

Background: Proline-rich tyrosine kinase 2 (Pyk2) is implicated in synaptic plasticity; however, it remains unclear how Pyk2 is inactivated within neurons.
Results: Striatal-enriched protein-tyrosine phosphatase (STEP) directly binds to and dephosphorylates Pyk2 at Tyr402.
Conclusion: STEP inactivates Pyk2 and its downstream signaling pathways.
Significance: These results identify an important regulatory mechanism for Pyk2 signaling that is critical for understanding the molecular mechanisms underlying synaptic plasticity.
Keywords: Neuroscience, Signal Transduction, Synaptic Plasticity, Tyrosine Protein Kinase (Tyrosine Kinase), Tyrosine Protein Phosphatase (Tyrosine Phosphatase), KCl Depolarization, Pyk2, STEP
Abstract
Proline-rich tyrosine kinase 2 (Pyk2) is a member of the focal adhesion kinase family and is highly expressed in brain and hematopoietic cells. Pyk2 plays diverse functions in cells, including the regulation of cell adhesion, migration, and cytoskeletal reorganization. In the brain, it is involved in the induction of long term potentiation through regulation of N-methyl-d-aspartate receptor trafficking. This occurs through the phosphorylation and activation of Src family tyrosine kinase members, such as Fyn, that phosphorylate GluN2B at Tyr1472. Phosphorylation at this site leads to exocytosis of GluN1-GluN2B receptors to synaptic membranes. Pyk2 activity is modulated by phosphorylation at several critical tyrosine sites, including Tyr402. In this study, we report that Pyk2 is a substrate of striatal-enriched protein-tyrosine phosphatase (STEP). STEP binds to and dephosphorylates Pyk2 at Tyr402. STEP KO mice showed enhanced phosphorylation of Pyk2 at Tyr402 and of the Pyk2 substrates paxillin and ASAP1. Functional studies indicated that STEP opposes Pyk2 activation after KCl depolarization of cortical slices and blocks Pyk2 translocation to postsynaptic densities, a key step required for Pyk2 activation and function. This is the first study to identify Pyk2 as a substrate for STEP.
Introduction
Proline-rich tyrosine kinase 2 (Pyk2),4 also known as cell adhesion kinase β, is a member of the focal adhesion kinase family and is highly expressed in the central nervous system (CNS) (1–3). Pyk2 is activated in response to increases in intracellular calcium levels (1, 4, 5), neuronal membrane depolarization (6, 7), hyperosmolarity (7), and activation of protein kinase C (1, 8, 9).
Upon stimulation, Pyk2 undergoes rapid autophosphorylation at Tyr402, creating a docking site for the SH2 domain of Src family kinase members Src and Fyn. Pyk2 phosphorylates and activates Src and Fyn (10–12), either one of which then phosphorylates Pyk2 at Tyr579/580 within its activation loop (13, 14). Pyk2 activation results in postsynaptic translocation and association of Pyk2 with the SH3 domain of postsynaptic density (PSD)-95 (15), which is essential for Pyk2 activation and the induction of long term potentiation (LTP) (15). Src also phosphorylates Pyk2 at Tyr881, leading to recruitment of the Grb2-SOS complex and subsequent activation of mitogen-activated protein kinase (MAPK) (1, 16). Thus, Pyk2 activates the Src family kinase and MAPK signaling pathways, both of which are required for the induction and maintenance of LTP (17, 18). Overexpression of a catalytically inactive mutant of Pyk2 (K457A) or disruption of Pyk2/PSD-95 interactions disrupts LTP, underscoring the importance of Pyk2 in regulating synaptic plasticity (15, 17).
Considerably more is known about the kinases that activate Pyk2 within the CNS compared with the tyrosine phosphatases that inactivate it. Several protein-tyrosine phosphatases (PTPs) have been reported to associate with Pyk2 outside of the CNS and regulate its activity (19–21). SHP1 dephosphorylates the autophosphorylation site Tyr402 of Pyk2 and down-regulates integrin-mediated immune cell adhesion and inflammation (19). PTP-PEST also targets Tyr402 as well as Tyr579/580 in the activation loop of Pyk2 and regulates cell motility (20, 22). However, a physiologically relevant PTP in the brain has not yet been reported.
Here we focus on the brain-enriched striatal-enriched protein-tyrosine phosphatase (STEP) as a possible regulator of Pyk2 in neurons. Previous studies have established that STEP normally opposes the development of synaptic strengthening by dephosphorylating and inactivating ERK1/2 (23, 24) and Fyn (25). STEP also regulates glutamate receptor internalization. Dephosphorylation of the GluN2B subunit of the N-methyl-d-aspartate receptor (formerly NR2B) and the GluA2 subunit of the AMPA receptor (formerly GluR2) results in internalization of GluN1-GluN2B (26) and GluA1-GluA2 receptors, respectively (27). STEP is a critical regulator of synaptic function, and the identification of additional substrates will clarify its normal function. Here we identify Pyk2 as a novel substrate for STEP using biochemical, molecular, and immunocytochemical techniques in wild type (WT) and STEP knock-out (KO) mice.
EXPERIMENTAL PROCEDURES
Antibodies and Other Reagents
All antibodies used in this study are listed in Table 1. NMDA and ionomycin were purchased from Sigma-Aldrich, and M-PER mammalian lysis buffer was from Pierce.
TABLE 1.
Primary and secondary antibodies used in this study
CaMKII, Ca2+/calmodulin-dependent protein kinase II.
| Antibody | Format | Immunogen | Host | Dilution | Source |
|---|---|---|---|---|---|
| Anti-Tyr(P)402 Pyk2 | Whole IgG, unconjugated | Human synthetic phosphopeptide | Rabbit | 1:1,000 | Invitrogen |
| Anti-Pyk2 N terminus | Whole IgG, unconjugated | N terminus of rat sequence | Rabbit | 1:500 | As described (15) |
| Anti-Pyk2 (H-102) | Whole IgG, unconjugated | N terminus of human Pyk2 | Rabbit | 1:1,000 | Santa Cruz Biotechnology |
| Anti-STEP | IgG1, unconjugated | Rat synthetic peptide | Mouse | 1:1,000 | As described (68) |
| Anti-Fyn | Whole IgG, unconjugated | Human synthetic peptide | Rabbit | 1:600 | Millipore |
| Anti-Tyr(P)1472 NR2B | Whole IgG, unconjugated | Synthetic phosphopeptide | Rabbit | 1:1,000 | Phosphosolutions, Denver, CO |
| Anti-NR2B | Whole IgG, unconjugated | C terminus of mouse NR2B | Rabbit | 1:1,000 | Millipore |
| Anti-Tyr(P)118 paxillin | Whole IgG, unconjugated | Human synthetic phosphopeptide | Rabbit | 1:1,000 | Cell Signaling Technology, Danvers, MA |
| Anti-paxillin | Whole IgG, unconjugated | Human synthetic peptide | Rabbit | 1:1,000 | Cell Signaling Technology |
| Anti-Tyr(P)782 ASAP1 | Whole IgG, unconjugated | Mouse synthetic phosphopeptide | Rabbit | 1:1,000 | Rockland, Gilbertsville, PA |
| Anti-ASAP1 | Whole IgG, unconjugated | Mouse synthetic peptide | Mouse | 1:100 | Rockland |
| Anti-Tyr(P)204 ERK1/2 | Whole IgG, unconjugated | Human synthetic phosphopeptide | Mouse | 1:1,000 | Santa Cruz Biotechnology |
| Anti-ERK2 | Whole IgG, unconjugated | C terminus of rat sequence | Rabbit | 1:5,000 | Santa Cruz Biotechnology |
| Anti-PSD-95, clone K28/43 | IgG2aκ, unconjugated | Fragment of human PSD-95 protein | Mouse | 1:10,000 | Millipore |
| Anti-β-actin | Whole IgG, unconjugated | Human synthetic peptide | Rabbit | 1:1,000 | Sigma-Aldrich |
| Anti-GAPDH, clone 6c5 | IgG1, unconjugated | Purified protein from rabbit muscle | Mouse | 1:20,000 | Millipore |
| Anti-tubulin | IgG1, unconjugated | Purified chick brain tubulin | Mouse | 1:500 | Sigma-Aldrich |
| Anti-synaptophysin | Unpurified mouse IgG1, unconjugated | Rat retina synaptophysin | Mouse | 1:2,000 | Millipore |
| Anti-CaMKII | Whole IgG, unconjugated | Mouse synthetic peptide | Rabbit | 1:2,000 | Santa Cruz Biotechnology |
| Anti-histone H1 | IgG2a, unconjugated | Human synthetic peptide | Mouse | 1:2,000 | Santa Cruz Biotechnology |
| Anti-rabbit | Whole IgG peroxidase-conjugated | Rabbit Fc | Donkey | 1:10,000 | Amersham Biosciences |
| Anti-mouse | Whole IgG peroxidase-conjugated | Mouse Fc | Sheep | 1:10,000 | Amersham Biosciences |
| Anti-rabbit | Whole IgG Alexa Fluor 594-conjugated | Rabbit Fc | Goat | 1:600 | Molecular Probes |
| Anti-mouse | Whole IgG Alexa Fluor 488-conjugated | Mouse Fc | Goat | 1:600 | Molecular Probes |
Constructs and Purification of Fusion Proteins
PCR-amplified open reading frames of STEP61 or STEP46 were subcloned into pcDNA3-His/V5 (Invitrogen) or pGEX4T1 vector (GE Healthcare). Trans-activator of transcription (TAT)-STEP46 was subcloned into the pTrcHis2-TOPO construct (Invitrogen) as described (27, 28). Point mutations were introduced using site-directed mutagenesis (kit from Stratagene, Santa Clara, CA), and all constructs were sequenced prior to use. Deletion constructs of STEP (polyproline-rich 1 and 2 (PR1 and PR2) and the kinase-interacting motif (KIM) domains) and pCMV-hFyn constructs were described previously (25). GST-tagged Pyk2 constructs (15) were PCR-amplified and subcloned into pcDNA3-His/V5 or pTrcHis2 construct (Invitrogen). See Fig. 1, which was made with the assistance of DOG software as described (29), for schematic domain structures of constructs used in this study.
FIGURE 1.

Schematic domain structures used in this study. FERM, band 4.1/ezrin/radixin/moesin homology domain; FAT, focal adhesion-targeting domain; TM1 and TM2, putative transmembrane domains; PTP, protein-tyrosine phosphatase domain; asterisk, cysteine residue in the catalytic core; UN, unique N-terminal domain. Regulatory phosphorylation sites are shown. Numbering refers to amino acid residues within the proteins.
Fusion constructs were expressed in Escherichia coli BL21(DE3) and purified on glutathione-Sepharose (GE Healthcare) or TALON metal affinity resin beads (Clontech) following the manufacturers' instructions. GST fusion proteins were eluted with 10 mm reduced glutathione in phosphate-buffered saline (PBS), pH 8. His-tagged proteins were eluted in PBS, pH 7.4, 150 mm imidazole. All proteins were concentrated using Amicon Ultra-4 filter units with 30- or 50-kDa cutoffs (Millipore, Billerica, MA). For some assays, STEP46 fusion proteins, which lack the N-terminal 172 amino acids of STEP61, were used, because of technical difficulty in purification of large amounts of the insoluble protein STEP61.
