

Background: Phospho-telokin is a cyclic nucleotide-dependent protein kinase substrate that leads to smooth muscle relaxation.
Results: Phospho-telokin activates inhibited phosphorylated myosin phosphatase.
Conclusion: Phospho-telokin binds to the phosphorylated myosin phosphatase facilitating its binding to phosphomyosin and myosin dephosphorylation.
Significance: This mechanism may play a role in cyclic nucleotide-mediated relaxation of telokin expressing smooth muscles in the gut and vasculature.
Keywords: Cell Signaling, Cyclic AMP (cAMP), Cyclic GMP (cGMP), Cyclic Nucleotides, Phosphatase, Smooth Muscle, Myosin Phosphatase, Telokin
Abstract
Phospho-telokin is a target of elevated cyclic nucleotide concentrations that lead to relaxation of gastrointestinal and some vascular smooth muscles (SM). Here, we demonstrate that in telokin-null SM, both Ca2+-activated contraction and Ca2+ sensitization of force induced by a GST-MYPT1(654–880) fragment inhibiting myosin light chain phosphatase were antagonized by the addition of recombinant S13D telokin, without changing the inhibitory phosphorylation status of endogenous MYPT1 (the regulatory subunit of myosin light chain phosphatase) at Thr-696/Thr-853 or activity of Rho kinase. Cyclic nucleotide-induced relaxation of force in telokin-null ileum muscle was reduced but not correlated with a change in MYPT1 phosphorylation. The 40% inhibited activity of phosphorylated MYPT1 in telokin-null ileum homogenates was restored to nonphosphorylated MYPT1 levels by addition of S13D telokin. Using the GST-MYPT1 fragment as a ligand and SM homogenates from WT and telokin KO mice as a source of endogenous proteins, we found that only in the presence of endogenous telokin, thiophospho-GST-MYPT1 co-precipitated with phospho-20-kDa myosin regulatory light chain 20 and PP1. Surface plasmon resonance studies showed that S13D telokin bound to full-length phospho-MYPT1. Results of a protein ligation assay also supported interaction of endogenous phosphorylated MYPT1 with telokin in SM cells. We conclude that the mechanism of action of phospho-telokin is not through modulation of the MYPT1 phosphorylation status but rather it contributes to cyclic nucleotide-induced relaxation of SM by interacting with and activating the inhibited full-length phospho-MYPT1/PP1 through facilitating its binding to phosphomyosin and thus accelerating 20-kDa myosin regulatory light chain dephosphorylation.
Introduction
Although the major mechanism for initiating smooth muscle (SM)2 contraction is the rise in [Ca2+] resulting in an increase in 20-kDa myosin regulatory light chain (MLC20) phosphorylation at Ser-19, the magnitude of developed force can be further modulated through signaling pathways that shift the balance between myosin light chain kinase and myosin light chain phosphatase (MLCP) activities. Modulation of the activities of these enzymes occurs under physiological and pathophysiological conditions resulting in sensitization or desensitization of the contractile machinery to [Ca2+] and altered force output (1–3). Modulation of MLCP activity by inhibitory phosphorylation of its myosin phosphatase targeting subunit, MYPT1, at Thr-696 and Thr-853 (mammalian sequence) and of the phosphatase inhibitory protein, CPI-17, at Thr-38 plays a dominant role in the agonist-induced inhibition of MLCP resulting in increased MLC20 phosphorylation and force at constant [Ca2+] termed Ca2+ sensitization (4–13). Although MYPT1 is spontaneously phosphorylated at Thr-696 in resting SM, a further increase in MYPT1 Thr-696 phosphorylation is associated with hypertension and vasospasm (14–17). Other possible mechanisms of phosphorylation-dependent inhibition of MLCP include an allosteric effect of MYPT1 phosphorylation on MLCP activity (18), dissociation of MLCP holoenzyme from myosin (12), modulation of the MYPT1 “docking” function, and/or its translocation to the plasma membrane (19–21), although the population of MLCP undergoing translocation in intact SM tissues is unknown. Recently, using a GST-MYPT1 fragment (residues 654–853) having the inhibitory phosphorylation sites, we proposed a phosphorylation-dependent MLCP autoinhibition model where each phospho-Thr-696 and -Thr-853 docks directly at the active sites and inhibits MLCP accounting for Ca2+ sensitization of SM contraction (22). In addition to MYPT1 phosphorylation, the specific activity of MLCP depends on the extended (6 S) and folded (10:S) conformation of myosin (4, 23).
