SH2 domain of SOCS3 and the tyrosine kinase domain of Brk (Fig. 4). The kinase activity of Brk contributes to this interaction since SOCS3 binding to a kinase dead mutant is reduced (Fig. 5). However, the binding of SOCS3 to Brk is not sufficient to inhibit Brk activity. Inhibition requires the kinase inhibitory region (KIR) that is present at the amino-terminal region of SOCS3. Point mutations of the KIR domain abrogate the ability of SOCS3 to inhibit Brk (Fig. 2). In cytokine signaling, the SOCS3 KIR domain also appears to inhibit JAKs. The KIR domain was originally thought to act as a pseudo substrate (40), but recent evidence suggests the KIR domain is a noncompetitive inhibitor of JAK2 and that SOCS3 binds to both JAK2 and the tyrosine phosphorylated cell surface receptor (27). From our studies it appears that the SH2 domain of SOCS3 binds phosphotyrosines in Brk and the KIR domain inhibits Brk kinase activity. The SOCS3 KIR domain may also serve as a low affinity supplementary binding site for Brk in conjunction with the SH2 domain.
All SOCS proteins have a conserved C-terminal SOCS box. The SOCS box is able to bind to elongin C, a component of the ECS-type E3 ubiquitin ligase complex (38). SOCS proteins can thereby target associated proteins for ubiquitination. Ubiquitination can be formed by isopeptide linkages with seven different lysines on ubiquitin, can occur as homotypic chains or mixed chains, and can occur as polymers or monomers (53). Polyubiquitination via lysine 48 on ubiquitin can target proteins for proteosomal degradation, and SOCS proteins have been found to regulate the ubiquitination and half-life of particular JAKs, receptors, and signaling molecules. For this reason we evaluated the effect of SOCS3 on Brk ubiquitination and degradation. Expression of SOCS3 with Brk was found to reduce the protein levels of Brk by less than 2-fold (Fig. 6). This reduction in Brk protein was dependent on the presence of the SOCS box in SOCS3. A 2-fold reduction in the level of Brk protein is not sufficient for the dramatic inhibitory effect of SOCS3 on Brk activity. Therefore the primary inhibitory effect of SOCS3 on Brk activity appears to be mediated by the KIR domain.
The results in this study suggest a primary a mechanism of action of SOCS3 on Brk activity. The SH2 domain of SOCS3 appears to bind to phosphotyrosines in the Brk tyrosine kinase domain. This binding positions the SOCS3 KIR domain so that it inhibits the ability of Brk to phosphorylate STAT3. The KIR domain may help to facilitate binding to Brk, but its primary role is inhibition of kinase activity. The SOCS box of SOCS3 is not necessary for inhibition of Brk although it appears to play a modest role in Brk stability. In addition to JAKs, SOCS3 has been shown to inhibit the activity of focal adhesion kinase (FAK) (46). Distinct from inhibition of Brk, the primary mechanism of SOCS3 action on FAK appeared to be ubiquitin-mediated degradation.
Accumulating evidence suggests the SOCS proteins manifest the signs of tumor suppressors (47). Clinical studies have found disruption of SOCS genes by mutation or transcriptional silencing by methylation correlates with tumorigenesis. In addition, aberrant phosphorylation of SOCS proteins in some cancers has been shown to inhibit the ability of SOCS proteins to associate with elongin C and the E3 ligase complex. Increased expression of SOCS3 in breast tumor cells that express Brk results in a decrease in their proliferation (Fig. 7). Although originally discovered to inhibit JAK/STAT cytokine signaling, the SOCS proteins appear to have a more global role in the regulation of proliferation. The negative effect of SOCS3 on Brk may be required to maintain a normal balance necessary to inhibit uncontrolled proliferative signals.
Acknowledgments
We thank past and present members of the N.C.R. laboratory for their support, especially Dr. Ling Liu. We also greatly appreciate the generosity of Dr. W. Todd Miller for Brk reagents and advice.
This work was supported, in whole or in part, by National Institutes of Health Grant R01CA122910 and the Carol Baldwin Foundation for Breast Cancer Research (to N. C. R.).
- Brk
- breast tumor kinase
- STAT
- signal transducer and activator of transcription
- SOCS
- suppressor of cytokine signaling
- KIR
- kinase inhibitory domain
- FAK
- focal adhesion kinase.