Brain Slices and Treatments
The Yale University Institutional Animal Care and Use Committee approved all animal procedures used in the present study. Coronal corticostriatal slices (300 μm) were prepared from male WT or STEP KO mice (8–10 weeks on a C57B6 background as described (30)). Slices were sectioned in ice-cold oxygenated artificial cerebrospinal fluid (aCSF) (124 mm NaCl, 4 mm KCl, 26 mm NaHCO3, 1.5 mm CaCl2, 1.25 mm KH2PO4, 1.5 mm MgSO4, 10 mm d-glucose). After recovery in aCSF for 60 min, slices were treated with 40 mm KCl for 2 min. Some slices were preincubated with 2 μm TAT-STEP46 or TAT-STEP46 C/S (27, 2) followed by KCl stimulation. Slices were placed on dry ice immediately after stimulations and homogenized.
Preparations of Brain Lysates and PSD Fractions
Slices were homogenized in TEVP buffer (10 mm Tris, pH 7.4, 5 mm NaF, 1 mm Na3VO4, 1 mm EDTA, 1 mm EGTA, protease inhibitor mixture (Roche Applied Science)), and P2 fractions were obtained as described previously (28). P2 fractions were extracted in detergent (0.5% Triton X-100)-containing buffers as described (31). The insoluble fractions (PSD) were lysed in radioimmune precipitation assay buffer with 1% SDS buffer followed by brief sonication.
Preparation of Synaptoneurosomal Fractions
Synaptoneurosomes from WT or STEP KO mice whole brains were obtained as described previously (27). Briefly, tissues were homogenized in homogenization buffer (20 mm HEPES/NaOH, pH 7.4, 124 mm NaCl, 1.06 mm KH2PO4, 26 mm NaHCO3, 1.3 mm MgCl2, 2.5 mm CaCl2, 10 mm glucose, Complete protease inhibitor tablets (Roche Applied Science)). Homogenates were centrifuged at 2000 × g for 1 min. The supernatant was passed through two 100-μm nylon mesh filters (Sefar America, Richfield, MN) followed by a 5-μm nitrocellulose filter (Millipore) and centrifuged at 1000 × g at 4 °C for 10 min. Pellets were washed, resuspended in homogenization buffer, and dispensed into 100-μl aliquots. All aliquots were preincubated at 37 °C for 10 min before stimulation with 40 mm KCl for various times as indicated. Some aliquots were pretreated with cyclosporin A (CsA; 100 nm) for 20 min before the addition of KCl. Thirty microliters of each aliquot were mixed with 6× SDS sample buffer and subjected to Western blotting.
Cell Culture and Transfection
HEK293 cells or SYF cells (a generous gift from A. M. Bennett, Yale University School of Medicine) were grown in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics. HEK293 or SYF cells were seeded at 1 × 106 cells/35-mm dish 24 h before transfection. The next day, pcDNA3-STEP WT, mutants, or truncation constructs were co-transfected with pcDNA3-Pyk2 or pCMV-Fyn using Lipofectamine 2000 (Invitrogen) following the manufacturer's protocol. After 6 h of incubation at 37 °C, the medium was replaced, and cells were incubated for another 36–48 h. Cells were lysed in M-PER mammalian extraction reagent (Pierce) and subjected to immunoprecipitation or Western blotting.
Immunoprecipitation and Pulldown Assays
Mouse brain homogenates or cell lysates were subjected to immunoprecipitation with anti-STEP or anti-Pyk2 antibodies in M-PER reagents overnight at 4 °C. The 2nd day, Protein A/G PLUS-Agarose beads (Santa Cruz Biotechnology) were added and incubated for another 4 h. Beads were collected by centrifugation and washed three times with lysis buffer. Immunoprecipitates were resuspended in SDS sample buffer after the final wash and analyzed by immunoblotting with anti-STEP, anti-Pyk2, or anti-Fyn antibody.
For pulldown assays, GST fusion proteins were adsorbed to glutathione-Sepharose beads for 2 h at 4 °C and then incubated with WT or STEP KO mouse brain homogenates (100 μg) in radioimmune precipitation assay buffer overnight at 4 °C. In some experiments, the catalytically inactive GST-STEP61 C472S protein was mixed with 1 μg of His-Pyk2 and increasing amounts of His-Fyn. Beads were washed and subjected to SDS-PAGE followed by immunoblotting.
In Vitro Binding
GST-tagged Pyk2 constructs (either full-length Pyk2 (amino acids 1–1009) or with amino acid deletions to give amino acids 1–710, 671–1009, 671–875, 728–849, or 876–1009) or GST alone was immobilized on glutathione-Sepharose beads. After washes, His-tagged STEP46 (500 nm) was added for 2 h at 4 °C. The samples were washed and subjected to SDS-PAGE. Prior to immunoblotting with anti-STEP antibody, the membranes were stained with Ponceau S stain to visualize GST fusion proteins.
Fluorescence Polarization (FP) and Competitive Binding Assays
FP-based binding assays were performed in a Synergy 2 plate reader (BioTek, Winooski, VT) with Gen5 software using 96-well black polystyrene plates (Corning, Corning, NY). Fluorescein isothiocyanate (FITC)-labeled peptides were obtained from CHI Scientific (Maynard, MA). The sequences are shown in Table 2. Increasing concentrations of recombinant proteins (either GST-STEP61, GST-STEP46, or GST-Pyk2(671–875)) were incubated with FITC-labeled peptide at a final concentration of 1 μm in FP buffer (50 mm HEPES, pH 7.4, 100 mm KCl, 1 mm MgCl2, 0.05 mm EGTA, 5 mm nitrilotriacetic acid). Parallel and perpendicular intensities were determined with 485/20λ excitation and 528/20λ emission filters, and FP was calculated as described by Lim et al. (32) For each graph, signals were normalized by setting the lowest value as zero and the highest value as 100%.
TABLE 2.
Sequences of peptides used in fluorescence polarization and pulldown assay
| Name | Sequence | Modification |
|---|---|---|
| STEP_PR2 | HLLKAPPEPPAPLPPEDRRQ | N terminus, FITC; C terminus, amide |
| STEP_KIM | TVKSMGLQERRGSNVSLTLDM | N terminus, FITC; C terminus, amide |
| Pyk2_671–694 | RFTELVCSLSDIYQMERDIAIEQE | N terminus, FITC; C terminus, amide |
| Pyk2_689–712 | IAIEQERNARYRPPKILEPTAFQE | N terminus, FITC; C terminus, amide |
| Pyk2_707–723 | PTAFQEPPPKPSRPKYK | N terminus, FITC; C terminus, amide |
| Pyk2_850–866 | TEFTGPPQKPPRLGAQS | N terminus, FITC; C terminus, amide |
| TAT-Myc | YGRKKRRQRRREQKLISEEDL | None |
Competitive binding by peptides was also examined in GST pulldown assays. GST-STEP61 or GST-Pyk2(671–875) was bound to glutathione-Sepharose beads. Mouse brain lysates were preincubated with increasing concentrations of peptides for 2 h at 4 °C and then mixed with protein-Sepharose complexes overnight at 4 °C. Precipitates were washed extensively and resuspended in SDS sample buffer.
Dephosphorylation of Pyk2 by STEP in Vitro
Mouse brain lysates were incubated at 65 °C for 20 min to inactivate endogenous kinases and phosphatases. The same amount of lysates (100 μg) was incubated with GST-STEP46 (active) or GST-STEP46 C/S (inactive) fusion proteins in dephosphorylation buffer (25 mm HEPES, pH 7.3, 5 mm EDTA, 5 mm DTT) for 30 min at 30 °C.
Dephosphorylation of Pyk2 by STEP was also carried out with Pyk2 immunoprecipitated from mouse brain lysates using anti-Pyk2 antibody and protein A/G-agarose. The precipitates were allowed to undergo autophosphorylation in the presence or absence of ATP and/or pervanadate. In some assays, active STEP (10 μm) was added in the presence or absence of pervanadate (1 mm). Phosphorylation of Pyk2 at Tyr402 was accessed with a phosphospecific antibody. The Tyr(P)402 signal was normalized to total Pyk2 levels.
Immunoblotting
Samples were resolved by SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad) as described previously (28). Membranes were blocked in TBS-Tween 20 + 5% bovine serum albumin (BSA) and incubated with primary antibodies overnight at 4 °C. The next day membranes were washed and incubated in peroxidase-conjugated secondary antibodies (Amersham Biosciences) for 2 h at RT. Blots were developed using a chemiluminescent substrate kit (Pierce), and immunoreactivity was captured by a G:BOX with the image program GeneSnap (Syngene, Cambridge, UK). All densitometric quantifications were performed using the Genetools program (Syngene).
Immunofluorescence
Primary hippocampal neurons were isolated from rat E18 embryos as described previously (28). Neurons were plated on poly-d-lysine-coated slides (2.5 × 104 cells/cm2; Thermo Scientific) in Neurobasal medium supplemented with 2% B27 (Invitrogen) and grown for 14–21 days. Cells were fixed in 4% paraformaldehyde with 4% sucrose and permeabilized with 0.2% Triton X-100 in PBS as described previously (28). After blocking with 10% normal goat serum and 1% BSA for 1 h, cells were stained with anti-Pyk2 and anti-STEP antibodies overnight at 4 °C. Goat anti-rabbit Alexa Fluor 594- or goat anti-mouse Alexa Flour 488-conjugated secondary antibodies (Molecular Probes, Eugene, OR) were used to detect the primary antibodies. To exclude the possibility of nonspecific antibody staining, immunodepleted antibodies were also used as controls. Anti-STEP or anti-Pyk2 antibody was subjected to four sequential immunodepletion rounds with GST-STEP61 or GST-Pyk2, which was preadsorbed to glutathione-Sepharose beads. Secondary antibodies alone were also included as controls. To visualize Pyk2 translocation to PSD fractions upon KCl stimulations, anti-Pyk2 and anti-PSD-95 antibodies were used to indicate colocalization. Microscopy was performed with a Zeiss Axiovert 2000 microscope with an apotome (Applied Scientific Instruments, Eugene, OR) using a 100× objective lens. A punctum was defined as a continuous group of pixels corresponding to 0.5–3.0 μm. Colocalization analysis was performed on randomly selected images (n = 17) using NIH Image J software with the colocalization analysis plug-in at default settings (ratio, 50%; threshold for each channel, 50).
Statistical Analysis
All experiments were repeated at least three times. Data were expressed as means ± S.E. For some experiments, statistical significance was determined by Student's t test, whereas in others, data were analyzed with one-way ANOVA with post hoc Tukey test. p values <0.05 were considered significant.