Conversely, NO, atrial natriuretic factors, β-adrenergic agents, and vasoactive intestinal peptide through cAMP/cGMP can reverse Ca2+-sensitized force inducing relaxation through reduction of Ca2+ and reduction of Ca2+ sensitivity, termed Ca2+ desensitization (1, 3, 24, 25). Lines of evidence indicate an involvement of PKA and PKG1a in Ca2+ desensitization and SM relaxation, although the underlying mechanism(s) remains controversial. De-inhibition can occur through MYPT1 Ser-695 phosphorylation blocking the inhibitory phosphorylation at Thr-696 (26, 27). However, in cerebral arteries Thr-696 phosphorylation is independent of Ser-695 phosphorylation (20). Consistently, the force produced by the MYPT1 fragment with Ala substitution at Ser-695 is relaxed in response to 8-Br-cGMP treatment to the same extent as the Ser-695-containing fragment (22) suggesting additional mechanisms mediating PKA/PKG signals. Prostate apoptosis response (Par)-4 has been reported to activate MLCP by binding to its leucine zipper (LZ) and preventing inhibitory phosphorylation that is reversed by phosphorylation of Par-4 by zipper-interacting kinase (28). Direct stimulation of MLCP in arterial SM is also reported to occur through interaction between the LZ motifs of PKG1α and MYPT1 (29) and/or amino acids 888–982 of MYPT1 and fragment PKG1α (30). However, it is not clear whether PKG binding to MYPT1 significantly contributes to PKG/PKA-induced relaxation of gastrointestinal SM or whether Par-4 plays any role. We have found that telokin, also known as kinase-related protein, is a PKA/PKG target that activates MLCP activity (25, 31–33). Telokin is an SM-specific, 17-kDa acidic protein whose sequence is identical to the noncatalytic C terminus of SM myosin light chain kinase and is independently expressed in SM through a promoter located within an intron of the MYLK gene (33–36). Telokin is abundantly expressed in phasic SMs such as the ileum, bladder, and portal vein, where it is equivalent to the myosin concentration (25, 31–33). In intact ileum, telokin is the major protein phosphorylated at Ser-13 during forskolin- or 8-Br-cGMP-induced relaxation (33, 37, 38). In permeabilized SM, recombinant S13D telokin relaxes submaximal Ca2+-activated force under clamped [Ca2+] conditions or accelerates the relaxation induced by decreasing internal [Ca2+] or by inclusion of 8-Br-cGMP into the solution (25, 32, 33). Genetic deletion of telokin causes Ca2+ sensitization of ileum SM contraction, accompanied by a 30% decrease in MLCP activity (32). Both force and phosphatase activity were rescued by exogenous telokin and further enhanced by PKG. cGMP-induced relaxation was attenuated by 50% in ileum smooth muscle strips from telokin KO mice (32). Normal arrays of thick filaments in telokin KO SM indicated that telokin is not required for filament formation, although it directly binds to MLC20 and the myosin heavy chain S2 region, causing stabilization of myosin filaments in vitro (39, 40). The above findings led us to hypothesize that telokin is involved in cyclic nucleotide-dependent activation of MLCP in SM tissues.
Contrary to these documented effects of telokin on mechanical behavior of SM, little is known about the molecular mechanism(s) responsible for the ability of telokin to activate MLCP activity, the focus of this study. Using mechanical measurements of contractile activity of permeabilized SMs combined with biochemical and binding studies, we demonstrate the following: 1) telokin desensitizes SM to Ca2+ in the absence of significant changes in the phosphorylation level of either Thr-696/Thr-853 MYPT1 sites; 2) the smaller magnitude of cyclic nucleotide-induced relaxation in the telokin-null versus the wild type SM is independent of changes in MYPT1 phosphorylation at Thr-696/853; 3) based on the lack of an effect of the Rho-associated coiled-coil containing protein kinase (ROCK) inhibitor on the ability of recombinant telokin to induce relaxation, we conclude that telokin functions independently of ROCK, and 4) telokin binds to and enhances MLCP activity of only phosphorylated inhibited MLCP. Based on these findings, we propose a model in which cGMP/cAMP-generated phospho-telokin enhances the binding affinity of phospho-MYPT1/PP1C for phosphomyosin inducing MLCP activation resulting in a fall in MLC20 phosphorylation and relaxation. It is expected that in gastrointestinal and bladder SM and in some blood vessels where telokin is abundant, telokin contributes to cGMP/cAMP-induced relaxation of SM. Abnormalities of this pathway may contribute to diseases such as gastroparesis and disturbed gastrointestinal motility.