REFERENCES
- 1. Harvey A. J., Pennington C. J., Porter S., Burmi R. S., Edwards D. R., Court W., Eccles S. A., Crompton M. R. (2009) Brk protects breast cancer cells from autophagic cell death induced by loss of anchorage. Am. J. Pathol. 175, 1226–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brauer P. M., Tyner A. L. (2010) Building a better understanding of the intracellular tyrosine kinase PTK6 - BRK by BRK. Biochim. Biophys. Acta 1806, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ostrander J. H., Daniel A. R., Lofgren K., Kleer C. G., Lange C. A. (2007) Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer Res. 67, 4199–4209 [DOI] [PubMed] [Google Scholar]
- 4. Mitchell P. J., Barker K. T., Martindale J. E., Kamalati T., Lowe P. N., Page M. J., Gusterson B. A., Crompton M. R. (1994) Cloning and characterization of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene 9, 2383–2390 [PubMed] [Google Scholar]
- 5. Gschwind A., Fischer O. M., Ullrich A. (2004) The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 4, 361–370 [DOI] [PubMed] [Google Scholar]
- 6. Hynes N. E., Lane H. A. (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354 [DOI] [PubMed] [Google Scholar]
- 7. Born M., Quintanilla-Fend L., Braselmann H., Reich U., Richter M., Hutzler P., Aubele M. (2005) Simultaneous overexpression of the Her2/neu and PTK6 tyrosine kinases in archival invasive ductal breast carcinomas. J. Pathol. 205, 592–596 [DOI] [PubMed] [Google Scholar]
- 8. Xiang B., Chatti K., Qiu H., Lakshmi B., Krasnitz A., Hicks J., Yu M., Miller W. T., Muthuswamy S. K. (2008) Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 12463–12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kamalati T., Jolin H. E., Mitchell P. J., Barker K. T., Jackson L. E., Dean C. J., Page M. J., Gusterson B. A., Crompton M. R. (1996) Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J. Biol. Chem. 271, 30956–30963 [DOI] [PubMed] [Google Scholar]
- 10. Kang S. A., Lee E. S., Yoon H. Y., Randazzo P. A., Lee S. T. (2010) PTK6 inhibits down-regulation of EGF receptor through phosphorylation of ARAP1. J. Biol. Chem. 285, 26013–26021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen C. H., Chen H. Y., Lin M. S., Li F. Y., Chang C. C., Kuo M. L., Settleman J., Chen R. H. (2008) Breast tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res. 68, 7779–7787 [DOI] [PubMed] [Google Scholar]
- 12. Chen H. Y., Shen C. H., Tsai Y. T., Lin F. C., Huang Y. P., Chen R. H. (2004) Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol. Cell Biol. 24, 10558–10572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Qiu H., Miller W. T. (2004) Role of the Brk SH3 domain in substrate recognition. Oncogene 23, 2216–2223 [DOI] [PubMed] [Google Scholar]
- 14. Qiu H., Miller W. T. (2002) Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J. Biol. Chem. 277, 34634–34641 [DOI] [PubMed] [Google Scholar]
- 15. Liu L., Gao Y., Qiu H., Miller W. T., Poli V., Reich N. C. (2006) Identification of STAT3 as a specific substrate of breast tumor kinase. Oncogene 25, 4904–4912 [DOI] [PubMed] [Google Scholar]
- 16. Weaver A. M., Silva C. M. (2007) Signal transducer and activator of transcription 5b: a new target of breast tumor kinase/protein tyrosine kinase 6. Breast Cancer Res. 9, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schindler C., Levy D. E., Decker T. (2007) JAK-STAT signaling: from interferons to cytokines. J. Biol. Chem. 282, 20059–20063 [DOI] [PubMed] [Google Scholar]
- 18. Yu H., Pardoll D., Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ludyga N., Anastasov N., Gonzalez-Vasconcellos I., Ram M., Höfler H., Aubele M. (2011) Impact of protein tyrosine kinase 6 (PTK6) on human epidermal growth factor receptor (HER) signaling in breast cancer. Mol. Biosyst. 7, 1603–1612 [DOI] [PubMed] [Google Scholar]