RESULTS
Inhibition of Tyrosine Phosphatases Leads to Activation of Pyk2
Our previous study showed that resin-bound anti-Pyk2 antibody facilitated Pyk2 dimerization and activation (15). We used this technique to activate Pyk2 in rat brain homogenates. We assessed autophosphorylation of Pyk2 in solution (Fig. 2A, No Resin) or bound to beads (Fig. 2A, Resin Bound) in the absence or presence of the general tyrosine phosphatase inhibitor pervanadate (1 mm). Similar to previous findings (15), Pyk2 antibody induced autophosphorylation of Pyk2 in lysate and on resins presumably by promoting dimerization of Pyk2. No autophosphorylation of Pyk2 was detected in lysate when pervanadate was absent. However, immunoprecipitated Pyk2 underwent limited autophosphorylation at Tyr402 in the absence of pervanadate. This phosphorylation could result from dissociation of a tyrosine phosphatase from a subpopulation of Pyk2 during washing procedures. Addition of pervanadate led to a significant increase in the tyrosine phosphorylation of Pyk2, suggesting that an unknown PTP antagonized the antibody-induced autophosphorylation of the subpopulation of Pyk2 that retained a PTP.
FIGURE 2.
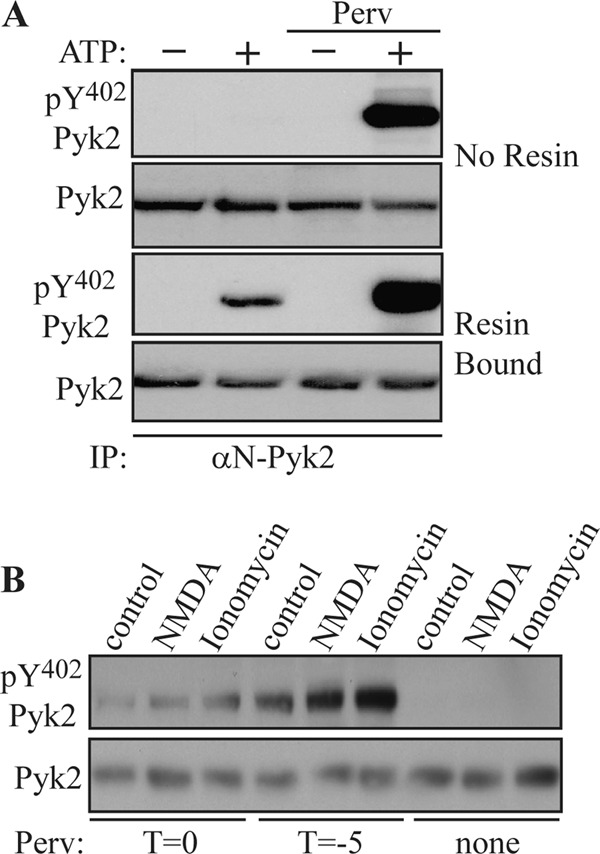
Phosphorylation of Pyk2 at Tyr402 is increased in presence of protein-tyrosine phosphatase inhibitor. A, rat brain extracts were incubated with 1 μg of αN-Pyk2 antibody in the absence of protein A-agarose. Mg-ATP was then added in the presence or absence of pervanadate (Perv) (1 mm) (top, No Resin). In another set of experiments, Pyk2 was first immunoprecipitated (IP) with αN-Pyk2 antibody and protein A-Sepharose and washed before the addition of Mg-ATP in the presence or absence of pervanadate (1 mm) (Resin Bound). After 1 h at 4 °C, EDTA (50 mm) was added to chelate the Mg2+ and halt further phosphorylation. Tyr(P)402 Pyk2 levels were examined by immunoblotting with phosphospecific antibody. B, primary hippocampal cultures were treated with NMDA (50 μm) or ionomycin (1 μm) for 15 min. Pervanadate (1 mm) was added 5 min prior to (T = −5) or at the time of stimulation with the absence of pervanadate serving as a control.
We used NMDA or ionomycin to activate Pyk2 in primary hippocampal neurons. Pretreatment of cultures with pervanadate for 5 min (T = −5) followed by stimulation with NMDA or ionomycin for 15 min led to a significant increase in the phosphorylation of Pyk2 at Tyr402. In contrast, NMDA or ionomycin failed to induce Pyk2 phosphorylation in the absence of pervanadate, indicating that activation of Pyk2 was blocked by PTPs or that PTPs rapidly dephosphorylated Pyk2 at Tyr402 (Fig. 2B). Taken together, these results suggest that activation of Pyk2 was counteracted by an unknown PTP in the brain.
STEP Binds to Pyk2
To evaluate whether STEP interacted with Pyk2, STEP was immunoprecipitated from mouse brain lysates. STEP KO lysates were used as a negative control. Pyk2 co-immunoprecipitated with STEP from WT mouse brains but not with mouse IgG control or from STEP KO mouse brains (Fig. 3A, upper panel). Fyn also co-immunoprecipitated with STEP, consistent with previous findings (25). We confirmed the interaction of STEP and Pyk2 by immunoprecipitating Pyk2 and probing with anti-STEP antibody (Fig. 3A, lower panel). We confirmed that Pyk2 interacts with Fyn (33). We also observed that Pyk2 associated with Fyn in the STEP KO lysates (Fig. 3A).
FIGURE 3.

Pyk2 is associated with STEP in vivo. A, total brain homogenates from WT or STEP KO mice were immunoprecipitated (IP) with anti-STEP antibody (upper panel) or anti-Pyk2 antibody (lower panel). Immunoprecipitates were resolved by SDS-PAGE and immunoblotted with anti-Pyk2 or anti-Fyn antibody. Blots were reprobed with anti-STEP antibody. B, primary hippocampal neurons were labeled with anti-STEP (green) and anti-Pyk2 (red) antibodies, and nuclei were stained (DAPI; blue). Some cultures were stained with immunodepleted anti-STEP (ID 4) or anti-Pyk2 (ID 4) or secondary antibodies only. The specificity of immunodepleted antibodies was assessed using hippocampal neuronal lysates with each fraction. GAPDH was probed as the loading control.
To corroborate the association of Pyk2 and STEP, the cellular distribution of these proteins was determined by immunofluorescence microscopy in primary hippocampal neurons. Neurons were stained simultaneously with anti-Pyk2 (H102) and anti-STEP antibodies followed by Alexa Fluor-488-coupled anti-mouse and Alexa Fluor-594-coupled anti-rabbit secondary antibodies. Pyk2 and STEP colocalized throughout the soma as well as along neurites as indicated in the merged image (Fig. 3B). To establish that the staining was specific, immunodepleted anti-STEP and anti-Pyk2 or secondary antibodies alone were also examined (Fig. 3B). Collectively, the above results demonstrated that STEP, Pyk2, and Fyn associate with each other in vivo, although this does not indicate that they are all present in the same complexes, an issue addressed further below.
STEP Dephosphorylates Pyk2
Phosphorylation at Tyr402 is essential for Pyk2 activation. To determine whether STEP dephosphorylates Pyk2 at Tyr402, we performed in vitro autophosphorylation assays with immunoprecipitated Pyk2 from mouse brain lysates. Pyk2 was incubated with Mg-ATP in the presence or absence of pervanadate. Due to pervanadate inhibition of endogenous PTPs, including STEP, during lysis and immunoprecipitation procedures, no difference was seen in autophosphorylation of Pyk2 (Fig. 4A, lane 2 versus lane 4). Addition of active STEP readily led to dephosphorylation of Pyk2 (Fig. 4A, lane 5), which was prevented by the presence of pervanadate (lane 6). These results indicated that autophosphorylation of Pyk2 at Tyr402 was reversed by active STEP. Inhibition of STEP activity by pervanadate restored Pyk2 autophosphorylation. These results suggest that STEP is capable of dephosphorylating Pyk2 at Tyr402 in vitro.
FIGURE 4.

STEP dephosphorylates Pyk2 at Tyr402. A, Pyk2 was immunoprecipitated from mouse brain lysates using anti-Pyk2 antibody and protein A/G-agarose in the presence of pervanadate (Perv) (1 mm). Mg-ATP, STEP (10 μm), and pervanadate (1 mm) were added as indicated. The Tyr(P)402 signal was normalized to total Pyk2 levels. Error bars indicate the standard error of the mean (SEM) of at least three independent experiments (in this and subsequent panels). Asterisks indicate statistical significance as compared with control (**, p < 0.01; one-way ANOVA with post hoc Tukey test; n = 4). B, total brain lysates were heated at 65 °C for 20 min to inactive endogenous kinases and phosphatases. Treated lysates (50 μg) were incubated with purified GST-STEP46 WT (active) or GST-STEP46 C/S (inactive) at the indicated concentrations and blotted with Tyr(P)402 Pyk2 or Tyr(P)1472 NR2B. Quantitative analyses for each were normalized to GAPDH (*, p < 0.05; **, p < 0.01; one-way ANOVA with post hoc Tukey test; n = 3). C, full-length pcDNA3-Pyk2 (1 μg) construct was co-transfected with pcDNA3-STEP61 WT (active) or pcDNA3-STEP61 C472S (inactive) construct into HEK293 cells. Thirty-six hours post-transfection, cells were lysed in radioimmune precipitation assay buffer and blotted with antibodies as indicated in the figure.
We confirmed STEP dephosphorylation of Pyk2 by mixing increasing amounts of active STEP (GST-STEP46) or enzymatically inactive STEP (GST-STEP46 C/S) with equal amounts of heat-inactivated brain lysates. Active STEP caused a significant reduction in Pyk2 phosphorylation, whereas inactive STEP did not (Fig. 4B, 1000 nm; 74.8 ± 2.5% of no STEP control; p < 0.01). Tyr(P)1472 GluN2B is a known substrate of STEP, and there was a significant decrease in Tyr(P)1472 GluN2B levels in the presence of active, but not inactive, STEP (Fig. 4B, 1000 nm; 68.8 ± 4.6% of no STEP control; p < 0.01). These results confirmed that Pyk2 is dephosphorylated at Tyr402 by STEP.
To test whether STEP was able to dephosphorylate Pyk2 in intact cells, HEK293 cells were transfected with Pyk2 along with either active WT STEP61 or a catalytically inactive STEP61 C472S construct. Phosphorylation of Pyk2 at Tyr402 was reduced when HEK293 cells were co-transfected with WT STEP61 but not the inactive STEP61 protein (Fig. 4C).
STEP Interacts Directly with Pyk2
To map the binding regions involved in STEP/Pyk2 interaction, several STEP61 deletion mutants were co-transfected with full-length Pyk2 into HEK293 cells. These proteins were immunoprecipitated with anti-STEP antibody, and Pyk2 levels were examined with anti-Pyk2 (H102) antibody (Fig. 5A). The full-length substrate-trapping mutant of STEP61 (25) showed high affinity for Pyk2, whereas deletion of either the PR2 or KIM domain decreased the amount of Pyk2 pulled down. The substrate-trapping STEP46 mutant that lacks the N-terminal region also interacted but pulled down less Pyk2 (Fig. 5A). These results suggest that the PR2 and KIM domains are involved in the interaction of STEP with Pyk2. We confirmed the involvement of the KIM domain in STEP/Pyk2 interactions by GST pulldown assays (supplemental Fig. S1).