EXPERIMENTAL PROCEDURES
Tissues, Force Measurements, and Protein Production
Mice were anesthetized and killed according to a protocol approved by the Animal Care and Use Committee at the University of Virginia. Force measurements from longitudinal ileum SM from WT and telokin KO mice, β-escin permeabilization protocols and solutions, MLC20 and MLCP phosphorylation, preparation and blots of phosphorylated turkey gizzard myosin, S13D telokin, and the recombinant GST-MYPT1 fragments used for mechanical and binding studies are described in the supplemental material.
MLCP Assays
Homogenates of SM were prepared as described previously (22) and cleared by spinning (13,000 × g for 10 min), and MLCP activity was measured in the supernatant as described in the supplemental material.
Binding Assays
Pulldown assays were used to evaluate binding between proteins MYPT1, GST-MYPT1(654–880) fragment, PP1C, and SM myosin as a function of the phosphorylation status of MYPT1 and myosin in the presence or absence of the phospho-mimic mutant, S13D telokin, and are detailed in the supplemental material.
Reconstituted full-length FLAG-tagged MYPT1-PP1 complex was prepared by the mammalian cell co-expression system3 as described in the supplemental material. Note that that this full-length FLAG-tagged MYPT1-PP1 complex was used in all the SPR assays.
SPR Assays
SPR experiments were carried out on the Biacore 3000 (Biacore, Uppsala, Sweden) at 25 °C as detailed in the supplemental material. Using CM3 chips, we found that thiophosphorylated full-length MYPT1 directly binds strongly to the CM3 chip matrix and cannot be removed by 100 mm glycine at pH 2. Therefore, in the experiments designed to test the direct binding of S13D telokin and thiophosphorylated full-length MYPT1, no additional immobilization method was used. The amount of bound protein was estimated as the difference between the steady SPR signal after ∼2 min of washing away the unbound proteins after completion of injection and the basal SPR signal immediately before injection.
Indirect Immunofluorescence Staining and Proximity Ligation Assay (PLA)
Serum-starved subconfluent myosin light chain kinase/telokin-null aortic SMCs transfected with FLAG-telokin and stimulated with 1 μm sphingosine 1-phosphate with or without 10 μm Rho kinase inhibitor Y-27632 were fixed, and PLA was carried out as detailed in the supplemental material.
Reagents
Reagents are given in the supplemental material.
Statistics
Means ± S.E. were calculated from three to five independent experiments for each condition. Student's t test or paired t tests were calculated using Sigmaplot 9.0 for comparison of means. p values <0.05 were used as a statistical significant threshold.
RESULTS
Telokin Relaxes Ca2+-activated Contractions without Changing the Phosphorylation Level of MYPT1 and Acts Independently of the RhoA/ROCK Pathway
We have previously shown that the S13D phospho-mimic telokin is 2-fold more potent in relaxing SM than S13A telokin (33, 38), and we use this stable phospho-mimic to test the ability of telokin to modify the MYPT1 phosphorylation status. Isometric force of β-escin-permeabilized WT mouse ileum SM increased in submaximal Ca2+ (pCa 6.3) and was accompanied by a 2-fold increase in MYPT1 Thr-696 and Thr-853 phosphorylation (Fig. 1). This increase in MYPT1 phosphorylation was approximately half of the magnitude reached in the presence of 100 μm GTPγS (data not shown). Addition of 10 μm phospho-mimic S13D telokin mutant triggered a rapid relaxation to 42 ± 7% (n = 6) of the pCa 6.3-induced force without significant changes in the levels of MYPT1 phosphorylation: Thr-696 = 1.12 ± 0.08 and Thr-853 = 0.89 ± 0.06 relative to that at pCa 6.3 (given a value of 1). Thus, at the clamped [Ca2+], telokin can induce relaxation without changing MYPT1 phosphorylation. An involvement of ROCK in telokin-induced relaxation was tested using a specific ROCK inhibitor Y-27632 (Fig. 1B). Ileum SM cells permeabilized with β-escin were activated with pCa 6.3 and treated with Y-27632 (10 μm), and then S13D telokin was added to initiate relaxation. Addition of Y-27632 induced only a small reduction or no reduction in Ca2+-induced contraction (Fig. 1B), although surprisingly MYPT1 phosphorylation at Thr-853 but not Thr-696 was significantly decreased by 40% (1.0 to 0.58 ± 0.06, n = 4) by Y-27632 treatment without relaxation. The subsequent addition of S13D telokin to the muscles markedly reduced Ca2+-activated force by 35 ± 5% (n = 8) (Fig. 1B). This magnitude of relaxation was not significantly different from that in the absence of Y-27632 (42 ± 7% versus 35 ± 5%, n = 8). The MYPT1 Thr-696 level of phosphorylation did not change significantly following ROCK inhibition (1.06 ± 0.07, n = 4, relative to the pCa 6.3 values of 1.0). Neither Thr-696 nor Thr-853 phosphorylation was significantly affected by the subsequent addition of S13D telokin to Y-27632-treated muscles, 1.06 ± 0.07 versus 0.91 ± 0.05 and 0.58 ± 0.06 versus 0.59 ± 0.04 respectively (n = 4) (Fig. 1B). These data suggest that signals mediating telokin-induced SM relaxation are independent of changes in MYPT1 phosphorylation or ROCK activity.