- 20. Levy D. E., Darnell J. E., Jr. (2002) Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662 [DOI] [PubMed] [Google Scholar]
- 21. Wegenka U. M., Buschmann J., Lütticken C., Heinrich P. C., Horn F. (1993) Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the post-translational level. Mol. Cell Biol. 13, 276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lütticken C., Wegenka U. M., Yuan J., Buschmann J., Schindler C., Ziemiecki A., Harpur A. G., Wilks A. F., Yasukawa K., Taga T., et al. (1994) Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 263, 89–92 [DOI] [PubMed] [Google Scholar]
- 23. Akira S., Nishio Y., Inoue M., Wang X. J., Wei S., Matsusaka T., Yoshida K., Sudo T., Naruto M., Kishimoto T. (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71 [DOI] [PubMed] [Google Scholar]
- 24. Taga T., Kishimoto T. (1997) Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 15, 797–819 [DOI] [PubMed] [Google Scholar]
- 25. Starr R., Willson T. A., Viney E. M., Murray L. J., Rayner J. R., Jenkins B. J., Gonda T. J., Alexander W. S., Metcalf D., Nicola N. A., Hilton D. J. (1997) A family of cytokine-inducible inhibitors of signaling. Nature 387, 917–921 [DOI] [PubMed] [Google Scholar]
- 26. Croker B. A., Krebs D. L., Zhang J. G., Wormald S., Willson T. A., Stanley E. G., Robb L., Greenhalgh C. J., Forster I., Clausen B. E., Nicola N. A., Metcalf D., Hilton D. J., Roberts A. W., Alexander W. S. (2003) SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 4, 540–545 [DOI] [PubMed] [Google Scholar]
- 27. Babon J. J., Kershaw N. J., Murphy J. M., Varghese L. N., Laktyushin A., Young S. N., Lucet I. S., Norton R. S., Nicola N. A. (2012) Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 36, 239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Boyle K., Zhang J. G., Nicholson S. E., Trounson E., Babon J. J., McManus E. J., Nicola N. A., Robb L. (2009) Deletion of the SOCS box of suppressor of cytokine signaling 3 (SOCS3) in embryonic stem cells reveals SOCS box-dependent regulation of JAK but not STAT phosphorylation. Cell Signal. 21, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grivennikov S. I., Karin M. (2010) Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Iliopoulos D., Hirsch H. A., Struhl K. (2009) An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He B., You L., Uematsu K., Zang K., Xu Z., Lee A. Y., Costello J. F., McCormick F., Jablons D. M. (2003) SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. U.S.A. 100, 14133–14138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu L., McBride K. M., Reich N. C. (2005) STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-α3. Proc. Natl. Acad. Sci. U.S.A. 102, 8150–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Treier M., Staszewski L. M., Bohmann D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the delta domain. Cell 78, 787–798 [DOI] [PubMed] [Google Scholar]
- 34. Campanero M. R., Flemington E. K. (1997) Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc. Natl. Acad. Sci. U.S.A. 94, 2221–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Senn J. J., Klover P. J., Nowak I. A., Zimmers T. A., Koniaris L. G., Furlanetto R. W., Mooney R. A. (2003) Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 278, 13740–13746 [DOI] [PubMed] [Google Scholar]
- 36. He B., You L., Uematsu K., Matsangou M., Xu Z., He M., McCormick F., Jablons D. M. (2003) Cloning and characterization of a functional promoter of the human SOCS-3 gene. Biochem. Biophys. Res. Commun. 301, 386–391 [DOI] [PubMed] [Google Scholar]
- 37. Croker B. A., Kiu H., Nicholson S. E. (2008) SOCS regulation of the JAK/STAT signaling pathway. Semin Cell Dev. Biol. 19, 414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piessevaux J., Lavens D., Peelman F., Tavernier J. (2008) The many faces of the SOCS box. Cytokine Growth Factor Rev. 19, 371–381 [DOI] [PubMed] [Google Scholar]