FIGURE 5.
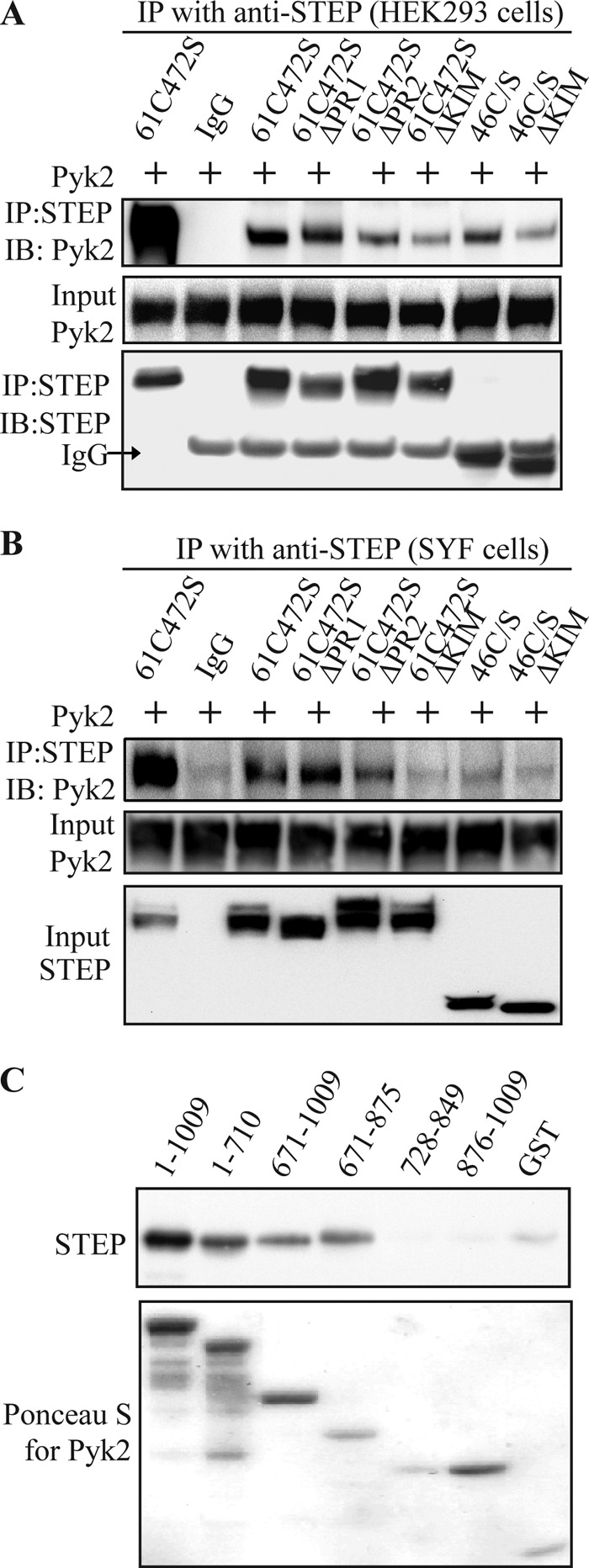
Interacting regions in STEP and Pyk2. A, HEK293 cells were transfected with full-length Pyk2 along with various STEP mutants (Fig. 1). STEP was immunoprecipitated, and Pyk2 levels were assessed with anti-Pyk2 antibody. B, SYF cells were co-transfected with the same STEP and Pyk2 constructs used in A. Association of Pyk2 with STEP was visualized by immunoprecipitation (IP) of STEP and probing for Pyk2. C, in vitro binding of STEP and Pyk2. GST-tagged Pyk2 fragments (Fig. 1) or GST alone was immobilized on glutathione-Sepharose. After washes, STEP (500 nm) was added for 2 h at 4 °C. Binding of STEP to various fragments was visualized with anti-STEP antibody (upper panel), and Pyk2 proteins were visualized by Ponceau S staining (lower panel). IB, immunoblotting.
Previous studies have shown that Fyn interacts with Pyk2 (34) and STEP (25). We wanted to test whether the association of STEP and Pyk2 required Fyn to be present. STEP and Pyk2 constructs were transfected into SYF cells, which are deficient for Src, Yes, and Fyn (34, 35). STEP61 was still able to pull down Pyk2, whereas deletion of PR2 or KIM domain impaired STEP/Pyk2 association (Fig. 5B). These results indicate that STEP is able to interact with Pyk2 in the absence of Fyn.
We next examined the domains within Pyk2 necessary for the association of Pyk2 with STEP. His-tagged STEP46 C/S was incubated with different fragments of GST-tagged Pyk2 coupled to glutathione-Sepharose beads. Full-length Pyk2 (amino acids 1–1009) was effective in binding to STEP as was an ∼200-amino acid sequence of Pyk2 (amino acids 671–875; Figs. 5C and 1). This region also binds PSD-95 (15) (see below).
To determine whether residues 671–875 of Pyk2 or the KIM and PR2 domains of STEP are required for binding, we utilized FP assays. Binding affinities of the FITC-conjugated peptides KIM and PR2 of STEP and residues 671–694, 689–712, 707–723, and 850–866 of Pyk2 were determined for GST-Pyk2(671–875) and for GST-STEP61 and -STEP46, respectively (Fig. 6A). Pyk2(671–694) but none of the other peptides bound STEP in a saturating and hence specific manner. It showed higher affinity for STEP61 (Kd = 2.63 μm) than STEP46 (Kd = 11.83 μm), suggesting involvement of the unique N terminus of STEP61 in these interactions, which is consistent with our findings in Fig. 5 and supplemental Fig. S1. The PR2 domain of STEP61 was also able to bind to Pyk2 directly (Fig. 6A) albeit with low affinity as often observed for peptides derived from proline-rich segments.
FIGURE 6.

PR2 and KIM domains of STEP61 and residues 671–694 of Pyk2 mediate STEP/Pyk2 interaction. A, FITC-conjugated Pyk2-derived peptides 671–694, 689–712, 707–723, and 850–866 were titrated with GST-STEP61 (left) and -STEP46 (middle), and STEP-derived PR2 peptide with GST-Pyk2(671–875) (right). Binding was monitored by FP. For each graph, the signals were normalized, setting as zero the lowest value and as 100% the highest value measured, and fitted to saturation curves. Calculated binding affinities of 671–694 for GST-STEP61 and -STEP46 and of PR2 for GST-Pyk2 671–875 are indicated. AU, absorbance units. Error bars indicate the SEM of at least three independent experiments (in this and subsequent panels). B and C, competition of PR2, KIM, and Myc (control) peptide with GST-STEP61 for Pyk2 and ERK2 (B) and of the Pyk2-derived peptides with GST-Pyk2(671–875) for STEP61 and PSD-95 (C) by increasing concentrations of peptides (as indicated) in pulldown assays with GST-STEP61 (B) and GST-Pyk2(671–875) (C) using brain lysates as the source of native proteins. Pyk2 and ERK2 (B) and STEP61 and PSD-95 (C) were determined by immunoblotting (*, p < 0.05; **, p < 0.01; one-way ANOVA with post hoc Tukey test; n = 4).
The KIM peptide showed poor solubility and strong adsorption to the polystyrene plates used in the FP assay, preventing its use in this assay. Therefore, we examined the effect of this and the other peptides in GST pulldown assays (Fig. 6B). The results confirmed that both PR2 and KIM domains of STEP and residues 671–694 of Pyk2 are involved in the STEP/Pyk2 interaction. In contrast, the STEP/ERK2 interaction was not affected by the PR2 domain, although it was inhibited by the KIM peptide in agreement with earlier work indicating that KIM mediates STEP binding to ERK (24, 36). As a control, Myc peptide did not have any impact on binding. Furthermore, the pulldown data confirm that Pyk2(671–694) interacts with STEP (Fig. 6C). The data further indicate that STEP and PSD-95 bind to different motifs within the Pyk2(671–875) region (15) with PSD-95 interacting with Pyk2(707–723) but not Pyk2(671–694).
Fyn and Pyk2 Compete for STEP Binding
Because the N terminus and the KIM domain of STEP are needed for binding to both Fyn and Pyk2, we next examined whether we could competitively disrupt the interaction of one by increasing the concentration of the other in transfection experiments. We co-transfected STEP61 and full-length Pyk2 constructs with increasing amounts of Fyn. There was a decrease in the association of Pyk2 with STEP as Fyn levels were increased (Fig. 7A). Similarly, there was a decrease in the association of Fyn with STEP as Pyk2 levels were increased (Fig. 7B). These results suggest that Pyk2 and Fyn compete for binding with STEP61.
FIGURE 7.