FIGURE 1.

A, recombinant S13D telokin mutant markedly relaxes submaximal Ca2+-activated contraction in β-escin-permeabilized WT mouse ileum SM without significant changes of endogenous MYPT1 phosphorylation levels at Thr-696 and Thr-853 sites as shown in the typical Western blots and bar graph. B, preinhibition with ROCK inhibitor, Y-27632 (10 μm) reduces the phosphorylation level of the MYPT1 Thr-853 site without a significant effect on either the magnitude of S13D telokin-induced relaxation of force or MYPT1 phosphorylation at either Thr-695 or Thr-853 as shown in the typical Western blots and bar graph. Filtrate control is the pCa 6.3 buffer separated from the protein. G10 solution is the cytoplasmic buffer containing 10 mm EGTA and no added Ca2+ (32).
Telokin Amplifies 8-Br-cGMP-induced Relaxation without Change in Phosphorylation Status of Endogenous MYPT1
Cyclic GMP-induced reduction in MYPT1 Thr-696/853 phosphorylation is thought to disinhibit MLCP activity contributing to relaxation of force (15). We tested whether telokin is necessary for the 8-Br-cGMP-induced decrease in the inhibitory phosphorylation of endogenous MYPT1 using SM from our telokin-null transgenic and WT mice (Fig. 2). Force development was measured in ileum strips from WT and telokin KO mice permeabilized with α-toxin and activated by pCa 6.3 followed by a saturating amount of 8-Br-cGMP (10 μm). As reported previously (32), the 8-Br-cGMP-induced relaxation was significantly impaired in telokin-null tissues, compared with WT (Fig. 2A). Surprisingly, the different amplitudes in force relaxation of WT and telokin-null SM (0.51 ± 0.02 and 0.24 ± 0.03, respectively) were not accompanied by proportional changes in MYPT1 phosphorylation at Thr-696 or Thr-853 (WT versus telokin KO Thr-696 = 0.71 ± 0.03 and 0.66 ± 0.04, respectively, n = 8; Thr-853 = 0.61 ± 0.1 and 0.72 ± 0.15, respectively, n = 3) (Fig. 2A, lower panel). Thus, the approximate 40% 8-Br-cGMP-induced decrease in MYPT1 Thr-696/853 phosphorylation did not differ in WT and telokin-null tissues even though the magnitudes of relaxation differed by 2-fold. We have previously shown that neither the amplitude nor EC50 of 8-Br-cGMP-mediated relaxation in the WT or KO ileum differ significantly (32) and therefore do not contribute to this 2-fold difference. Altogether, these results suggest that the telokin-induced contribution to cGMP-mediated relaxation is not coupled with dephosphorylation of MYPT1, a potential cause of an increased MLCP activity.
FIGURE 2.

8-Br-cGMP (10 μm) induces a 2-fold greater relaxation of the submaximal Ca2+-activated force in α-toxin permeabilized ileum from WT compared with telokin-null mice (upper panel) in the absence of a commensurate difference in MYPT1 Thr-696 and Thr 853 phosphorylation as shown in the typical Western blots and bar graph (lower two panels). *, p < 0.05; n.s., not significant.