- 39. Babon J. J., McManus E. J., Yao S., DeSouza D. P., Mielke L. A., Sprigg N. S., Willson T. A., Hilton D. J., Nicola N. A., Baca M., Nicholson S. E., Norton R. S. (2006) The structure of SOCS3 reveals the basis of the extended SH2 domain function and identifies an unstructured insertion that regulates stability. Mol. Cell 22, 205–216 [DOI] [PubMed] [Google Scholar]
- 40. Sasaki A., Yasukawa H., Suzuki A., Kamizono S., Syoda T., Kinjyo I., Sasaki M., Johnston J. A., Yoshimura A. (1999) Cytokine-inducible SH2 protein-3 (CIS3/SOCS3) inhibits Janus tyrosine kinase by binding through the N-terminal kinase inhibitory region as well as SH2 domain. Genes Cells 4, 339–351 [DOI] [PubMed] [Google Scholar]
- 41. Nicholson S. E., De Souza D., Fabri L. J., Corbin J., Willson T. A., Zhang J. G., Silva A., Asimakis M., Farley A., Nash A. D., Metcalf D., Hilton D. J., Nicola N. A., Baca M. (2000) Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc. Natl. Acad. Sci. U.S.A. 97, 6493–6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bergamin E., Wu J., Hubbard S. R. (2006) Structural basis for phosphotyrosine recognition by suppressor of cytokine signaling-3. Structure 14, 1285–1292 [DOI] [PubMed] [Google Scholar]
- 43. De Souza D., Fabri L. J., Nash A., Hilton D. J., Nicola N. A., Baca M. (2002) SH2 domains from suppressor of cytokine signaling-3 and protein tyrosine phosphatase SHP-2 have similar binding specificities. Biochemistry 41, 9229–9236 [DOI] [PubMed] [Google Scholar]
- 44. Orr S. J., Morgan N. M., Buick R. J., Boyd C. R., Elliott J., Burrows J. F., Jefferies C. A., Crocker P. R., Johnston J. A. (2007) SOCS3 targets Siglec 7 for proteasomal degradation and blocks Siglec 7-mediated responses. J. Biol. Chem. 282, 3418–3422 [DOI] [PubMed] [Google Scholar]
- 45. Rui L., Yuan M., Frantz D., Shoelson S., White M. F. (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J. Biol. Chem. 277, 42394–42398 [DOI] [PubMed] [Google Scholar]
- 46. Liu E., Côté J. F., Vuori K. (2003) Negative regulation of FAK signaling by SOCS proteins. EMBO J. 22, 5036–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elliott J., Hookham M. B., Johnston J. A. (2008) The suppressors of cytokine signaling E3 ligases behave as tumour suppressors. Biochem. Soc. Trans. 36, 464–468 [DOI] [PubMed] [Google Scholar]
- 48. Haegebarth A., Bie W., Yang R., Crawford S. E., Vasioukhin V., Fuchs E., Tyner A. L. (2006) Protein tyrosine kinase 6 negatively regulates growth and promotes enterocyte differentiation in the small intestine. Mol. Cell Biol. 26, 4949–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kortylewski M., Kujawski M., Wang T., Wei S., Zhang S., Pilon-Thomas S., Niu G., Kay H., Mule J., Kerr W. G., Jove R., Pardoll D., Yu H. (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11, 1314–1321 [DOI] [PubMed] [Google Scholar]
- 50. Bromberg J. F., Wrzeszczynska M. H., Devgan G., Zhao Y., Pestell R. G., Albanese C., Darnell J. E., Jr. (1999) Stat3 as an oncogene. Cell 98, 295–303 [DOI] [PubMed] [Google Scholar]
- 51. Chiarle R., Simmons W. J., Cai H., Dhall G., Zamo A., Raz R., Karras J. G., Levy D. E., Inghirami G. (2005) Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med. 11, 623–629 [DOI] [PubMed] [Google Scholar]
- 52. Ying M., Li D., Yang L., Wang M., Wang N., Chen Y., He M., Wang Y. (2010) Loss of SOCS3 expression is associated with an increased risk of recurrent disease in breast carcinoma. J. Cancer Res. Clin. Oncol. 136, 1617–1626 [DOI] [PubMed] [Google Scholar]
- 53. Ikeda F., Dikic I. (2008) Atypical ubiquitin chains: new molecular signals. 'Protein Modifications: Beyond the Usual Suspects' review series. EMBO Rep. 9, 536–542 [DOI] [PMC free article] [PubMed] [Google Scholar]