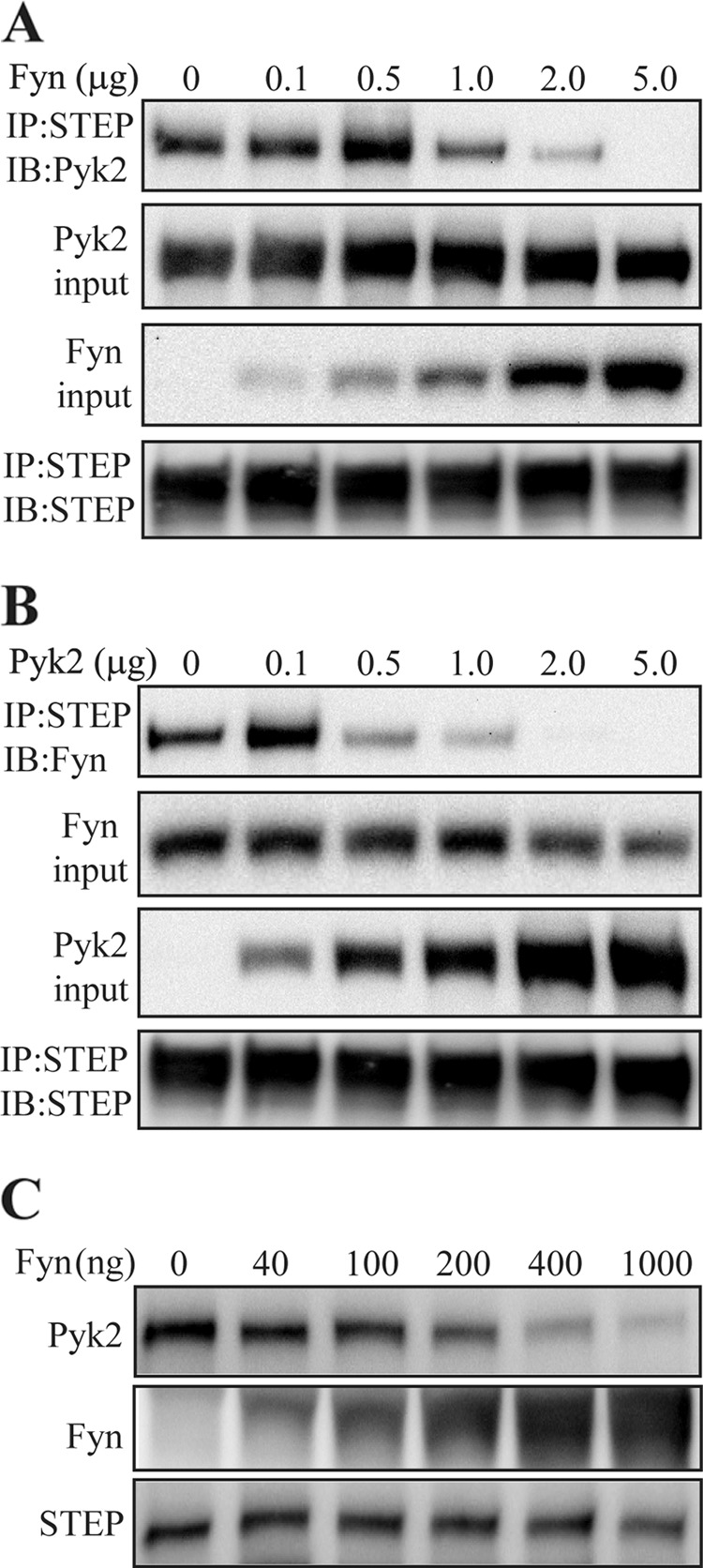
Fyn and Pyk2 compete for STEP binding. A, constant amounts (1 μg of cDNA) of full-length STEP and Pyk2 constructs were co-transfected with increasing amounts of Fyn construct into HEK293 cells. Thirty-six hours after transfection, cells were lysed before immunoprecipitation (IP) with anti-STEP antibody and immunoblotting (IB) with anti-Pyk2 antibody. Immunoblotting of lysates indicates increasing amounts of Fyn with increasing amounts of Fyn cDNA but unchanged amounts of Pyk2. Immunoblotting of STEP immunoprecipitates indicates unaltered amounts of STEP in all samples. B, constant amounts (1 μg of cDNA) of STEP and Fyn were co-transfected with increasing amounts of Pyk2. Co-immunoprecipitation of Fyn with STEP was measured by immunoblotting with anti-Fyn antibody. C, competitive binding was tested in vitro with all purified proteins. GST-tagged STEP was adsorbed to glutathione beads and incubated with a constant amount of Pyk2 (500 ng) and increasing amounts of Fyn as indicated.
In vitro competitive binding was also assessed with purified proteins. GST-tagged STEP61 C472S was incubated with His-tagged Pyk2 and increasing concentrations of His-tagged Fyn. Fyn competed in a concentration-dependent manner with Pyk2 for binding to STEP (Fig. 7C).
Increased Phosphorylation of Pyk2 in STEP KO Mice
Based on the findings that STEP binds to and dephosphorylates Pyk2 at Tyr402, we predicted that STEP KO mice would have elevated Pyk2 phosphorylation. We first determined the basal levels of Tyr phosphorylation of Pyk2 in synaptosomal (P2) fractions from STEP KO versus WT mice. There was a significant increase of Tyr(P)402 levels in STEP KO mice (Tyr(P)402, 130.2 ± 7.5% of WT; p < 0.01), whereas total Pyk2 levels remained unchanged (p > 0.05) (Fig. 8A). We also looked at additional tyrosine phosphorylation sites of Pyk2 and showed increases of Tyr(P)881 but not Tyr(P)579 or Tyr(P)580 levels in STEP KO mouse brains (supplemental Fig. S2), suggesting that STEP might also directly dephosphorylate Tyr881 or block the phosphorylation by kinases (such as Src and Fyn) at this site. Such blockage could be due to shifting Tyr402 toward the dephosphorylated state, which then would lead to less binding of Src family kinases to Pyk2 and thereby less phosphorylation of the other sites on Pyk2.
FIGURE 8.
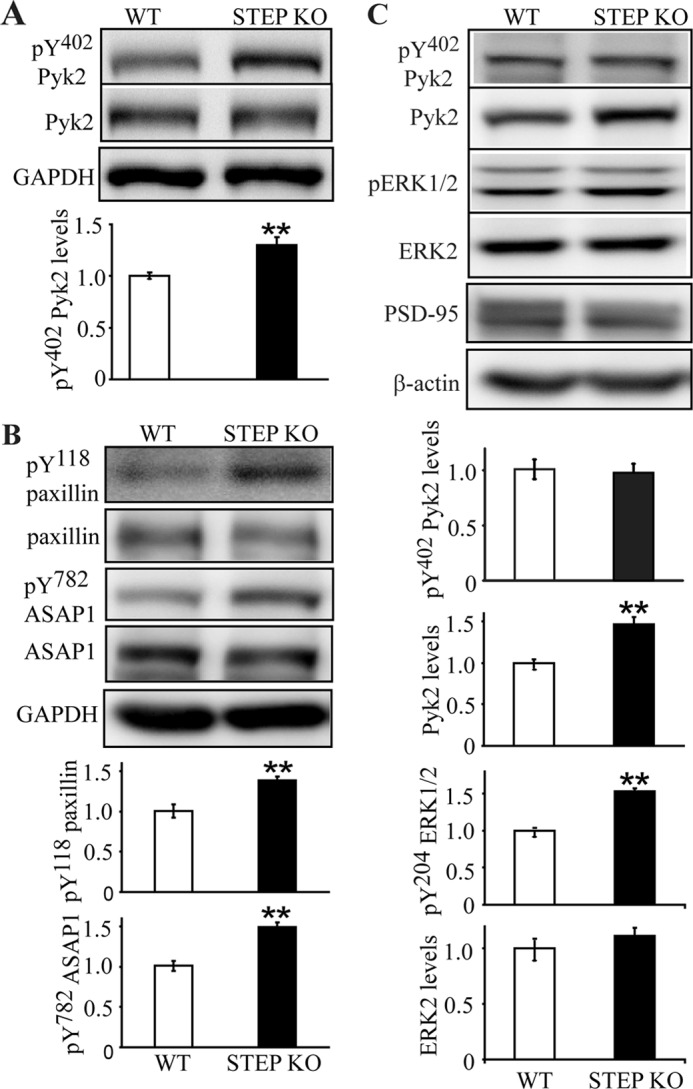
Basal phosphotyrosine levels of Pyk2 and Pyk2 substrates are elevated in STEP KO mouse brains. A, synaptosomal (P2) fractions from WT and STEP KO littermates were used to determine levels of tyrosine phosphorylation of Pyk2 at Tyr(P)402 and total Pyk2 levels. Error bars indicate the SEM of at least three independent experiments (in this and subsequent panels). Quantitative analyses for each were normalized to GAPDH (*, p < 0.05; **, p < 0.01; Student's t test; n = 4). B, phosphorylation of paxillin (Tyr(P)118) and ASAP1 (Tyr(P)782) were determined in P2 fractions with phosphospecific antibodies. Quantitations were normalized to total protein levels, respectively, and then to GAPDH. C, PSD fractions from WT and STEP KO mice were purified. Tyrosine phosphorylation of Pyk2 and ERK1/2 was compared between WT and STEP KO mice (**, p < 0.01; Student's t test; n = 4).
To further confirm that dephosphorylation of Pyk2 by STEP had a functional consequence on Pyk2 signaling, we probed for the tyrosine phosphorylation levels of paxillin, a well known substrate for Pyk2 and focal adhesion kinase (11, 37), and ASAP1, a recently identified Pyk2 substrate that is not phosphorylated by focal adhesion kinase (38, 39). Phosphorylation of paxillin at Tyr118 and of ASAP1 at Tyr782 was enhanced in STEP KO mouse brain lysates by 138.4 ± 5.1 and 149.5 ± 5.7% versus WT, respectively (Fig. 8B; p < 0.01).
Phosphorylation and activation of Pyk2 lead to its translocation to PSD fractions (15). We next examined whether translocation of Pyk2 was increased in STEP KO mice in which Pyk2 Tyr phosphorylation was increased. We measured phosphorylated and total Pyk2 levels in PSD fractions from WT and STEP KO mouse brains (Fig. 8C). Total Pyk2 levels were greater in the PSD from STEP KO mice (146.4 ± 9.4% of WT; p < 0.01). No change in Tyr(P)402 levels was seen when normalized to total Pyk2 levels. These data suggest that only Tyr402-phosphorylated Pyk2 associates with the PSD complex in a manner that is stable enough to endure purification of PSDs. However, catalytic activity of Pyk2 is not required for the translocation to postsynaptic sites in hippocampal cultures upon Ca2+ influx (15). Phospho-ERK1/2 levels were also increased in PSD from STEP KO mice (153.1 ± 4.0% of WT; p < 0.01; Fig. 8C). There was no change in ERK2 or PSD-95 (Fig. 8C) levels in PSD fractions. These data indicate that Pyk2 associates more stably with PSD fractions in STEP KO versus WT mice.
STEP Blocks KCl-induced Activation and Translocation of Pyk2 to PSD Fraction
Treatment of rat brain slices with KCl activates Pyk2 (40). We therefore examined the inhibition of Pyk2 by STEP in a KCl-treated brain slice model. We took advantage of efficient transduction of fusion proteins mediated by the human immunodeficiency virus, type 1 TAT domain, which is widely used to deliver exogenous proteins into various cell types (28, 41). Slices from WT or STEP KO mice were pretreated with WT TAT-STEP, inactive TAT-STEP C/S, or active STEP without TAT for 30 min followed by KCl stimulation and isolation of PSD fractions. KCl stimulation led to a significant increase in translocation of Pyk2 to the PSD fraction (Fig. 9A) (146.8 ± 2.5% of control; p < 0.01). Preincubation with TAT-STEP46 WT antagonized the Pyk2 translocation to PSDs (99.8 ± 4.9% of control; p > 0.05). The inactive TAT-STEP46 C/S or active STEP without TAT did not antagonize the activation or translocation of Pyk2 (148.7 ± 6.6% of control and 152.1 ± 8.8% of control, respectively; p < 0.01). Phospho-Pyk2 levels remained constant in the PSD fractions when normalized to total Pyk2 levels regardless of treatments, suggesting that PSD-localized Pyk2 is mostly, if not all, phosphorylated at Tyr402.
FIGURE 9.