Reduction in 8-Br-cGMP-induced Relaxation in Telokin-null SM Is Not Due to Changes in Expression of the Cyclic GMP-sensitive Leucine Zipper MYPT1 Isoform
Direct stimulation of MLCP in arterial SM is reported to occur through interaction between the LZ motifs of PKG1α and MYPT1 (29). We therefore examined whether the impaired 8-Br-cGMP-induced relaxation of telokin-null ileum SM was due to a reduction in expression of the LZ-positive MYPT1 isoform using immunoblotting with an antibody recognizing the specific isoform (Fig. 3A). Even when densitometric data of immunoblotting from bladder and ileum samples (Fig. 3A) were pooled to increase sample size, the relative amounts of LZ MYPT1 in WT and KO normalized against myosin heavy chain were 1.0 ± 0.0 and 1.1 ± 0.3, respectively, without a significant difference. Also, expression of total MYPT1 was not significantly different in WT and KO samples; the relative amounts normalized against myosin heavy chain were 1.0 ± 0.0 and 0.9 ± 0.2, respectively. Furthermore, mouse ileum and bladder SM were low LZ MYPT1 expressors compared with aorta (Fig. 3B). Therefore, changes in expression of LZ MYPT1 do not account for the reduced 8-Br-cGMP-induced relaxation of telokin-null SM compared with WT.
FIGURE 3.

A, LZ isoform of MYPT1 is not increased in telokin-null SM. Western blots of total MYPT1 and LZ MYPT1 isoforms in WT and telokin KO mouse bladder and ileum SM. For detection of the LZ MYPT1 isoform, 5-fold greater protein loads were necessary. SM myosin stained with Coomassie Blue was used as a loading control. B, comparison of total MYPT1 and LZ isoform content in mouse bladder, ileum, and thoracic aorta (normalized to myosin) (n = 3) and (lower panel) corresponding Western blots. The level of MYPT1 and LZ+ isoform in thoracic aorta was taken as 1.
S13D Telokin Is Capable of Neutralizing Ca2+-sensitized Force Induced by a GST-tagged MYPT1(654–880) Fragment without Decreasing Its Phosphorylation Level
Recently, we have shown that a GST-tagged MYPT1(654–880) fragment, including the two phosphorylation sites (Thr-696/853), is spontaneously phosphorylated by endogenous ROCK when it is added to permeabilized ileum muscle strips in the presence of ATP. This phosphorylated fragment but not MYPT1 peptide T696A or GST alone bound to and inhibited endogenous MLCP activity resulting in Ca2+-sensitized force (22). Furthermore, S13D telokin-induced relaxation was 2-fold greater than S13A and was not increased by 8-Br-cGMP stimulation (22). In this study, addition of 5 μm GST-MYPT1(654–880) produced a large increase in force at constant [Ca2+] (pCa 6.5) as expected (Fig. 4A). This Ca2+ sensitization of force was relaxed by addition of 15 μm S13D telokin with no significant effect on the phosphorylation level of the GST-MYPT1 fragment at the Thr-696 site (Fig. 4B) (1.0 ± 0.3, n = 6, phosphorylation levels are normalized to the level in the presence of GST-MYPT1 fragment, taken as 1), whereas Thr-853 phosphorylation significantly increased (1.45 ± 0.2 versus 1.0 at pCa 6.5, n = 6), suggesting that S13D telokin does not induce relaxation by decreasing the inhibitory phosphorylation of the GST-MYPT1 fragment. The increase in Thr-853 phosphorylation is not understood and remains to be explored. S13A telokin also relaxed Ca2+-sensitized force (data not shown) but is significantly less potent as shown previously (22). This relaxation occurred without a decrease in MYPT1 phosphorylation of the fragment at either Thr-696 or Thr-853 (Fig. 4B). Relaxation induced by S13D or S13A telokin did not alter phosphorylation of endogenous MYPT1 at either Thr-696 or Thr-853 (Fig. 4C). Thus, exogenous telokin is capable of neutralizing the Ca2+-sensitized force induced by phospho-MYPT1 without decreasing the phosphorylation level of endogenous MYPT1 at Thr-696/853 and of GST-MYPT1(654–880) at Thr-696.
FIGURE 4.
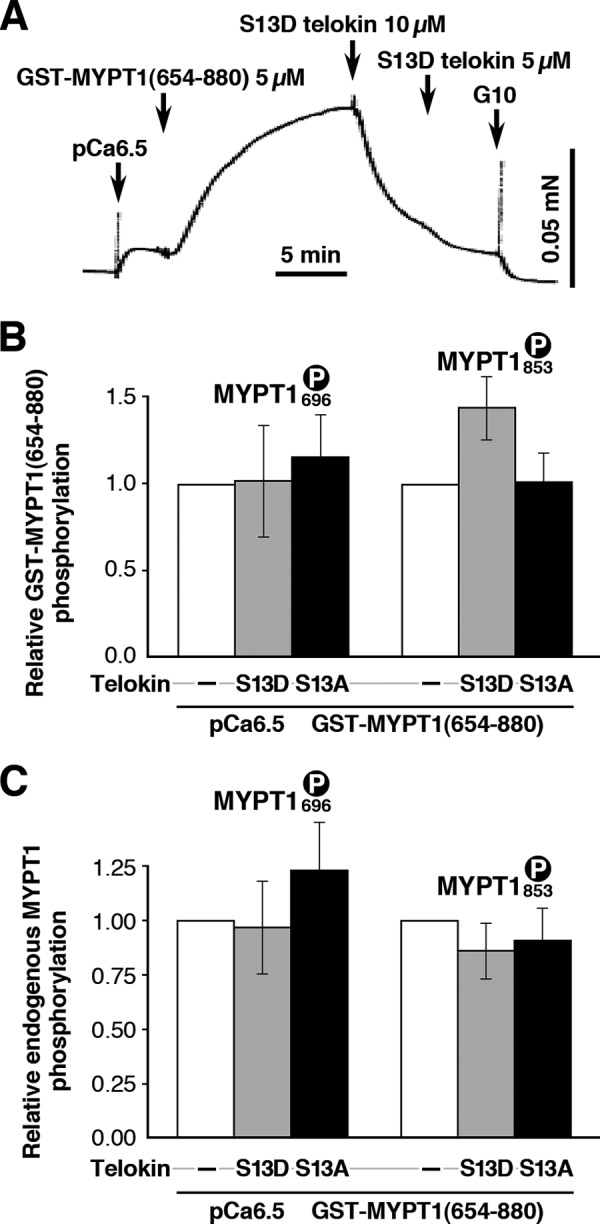
A and B, recombinant S13D telokin completely abolishes the contraction induced by GST-MYPT1(654–880) fragment in β-escin-permeabilized mouse ileum SM without a significant change in the Thr-696 phosphorylation of GST-MYPT1(654–880). A paradoxical increase in Thr-853 phosphorylation level occurred. Addition of S13A telokin, known to be 2-fold less effective than S13D telokin, also relaxed the contraction induced by GST-MYPT1(654–880) fragment to a lesser extent (data not shown) without a significant change in either the Thr-696 or Thr-853 phosphorylation of GST-MYPT1(654–880). Phosphorylation levels after S13D telokin treatment normalized to the levels at pCa 6.3 + GST-MYPT1(654–880). C, relaxation induced by S13D or S13A telokin did not alter the phosphorylation level of endogenous MYPT1 at Thr-696 or Thr-853.
S13D Telokin Protein Restores MLCP Activity Inhibited by Thiophosphorylation of MYPT1
We examined the effects of exogenous S13D telokin on the activity of endogenous MLCP in SM ileum homogenates. The SM homogenates were incubated with ATPγS and constitutively active ROCK to thiophosphorylate WT endogenous MYPT1. The MLCP activity of the homogenate was assayed by adding phospho-SM myosin as substrate and measuring the rate of Pi release. As shown in Fig. 5, pre-thiophosphorylation of the homogenates (phospho-MYPT1) significantly reduced MLCP activity compared with unphosphorylated homogenate (unphospho-MYPT1), 5.6 ± 1.2 versus 9.9 ± 2 pmol/min·mg, respectively, n = 7, p < 0.05. Importantly, the addition of S13D telokin to the thiophosphorylated MYPT1 reversed the inhibition of MLCP activity (8.6 ± 1.8 versus 5.6 ± 1.2 pmol/min·mg, n = 7, p < 0.05). Thus, S13D telokin reverses the phosphorylation-dependent inhibition of MLCP.
FIGURE 5.
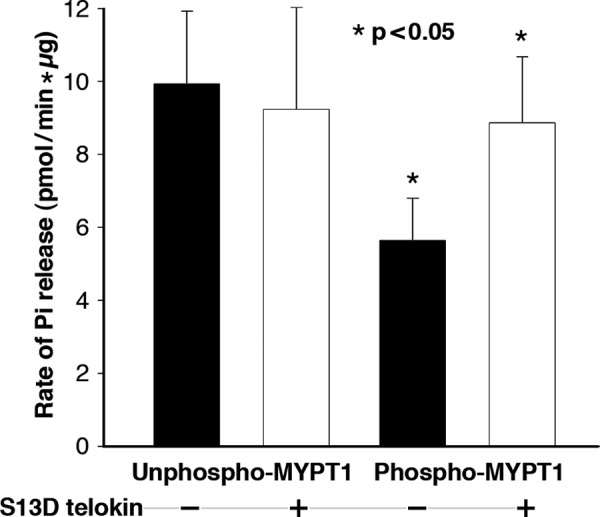
Recombinant S13D telokin restores MLCP activity of inhibited thiophosphorylated MYPT1 (*, p < 0. 05) but has no effect on MLCP activity when MYPT1 is unphosphorylated. MLCP activity was measured as rate of Pi release from prephosphorylated SM myosin as substrate and is based on total protein measured in the homogenate.