Active TAT-STEP protein blocks phosphorylation and translocation of Pyk2 to PSD upon KCl depolarization. A, WT or STEP KO mouse brain slices were pretreated with TAT-STEP WT (active), TAT-STEP C/S (inactive), or active STEP without TAT for 30 min; stimulated with 40 mm KCl for 2 min; and frozen on dry ice before isolation of PSD fractions. Phosphorylation levels were probed with phosphospecific antibodies (Tyr(P)402 Pyk2 or Tyr(P)204 ERK1/2) and normalized to total Pyk2 and ERK1/2 levels, respectively, and then to β-actin as a loading control. Error bars indicate the SEM of at least three independent experiments (in this and subsequent panels). All values were compared with those in WT aCSF samples (**, p < 0.01; one-way ANOVA with post hoc Tukey test; n = 4). B, primary hippocampal neurons were pretreated with active TAT-STEP WT or inactive TAT-STEP C/S followed by KCl (40 mm; 2 min) stimulations. Colocalization of Pyk2 and PSD-95 was visualized using immunostaining with anti-Pyk2 (red) and anti-PSD-95 (green) antibodies. Arrowheads in the merged images indicate colocalized puncta. The number of Pyk2/PSD-95-colocalized puncta was counted per 10 μm of dendrites; 17 neurons were used for quantification per treatment. All values were compared with control (**, p < 0.01; one-way ANOVA with post hoc Tukey test; n = 17). pERK1/2, phospho-ERK1/2, pPyk2, phospho-Pyk2.
We repeated these experiments in STEP KO mice. aCSF-treated STEP KO slices showed higher levels of total Pyk2 (128.8 ± 6.1% of WT aCSF; p < 0.01) in the PSD, indicating elevated basal levels in the STEP KO mice. KCl stimulation induced further increases in PSD-localized Pyk2 (172.0 ± 8.6 versus 128.8 ± 6.1%; p < 0.01). Translocation of Pyk2 was antagonized by the addition of active WT TAT-STEP protein (111.9 ± 6.6 versus 172.0 ± 8.6%; p < 0.01) but not by inactive TAT-STEP C/S protein or active STEP without the TAT tag (Fig. 9A).
ERK1/2 is a well established STEP substrate that is activated by KCl depolarization (40) and was examined as a positive control in these experiments. PSD fractions from STEP KO slices showed greater basal phosphorylation levels of ERK1/2 (149.8 ± 9.4% of WT aCSF), confirming earlier findings (28, 30). KCl stimulations led to an increase of ERK1/2 phosphorylation in both WT and STEP KO slices (WT, 235.3 ± 8.9% of aCSF control; KO, 355.6 ± 23.3% of WT aCSF control) that was blocked by WT TAT-STEP but not inactive TAT-STEP C/S or active STEP without TAT.
Blockade of Pyk2 translocation to the PSD fractions by STEP was also visualized using immunostaining. Primary hippocampal neurons were pretreated with active TAT-STEP WT or inactive TAT-STEP C/S followed by KCl stimulations, and the number of Pyk2- and PSD-95-colocalized puncta/10 μm of dendrites was counted. Consistent with previous findings (15), KCl stimulation led to increased colocalization of Pyk2 with PSD-95 (9.06 ± 0.46 puncta compared with 2.06 ± 0.39 puncta for control; p < 0.01). TAT-STEP WT blocked translocation of Pyk2 upon KCl stimulation (1.88 ± 0.37 puncta; p > 0.05), whereas inactive TAT-STEP C/S had no effect (7.65 ± 0.48 puncta; p < 0.01) (Fig. 9B).
Activation of STEP Results in Dephosphorylation of Pyk2 in Synaptoneurosomal Fractions
We examined whether Pyk2 phosphorylation would be stimulated by KCl depolarization in synaptoneurosomes and whether STEP modulated the dephosphorylation and inactivation of Pyk2 in these samples. Synaptoneurosomes were purified from WT or STEP KO mouse brain, and the purity of the preparation was examined by localization and expression of several marker proteins (27, 42). Synaptophysin (a presynaptic vesicular protein) and PSD-95 (a postsynaptic marker) were enriched in synaptoneurosomal fractions, whereas Ca2+/calmodulin-dependent protein kinase II (a pansynaptic protein) was detected in both synaptoneurosomal and cytosolic fractions. Histone H1 (a nuclear protein) was absent in synaptoneurosomal or cytosolic fractions, whereas tubulin (a non-synaptic protein) showed equal distribution in these compartments. We also confirmed the presence of Pyk2 and STEP61 in synaptoneurosomes (Fig. 10A).
FIGURE 10.

Activation of STEP leads to dephosphorylation of Pyk2 in synaptoneurosomes. A, expression of presynaptic and postsynaptic markers in synaptoneurosome preparation. SYP, synaptophysin; CaMKII, Ca2+/calmodulin-dependent protein kinase II. B, synaptoneurosomes from WT (upper panel) or STEP KO (lower panel) mice were stimulated with 40 mm KCl for the indicated durations. Phosphorylation of Pyk2 was determined with Tyr(P)402 Pyk2 antibody. C, CsA-pretreated synaptoneurosomes (100 nm; 10 min) from WT (upper panel) or STEP KO (lower panel) mouse brains were stimulated with 40 mm KCl as in B. D, quantitation of phospho-Pyk2 from B and C. Phosphorylation levels were first normalized to total protein and then to tubulin as a loading control. Error bars indicate the SEM of at least three independent experiments. All values were expressed as -fold changes compared with WT control levels (*, p < 0.05; **, p < 0.01; one-way ANOVA with post hoc Tukey test; n = 4).
Synaptosomes were stimulated for different times with KCl. Phosphorylation of Pyk2 was rapid (1 min, 156.4 ± 6.8% of control; 2 min, 174.7 ± 5.3% of control) and started decreasing at 5 min (5 min, 147.6 ± 5.6% of control; 30 min, 103.6 ± 3.0% of control) (Fig. 10, B and D), similar to earlier findings in hippocampal slices (39). Synaptoneurosomes from STEP KO mouse brains had higher levels of phospho-Pyk2 at base line (135.0 ± 6.0% of WT control; p < 0.01). KCl depolarization further enhanced Pyk2 phosphorylation (1 min, 168.1 ± 6.5% of WT control), and dephosphorylation of Pyk2 was impaired in STEP KO synaptoneurosomes (2 min, 190.5 ± 4.5%; 5 min, 190.3 ± 4.8%; 30 min, 178.6 ± 6.4%; p > 0.05; Fig. 10, B and D).
STEP activity is regulated by PKA phosphorylation, which decreases STEP activity, and by PP2B- and PP1-dependent dephosphorylation of Ser221 within the KIM domain, which increases STEP activity (23, 43, 44). Preincubation with a PP2B inhibitor, CsA, prevented dephosphorylation of Pyk2 at 30 min in WT slices (5 min, 184.2 ± 2.3%; 30 min, 175.2 ± 4.0%; p > 0.05; Fig. 10, C and D). In addition, CsA-pretreated synaptoneurosomes from WT and STEP KO mice showed a pattern similar to that of non-pretreated samples from the KO mice upon KCl stimulation except that CsA per se did not increase Pyk2 phosphorylation because PP2B would not be active under non-stimulated conditions (Fig. 10, C and D). Thus, CsA had no further effect in the absence of STEP. Taken together, these data indicate that STEP is required to dephosphorylate Pyk2 at Tyr402 in the KCl depolarization paradigm. Similarly, we observed a prolonged activation of ERK1/2 in STEP KO synaptoneurosomes or CsA-pretreated samples (supplemental Fig. S3), indicating that STEP opposed ERK activation in the KCl depolarization paradigm.
DISCUSSION
Pyk2 is a member of the non-receptor tyrosine kinase family and is structurally related to focal adhesion kinase. Within the CNS, Pyk2 is implicated in the development of synaptic plasticity and LTP through its ability to increase the conductance of N-methyl-d-aspartate receptor (17, 18) and activate MAPK pathways (13). In addition, Pyk2 is involved in cytoskeletal reorganization (45, 46) as well as the modulation of cell death pathways due to glutamate excitotoxicity (47–49). How Pyk2 is inactivated within neurons is an important unanswered question in the field.
Activation of Pyk2 typically involves membrane depolarization and calcium influx and is initiated by autophosphorylation at Tyr402 (1, 4–7), although additional kinases, including the Tyr receptor kinases EGF receptor and HER2/3, have also been implicated in this process (50, 51). Tyr402 serves as a docking site for the binding of Src and Fyn through their SH2 domains (13, 53). Phosphorylation at additional Pyk2 sites (Tyr579, Tyr580, and Tyr881) by Fyn or Src is required for the full activation of Pyk2 (13, 14). Recent mechanistic studies have demonstrated that Pyk2 interacts with PSD-95 and that translocation to PSDs is necessary for the full activation of Pyk2 and the induction of LTP (15, 17).
STEP is also present within the PSD as demonstrated by electron microscopy (31, 54) and biochemical analysis (28, 31). Unlike Pyk2, however, translocation of STEP to the PSD has not been reported. Thus, STEP might not control the initial translocation and activation of Pyk2 but rather may function to terminate Pyk2 signaling at the PSD. We also observed that the relative phosphorylation of Pyk2 increased in synaptoneurosome (Fig. 10D) but not PSD fractions (Figs. 8C and 9A). Based on our previous work (15), we speculate that Ca2+/calmodulin-dependent phosphorylation of Pyk2 at Tyr(P)402 may stabilize Pyk2 binding to PSD-95. Other possible binding partners at the PSD for stable, Tyr402 phosphorylation-dependent PSD association include Src, which binds to Tyr(P)402 (13) and is anchored via NADH dehydrogenase subunit 2 to NMDA receptors (55). The importance of these experiments is that STEP reverses Pyk2 phosphorylation and thereby suppresses its binding to PSD, hence the increase in both total Pyk2 and phospho-Pyk2 in STEP KO mice (Fig. 8C) or the decrease by TAT-STEP in the KCl paradigm (Fig. 9A).
We identified the domains in STEP and in Pyk2 that mediate their association. The KIM motif is required for the binding of STEP to all substrates tested to date (24, 25, 28, 35). This finding was confirmed with Pyk2 as removal of this domain disrupted the association of STEP with Pyk2. In addition, the unique N terminus of STEP61 appeared to facilitate the binding of STEP to Pyk2 or Fyn. STEP46, which lacks the N-terminal region, pulled down less Pyk2 or Fyn in GST pulldown assays (supplemental Fig. S1). There are two polyproline-rich domains within the N-terminal region (PR1 and PR2). In this study, we showed that the PR2 domain of STEP61 is also involved in the interaction of STEP with Pyk2. In contrast, the PR1 domain is required for the binding of STEP with Fyn, and deletion of the PR2 domain enhanced the binding of STEP to Fyn (25). The presence of the PR2 domain may facilitate the association of Pyk2 and impair Fyn binding to STEP.
The C-terminal region of Pyk2 that lies between the kinase domain and focal adhesion-targeting domain and contains two proline-rich domains is sufficient for the binding of Pyk2 to STEP. These proline-rich domains are necessary for the interaction of Pyk2 with the SH3 domain of PSD-95 and play a critical role in the translocation and activation of Pyk2 during synaptic plasticity (15). The results of our pulldown assays with peptides indicate that STEP and PSD-95 bind to different motifs within the C-terminal region of Pyk2 (residues 671–875). The relative selectivity of peptide 671–694 may have further implications in modulating STEP/Pyk2 interaction in synaptic plasticity, although the usefulness of a cell-penetrating form of this peptide (such as a TAT fusion peptide) awaits further characterization.