In the Absence of MYPT1, Telokin Does Not Change the Rate of SM Myosin Dephosphorylation by the Catalytic Subunit PP1C
Based on our finding that telokin modulates the activity of MLCP, we examined whether telokin affects the activity of PP1C, in the absence of MYPT1. Phosphorylated SM myosin was mixed with purified PP1C in the presence of exogenous S13D telokin or an equimolar amount of BSA as a control. The time course of MLC20 dephosphorylation in the presence and absence of telokin was not significantly different with the t½ of dephosphorylation being 2.5 ± 1 min in control and 3.5 ± 1.5 min, n = 3, in the presence of S13D telokin (supplemental Fig. S1). Results of this experiment and that in Fig. 5 suggest that telokin in itself does not accelerate the activity of MLCP, but rather it disinhibits the suppressed activity of MLCP having phosphorylated MYPT1 restore it to its uninhibited level.
Telokin Modulates Binding between MLCP and Myosin
Phosphorylated GST-MYPT1(654–880) fragment stably docks at the active site of PP1 associated with MYPT1 in cell lysates (22). Here, we used it to capture endogenous MLCP from SM lysates and examined the components of the complex (Fig. 6). Thiophospho- or unphospho-GST-MYPT1(654–880) immobilized on glutathione-Sepharose beads was mixed with mouse ileum lysates as a source of the native complex of endogenous MLCP, telokin, and myosin. The role of telokin was determined by comparison of the results of the assay using homogenates from WT and telokin-null (KO) SM tissue. Bound proteins were detected by immunoblotting using antibodies for MYPT1, PP1 (as an index of endogenous MLCP), telokin, and MLC20 (as an index of myosin bound). In both WT and telokin KO homogenates with nonphosphorylated myosin, thiophospho-GST-MYPT1 fragment efficiently captured endogenous PP1 but not MLC20 or telokin (Fig. 6A), suggesting that the endogenous MLCP complexed with phospho-GST-MYPT1 fragment does not bind to unphosphorylated myosin filaments in either the presence or absence of telokin under these conditions. This markedly contrasts to lysates from WT and telokin-null tissues when the lysates were pretreated with ATPγS in the presence of Ca2+ resulting in thiophosphorylation of endogenous MLC20 (Fig. 6B). Under this condition, all four involved proteins (GST-MYPT1 fragment, phospho-MLC20, telokin, and PP1) were found together in the precipitates, indicating that the GST-MYPT1 fragment beads captured the complex of endogenous MLCP and myosin filaments. Importantly, the binding of MLC20 depends on the presence or absence of telokin in the homogenate. In the presence of telokin in the lysates (Fig. 6B, lanes 1 and 2), MLC20 was co-precipitated with both phospho- and unphospho-GST-MYPT1 fragments. By contrast, without the endogenous telokin, thiophospho-MLC20 was complexed with only unphospho-GST-MYPT1 fragment but not phospho-GST-MYPT1 fragment (Fig. 6B, lanes 3 and 4). This binding of thiophospho-MLC20 to the complex of thiophospho-GST-MYPT1 fragment was restored by addition of S13D telokin to the telokin-null lysate (Fig. 6C, lanes 1 and 2). (Note that Fig. 6C, lane 4, replicates the conditions and findings in Fig. 6B, lane 4.) Therefore, telokin is necessary for phosphomyosin association with the endogenous MLCP, which is captured with the thiophospho-GST-MYPT1 fragment. These results suggest that the accessibility of the endogenous, phosphorylated, and inhibited MLCP to MLC20 depends on the presence of telokin. Because unphosphorylated GST-MYPT1(654–880) fragment does not directly bind to MLCP (22), the co-precipitation of the fragment with MLC20 (Fig. 6B, lanes 2 and 4) likely indicates the direct binding of the unphospho-GST-MYPT1 fragment to the S2 region of myosin and not to the MLCP-active site. Note that as expected, telokin did not alter the interaction of unphospho-GST-MYPT1 fragment with myosin (Fig. 6B, lanes 2 and 4). Myosin binding to unphosphorylated GST-MYPT1(654–880) (Fig. 6B, lanes 2 and 4) likely caused the PP1C binding to the beads, through the interaction between myosin and MYPT1.