An important observation in the present study was that STEP interacted with Pyk2 in the absence of Fyn, indicating that the binding of STEP to Pyk2 does not require the presence of Fyn. Similarly, Pyk2 and Fyn were co-immunoprecipitated from lysates obtained from STEP KO mice, indicating that they are able to associate in the absence of STEP. Competitive binding assays showed that increasing the amount of Fyn or Pyk2 resulted in displacement of Pyk2 or Fyn, respectively. Although these results suggest that Pyk2 and Fyn cannot simultaneously bind STEP directly and that distinct dyadic pools exist, our experiments do not rule out the possibility that these three molecules also form a tripartite complex perhaps via adaptor proteins.
The association of both Pyk2 and Fyn with STEP is of interest as it adds insight into the synergistic regulation of these enzymes. STEP dephosphorylates and inactivates both Pyk2 (this study) and Fyn (25), which are known to phosphorylate and activate each other. In this way, STEP inactivates two kinases involved in the regulation of synaptic plasticity. We also showed an elevated phosphorylation of Pyk2 at Tyr881 in STEP KO samples (supplemental Fig. S2). Because Tyr881 is involved in the activation of the MAPK pathway (1), these data suggest that STEP may regulate MAPK signaling via Pyk2. These findings are consistent with earlier studies that have shown that STEP normally opposes the development of synaptic plasticity (56, 57).
Another example of this type of coordinated modulation by STEP occurs in the internalization of N-methyl-d-aspartate receptor from synaptic membranes. Fyn phosphorylates Tyr1472 of the GluN2B subunit of the N-methyl-d-aspartate receptor, resulting in exocytosis of GluN1-GluN2B receptor complexes to synaptosomal surface membranes (58, 59). Tyr1472 of GluN2B is dephosphorylated by STEP, leading to internalization of GluN1-GluN2B receptors (26, 60). In addition, Fyn is dephosphorylated and inactivated by STEP. Thus, STEP dephosphorylates the Tyr residue on GluN2B targeted by Fyn and inactivates Fyn itself.
The regulation of STEP itself by dopamine signaling is relevant to this discussion. Dopamine D1 receptor stimulation leads to activation of PKA and phosphorylation of STEP at a regulatory serine residue within its KIM domain (42) so that STEP no longer interacts with its substrates. The Tyr phosphorylation and activity of STEP substrates are thereby increased in the case of Pyk2, Fyn, and ERK (23, 43), or the STEP substrates traffic to neuronal membranes in the case of GluN2B (60, 61). At the same time, PKA phosphorylates and activates DARPP-32, leading to the inhibition of PP1 (62–64). PP1 is the phosphatase that dephosphorylates STEP at the same regulatory serine residue within the KIM domain. In this case, a kinase (PKA) that phosphorylates and inactivates its substrate (STEP) is activated, while at the same time, the phosphatase (PP1) that normally activates STEP is inactivated.
STEP belongs to a subgroup of three highly homologous PTPs, which are the only proteins identified to date that contain a KIM domain. These PTPs include HePTP, which is enriched in hematopoietic cells (65), and STEP-like PTP (PTP-SL), which is enriched in cerebellum (66). STEP is not found in either of these tissues (67). As Pyk2 is present in both hematopoietic cells and the cerebellum, future studies are needed to test the prediction that the related PTPs regulate Pyk2 in these tissues.
In conclusion, the main finding of this study is that Pyk2 is a novel substrate of STEP. STEP directly binds to and dephosphorylates Pyk2 at Tyr402, and Pyk2 and two of its downstream targets are up-regulated in STEP KO mouse brain. The importance of STEP in the regulation of Pyk2 was further confirmed in a model for synaptic plasticity (KCl depolarization). The present study extends earlier findings that suggest that STEP opposes the development of synaptic strengthening by regulating signaling pathways that play important roles in normal physiological functions within the CNS.
Acknowledgment
We thank members of the laboratory for extensive discussions and suggestions.
This work was supported, in whole or in part, by National Institutes of Health Grants MH052711 and MH091037 (to P. J. L.) and AG017502 (to J. W. H.).

This article contains supplemental Figs. S1–S3.
- Pyk2
- proline-rich tyrosine kinase 2
- LTP
- long term potentiation
- STEP
- striatal-enriched protein-tyrosine phosphatase
- PSD
- postsynaptic density
- PR
- polyproline-rich
- SH
- Src homology
- PTP
- protein-tyrosine phosphatase
- TAT
- trans-activator of transcription
- KIM
- kinase-interacting motif
- aCSF
- artificial cerebrospinal fluid
- CsA
- cyclosporin A
- C/S
- C300S
- FP
- fluorescence polarization
- SOS
- Son-of-sevenless
- PEST
- proline (P), glutamic acid (E), serine (S) and threonine (T)
- SYF cells
- mouse embryo fibroblast cells deficient for Src, Yes and Fyn
- SEM
- standard error of the mean
- ANOVA
- analysis of variance.
REFERENCES
- 1. Lev S., Moreno H., Martinez R., Canoll P., Peles E., Musacchio J. M., Plowman G. D., Rudy B., Schlessinger J. (1995) Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376, 737–745 [DOI] [PubMed] [Google Scholar]
- 2. Sasaki H., Nagura K., Ishino M., Tobioka H., Kotani K., Sasaki T. (1995) Cloning and characterization of cell adhesion kinase β, a novel protein tyrosine kinase of the focal adhesion kinase subfamily. J. Biol. Chem. 270, 21206–21219 [DOI] [PubMed] [Google Scholar]
- 3. Menegon A., Burgaya F., Baudot P., Dunlap D. D., Girault J. A., Valtorta F. (1999) FAK+ and PYK2/CAKβ, two related tyrosine kinases highly expressed in the central nervous system: similarities and differences in the expression pattern. Eur. J. Neurosci. 11, 3777–3788 [DOI] [PubMed] [Google Scholar]
- 4. Girault J. A., Costa A., Derkinderen P., Studler J. M., Toutant M. (1999) FAK and PYK2/CAKβ in the nervous system: a link between neuronal activity, plasticity and survival? Trends Neurosci. 22, 257–263 [DOI] [PubMed] [Google Scholar]
- 5. Schaller M. D. (2008) Calcium-dependent Pyk2 activation: a role for calmodulin? Biochem. J. 410, e3–e4 [DOI] [PubMed] [Google Scholar]
- 6. Siciliano J. C., Toutant M., Derkinderen P., Sasaki T., Girault J. A. (1996) Differential regulation of proline-rich tyrosine kinase 2/cell adhesion kinase β (PYK2/CAKβ) and pp125FAK by glutamate and depolarization in rat hippocampus. J. Biol. Chem. 271, 28942–28946 [DOI] [PubMed] [Google Scholar]
- 7. Derkinderen P., Siciliano J., Toutant M., Girault J. A. (1998) Differential regulation of FAK+ and PYK2/Cakβ, two related tyrosine kinases, in rat hippocampal slices: effects of LPA, carbachol, depolarization and hyperosmolarity. Eur. J. Neurosci. 10, 1667–1675 [DOI] [PubMed] [Google Scholar]
- 8. Sabri A., Govindarajan G., Griffin T. M., Byron K. L., Samarel A. M., Lucchesi P. A. (1998) Calcium- and protein kinase C-dependent activation of the tyrosine kinase PYK2 by angiotensin II in vascular smooth muscle. Circ. Res. 83, 841–851 [DOI] [PubMed] [Google Scholar]
- 9. Lu W. Y., Jackson M. F., Bai D., Orser B. A., MacDonald J. F. (2000) In CA1 pyramidal neurons of the hippocampus protein kinase C regulates calcium dependent inactivation of NMDA receptors. J. Neurosci. 20, 4452–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schlaepfer D. D., Hauck C. R., Sieg D. J. (1999) Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71, 435–478 [DOI] [PubMed] [Google Scholar]
- 11. Avraham H., Park S. Y., Schinkmann K., Avraham S. (2000) RAFTK/Pyk2-mediated cellular signaling. Cell. Signal. 12, 123–133 [DOI] [PubMed] [Google Scholar]
- 12. Schauwienold D., Sastre A. P., Genzel N., Schaefer M., Reusch H. P. (2008) The transactivated epidermal growth factor receptor recruits Pyk2 to regulate Src kinase activity. J. Biol. Chem. 283, 27748–27756 [DOI] [PubMed] [Google Scholar]
- 13. Dikic I., Tokiwa G., Lev S., Courtneidge S. A., Schlessinger J. (1996) A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383, 547–550 [DOI] [PubMed] [Google Scholar]
- 14. Park S. Y., Avraham H. K., Avraham S. (2004) RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. J. Biol. Chem. 279, 33315–33322 [DOI] [PubMed] [Google Scholar]
- 15. Bartos J. A., Ulrich J. D., Li H., Beazely M. A., Chen Y., Macdonald J. F., Hell J. W. (2010) Postsynaptic clustering and activation of Pyk2 by PSD-95. J. Neurosci. 30, 449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blaukat A., Ivankovic-Dikic I., Grönroos E., Dolfi F., Tokiwa G., Vuori K., Dikic I. (1999) Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. J. Biol. Chem. 274, 14893–14901 [DOI] [PubMed] [Google Scholar]
- 17. Huang Y., Lu W., Ali D. W., Pelkey K. A., Pitcher G. M., Lu Y. M., Aoto H., Roder J. C., Sasaki T., Salter M. W., MacDonald J. F. (2001) CAKβ/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29, 485–496 [DOI] [PubMed] [Google Scholar]
- 18. Salter M. W., Kalia L. V. (2004) Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328 [DOI] [PubMed] [Google Scholar]
- 19. Kumar S., Avraham S., Bharti A., Goyal J., Pandey P., Kharbanda S. (1999) Negative regulation of PYK2/related adhesion focal tyrosine kinase signal transduction by hematopoietic tyrosine phosphatase SHPTP1. J. Biol. Chem. 274, 30657–30663 [DOI] [PubMed] [Google Scholar]
- 20. Lyons P. D., Dunty J. M., Schaefer E. M., Schaller M. D. (2001) Inhibition of the catalytic activity of cell adhesion kinase β by protein-tyrosine phosphatase-PEST-mediated dephosphorylation. J. Biol. Chem. 276, 24422–24431 [DOI] [PubMed] [Google Scholar]
- 21. Halfter U. M., Derbyshire Z. E., Vaillancourt R. R. (2005) Interferon-γ-dependent tyrosine phosphorylation of MEKK4 via Pyk2 is regulated by annexin II and SHP2 in keratinocytes. Biochem. J. 388, 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davidson D., Veillette A. (2001) PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 20, 3414–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paul S., Nairn A. C., Wang P., Lombroso P. J. (2003) NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 6, 34–42 [DOI] [PubMed] [Google Scholar]
- 24. Muñoz J. J., Tárrega C., Blanco-Aparicio C., Pulido R. (2003) Differential interaction of the tyrosine phosphatases PTP-SL, STEP and HePTP with the mitogen-activated protein kinases ERK1/2 and p38α is determined by a kinase specificity sequence and influenced by reducing agents. Biochem. J. 372, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen T. H., Liu J., Lombroso P. J. (2002) Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 277, 24274–24279 [DOI] [PubMed] [Google Scholar]
- 26. Zhang Y., Kurup P., Xu J., Carty N., Fernandez S. M., Nygaard H. B., Pittenger C., Greengard P., Strittmatter S. M., Nairn A. C., Lombroso P. J. (2010) Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer's disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 19014–19019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Venkitaramani D. V., Gladding C. M., Zhang Y., Kurup P., Molnar E., Collingridge G. L., Lombroso P. J. (2008) The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J. Neurosci. 28, 10561–10566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu J., Kurup P., Zhang Y., Goebel-Goody S. M., Wu P. H., Hawasli A. H., Baum M. L., Bibb J. A., Lombroso P. J. (2009) Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 29, 9330–9343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ren J., Wen L., Gao X., Jin C., Xue Y., Yao X. (2009) DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273 [DOI] [PubMed] [Google Scholar]
- 30. Venkitaramani D. V., Paul S., Zhang Y., Kurup P., Ding L., Tressler L., Allen M., Sacca R., Picciotto M. R., Lombroso P. J. (2009) Knockout of striatal enriched protein tyrosine phosphatase in mice results in increased ERK1/2 phosphorylation. Synapse 63, 69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goebel-Goody S. M., Davies K. D., Alvestad Linger R. M., Freund R. K., Browning M. D. (2009) Phospho-regulation of synaptic and extrasynaptic N-methyl-d-aspartate receptors in adult hippocampal slices. Neuroscience 158, 1446–1459 [DOI] [PubMed] [Google Scholar]
- 32. Lim I. A., Hall D. D., Hell J. W. (2002) Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J. Biol. Chem. 277, 21697–21711 [DOI] [PubMed] [Google Scholar]
- 33. Qian D., Lev S., van Oers N. S., Dikic I., Schlessinger J., Weiss A. (1997) Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J. Exp. Med. 185, 1253–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klinghoffer R. A., Sachsenmaier C., Cooper J. A., Soriano P. (1999) Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J. 18, 2459–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eminaga S., Bennett A. M. (2008) Noonan syndrome-associated SHP-2/Ptpn11 mutants enhance SIRPα and PZR tyrosyl phosphorylation and promote adhesion-mediated ERK activation. J. Biol. Chem. 283, 15328–15338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pulido R., Zúñiga A., Ullrich A. (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 17, 7337–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ostergaard H. L., Lou O., Arendt C. W., Berg N. N. (1998) Paxillin phosphorylation and association with Lck and Pyk2 in anti-CD3- or anti-CD45-stimulated T cells. J. Biol. Chem. 273, 5692–5696 [DOI] [PubMed] [Google Scholar]
- 38. Andreev J., Simon J. P., Sabatini D. D., Kam J., Plowman G., Randazzo P. A., Schlessinger J. (1999) Identification of a new Pyk2 target protein with Arf-GAP activity. Mol. Cell Biol. 19, 2338–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kruljac-Letunic A., Moelleken J., Kallin A., Wieland F., Blaukat A. (2003) The tyrosine kinase Pyk2 regulates Arf1 activity by phosphorylation and inhibition of the Arf-GTPase-activating protein ASAP1. J. Biol. Chem. 278, 29560–29570 [DOI] [PubMed] [Google Scholar]
- 40. Corvol J. C., Valjent E., Toutant M., Enslen H., Irinopoulou T., Lev S., Hervé D., Girault J. A. (2005) Depolarization activates ERK and proline-rich tyrosine kinase 2 (PYK2) independently in different cellular compartments in hippocampal slices. J. Biol. Chem. 280, 660–668 [DOI] [PubMed] [Google Scholar]
- 41. Schwarze S. R., Hruska K. A., Dowdy S. F. (2000) Protein transduction: unrestricted delivery into all cells? Trends. Cell Biol. 10, 290–295 [DOI] [PubMed] [Google Scholar]
- 42. Villasana L. E., Klann E., Tejada-Simon M. V. (2006) Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J. Neurosci. Methods 158, 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paul S., Snyder G. L., Yokakura H., Picciotto M. R., Nairn A. C., Lombroso P. J. (2000) Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase, STEP, through a PKA-mediated pathway. J. Neurosci. 20, 5630–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Valjent E., Pascoli V., Svenningsson P., Paul S., Enslen H., Corvol J. C., Stipanovich A., Caboche J., Lombroso P. J., Nairn A. C., Greengard P., Hervé D., Girault J. A. (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc. Natl. Acad. Sci. U.S.A. 102, 491–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Du Q. S., Ren X. R., Xie Y., Wang Q., Mei L., Xiong W. C. (2001) Inhibition of PYK2-induced actin cytoskeleton reorganization, PYK2 autophosphorylation and focal adhesion targeting by FAK. J. Cell Sci. 114, 2977–2987 [DOI] [PubMed] [Google Scholar]
- 46. Haglund K., Ivankovic-Dikic I., Shimokawa N., Kruh G. D., Dikic I. (2004) Recruitment of Pyk2 and Cbl to lipid rafts mediates signals important for actin reorganization in growing neurites. J. Cell Sci. 117, 2557–2568 [DOI] [PubMed] [Google Scholar]
- 47. Liu Y., Zhang G., Gao C., Hou X. (2001) NMDA receptor activation results in tyrosine phosphorylation of NMDA receptor subunit 2A (NR2A) and interaction of Pyk2 and Src with NR2A after transient cerebral ischemia and reperfusion. Brain Res. 909, 51–58 [DOI] [PubMed] [Google Scholar]
- 48. Ma J., Zhang G. Y., Liu Y., Yan J. Z., Hao Z. B. (2004) Lithium suppressed Tyr-402 phosphorylation of proline-rich tyrosine kinase (Pyk2) and interactions of Pyk2 and PSD-95 with NR2A in rat hippocampus following cerebral ischemia. Neurosci. Res. 49, 357–362 [DOI] [PubMed] [Google Scholar]
- 49. Guo J., Meng F., Fu X., Song B., Yan X., Zhang G. (2004) N-Methyl-d-aspartate receptor and L-type voltage-gated Ca2+ channel activation mediate proline-rich tyrosine kinase 2 phosphorylation during cerebral ischemia in rats. Neurosci. Lett. 355, 177–180 [DOI] [PubMed] [Google Scholar]
- 50. Andreev J., Galisteo M. L., Kranenburg O., Logan S. K., Chiu E. S., Okigaki M., Cary L. A., Moolenaar W. H., Schlessinger J. (2001) Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J. Biol. Chem. 276, 20130–20135 [DOI] [PubMed] [Google Scholar]
- 51. Collins M., Tremblay M., Chapman N., Curtiss M., Rothman P. B., Houtman J. C. (2010) The T cell receptor-mediated phosphorylation of Pyk2 tyrosines 402 and 580 occurs via a distinct mechanism than other receptor systems. J. Leukoc. Biol. 87, 691–701 [DOI] [PubMed] [Google Scholar]
- 52.Deleted in proof
- 53. Seabold G. K., Burette A., Lim I. A., Weinberg R. J., Hell J. W. (2003) Interaction of the tyrosine kinase Pyk2 with the N-methyl-d-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J. Biol. Chem. 278, 15040–15048 [DOI] [PubMed] [Google Scholar]
- 54. Oyama T., Goto S., Nishi T., Sato K., Yamada K., Yoshikawa M., Ushio Y. (1995) Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: a light and electron microscopic study with a complementary DNA-generated polyclonal antibody. Neuroscience 69, 869–880 [DOI] [PubMed] [Google Scholar]
- 55. Gingrich J. R., Pelkey K. A., Fam S. R., Huang Y., Petralia R. S., Wenthold R. J., Salter M. W. (2004) Unique domain anchoring of Src to synaptic NMDA receptors via the mitochondrial protein NADH dehydrogenase subunit 2. Proc. Natl. Acad. Sci. U.S.A. 101, 6237–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Braithwaite S. P., Paul S., Nairn A. C., Lombroso P. J. (2006) Synaptic plasticity: one STEP at a time. Trends Neurosci. 29, 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baum M. L., Kurup P., Xu J., Lombroso P. J. (2010) A STEP forward in neural function and degeneration. Commun. Integr. Biol. 3, 419–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dunah A. W., Sirianni A. C., Fienberg A. A., Bastia E., Schwarzschild M. A., Standaert D. G. (2004) Dopamine D1-dependent trafficking of striatal N-methyl-d-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 65, 121–129 [DOI] [PubMed] [Google Scholar]
- 59. Hallett P. J., Spoelgen R., Hyman B. T., Standaert D. G., Dunah A. W. (2006) Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J. Neurosci. 26, 4690–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Snyder E. M., Nong Y., Almeida C. G., Paul S., Moran T., Choi E. Y., Nairn A. C., Salter M. W., Lombroso P. J., Gouras G. K., Greengard P. (2005) Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 8, 1051–1058 [DOI] [PubMed] [Google Scholar]
- 61. Roche K. W., Standley S., McCallum J., Dune Ly C., Ehlers M. D., Wenthold R. J. (2001) Molecular determinants of NMDA receptor internalization. Nat. Neurosci. 4, 794–802 [DOI] [PubMed] [Google Scholar]
- 62. Hemmings H. C., Jr., Greengard P., Tung H. Y., Cohen P. (1984) DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature 310, 503–505 [DOI] [PubMed] [Google Scholar]
- 63. Nishi A., Snyder G. L., Nairn A. C., Greengard P. (1999) Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J. Neurochem. 72, 2015–2021 [DOI] [PubMed] [Google Scholar]
- 64. Svenningsson P., Nishi A., Fisone G., Girault J. A., Nairn A. C., Greengard P. (2004) DARPP-32: an integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 44, 269–296 [DOI] [PubMed] [Google Scholar]
- 65. Adachi M., Sekiya M., Isobe M., Kumura Y., Ogita Z., Hinoda Y., Imai K., Yachi A. (1992) Molecular cloning and chromosomal mapping of a human protein-tyrosine phosphatase LC-PTP. Biochem. Biophys. Res. Commun. 186, 1607–1615 [DOI] [PubMed] [Google Scholar]
- 66. Shiozuka K., Watanabe Y., Ikeda T., Hashimoto S., Kawashima H. (1995) Cloning and expression of PCPTP1 encoding protein tyrosine phosphatase. Gene 162, 279–284 [DOI] [PubMed] [Google Scholar]
- 67. Lombroso P. J., Murdoch G., Lerner M. (1991) Molecular characterization of a protein-tyrosine-phosphatase enriched in striatum. Proc. Natl. Acad. Sci. U.S.A. 88, 7242–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Boulanger L. M., Lombroso P. J., Raghunathan A., During M. J., Wahle P., Naegele J. R. (1995) Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J. Neurosci. 15, 1532–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]