FIGURE 6.
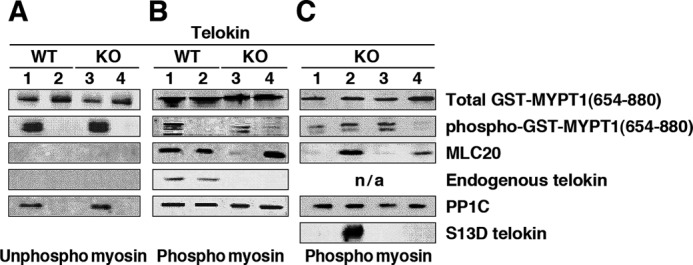
Telokin modulates binding between phospho-GST-MYPT1(654-880) and phosphomyosin. Pulldown assays were performed using thiophospho- or unphospho-GST-MYPT1(654–880) fragment and homogenate from WT or telokin KO smooth muscle (bladder) as source for endogenous PP1C and unphosphorylated (A) or phosphorylated (B) myosin. Appearance of MLC20 in pellet is used as an index of the amount of SM myosin bound to the complex of GST-MYPT1(654–880) fragment/PP1C. Importantly, a decreased amount of MLC20 was precipitated together with thiophosphorylated GST-MYPT1(654–880)/PP1C (lane 3 in B) when using SM homogenate from the telokin-null bladder compared with WT. The presence of WT telokin (lane 1 in B) resulted in precipitation of MLC20 by thiophosphorylated GST-MYPT1(654–880). Unphosphorylated GST-MYPT1(654–880)/PP1C bound phosphomyosin in both the presence or absence of telokin (lanes 2 and 4 in B). Results of a “rescue” experiment are shown in C where 10 μm recombinant S13D telokin (lane 2 in C) or, as a control, BSA (lane 1 in C) was added to the mixture of thiophosphorylated GST-MYPT1(654–880) fragment and homogenate from telokin KO ileum SM. The MLC20 band markedly increases in the presence of S13D telokin (lane 2 in C) but not in the presence of BSA (lane1 in C), as compared with that in the absence of S13D (lane 3). n/a, data not available.
We next tested whether recombinant S13D telokin directly binds to GST-MYPT1(654–880) using a GST pulldown assay (supplemental Fig. S2). S13D telokin was mixed with thiophospho- (supplemental Fig. S2, left) or unphospho- (supplemental Fig. S2, right) GST-MYPT1(654–880) in the presence of glutathione-Sepharose beads, and the pellets and supernatants were analyzed by immunoblotting. S13D telokin did not pellet together with either the phosphorylated or unphosphorylated GST-MYPT1 fragment indicative of a lack of strong binding between telokin and GST-MYPT1 fragment. As pulldown assays are not sufficiently sensitive to weak or transient protein-protein interactions, we next turned to an immunolocalization PLA and an SPR assay to determine whether telokin binds to phospho-MYPT1. In this case, we used full-length MYPT1 to reflect a more physiological state.
Telokin Interacts with Phospho-MYPT1 in SM Cells and in Vitro
The interaction of phospho-MYPT1 and telokin in situ was examined using a PLA that can detect the close proximity (<40 nm) of two antibodies (41), and unlike immunoprecipitation methods, it can detect both stable and transient protein-protein interactions. Using serum-starved telokin-null aortic SMCs (42) expressing FLAG-telokin, a significantly increased PLA signal was detected between endogenous phosphorylated (Thr-696) MYPT1 and FLAG-telokin upon stimulation with sphingosine-1-phosphate compared with control cells, and this was abolished by treatment with the ROCK inhibitor Y-27632 (Fig. 7). Note that the phospho-Thr-696 MYPT1 fluorescence is distributed periodically along stress fibers (Fig. 7C) and also at the cell periphery compared with unstimulated cells under identical labeling and imaging conditions (supplemental Fig. S3). In control experiments for possible nonspecificity of the phospho-Thr-696 MYPT1 antibody, co-expression of FLAG-telokin and Myc-MYPT1 in the presence of serum also showed a strong PLA signal for these very specific antibodies to the epitope tags (Fig. 7B). Thus, telokin and phosphorylated endogenous MYPT1 are in proximity to each other in SM cells under conditions of Ca2+ sensitization but not when ROCK is inhibited and presumably MYPT1 phosphorylation is low.
FIGURE 7.
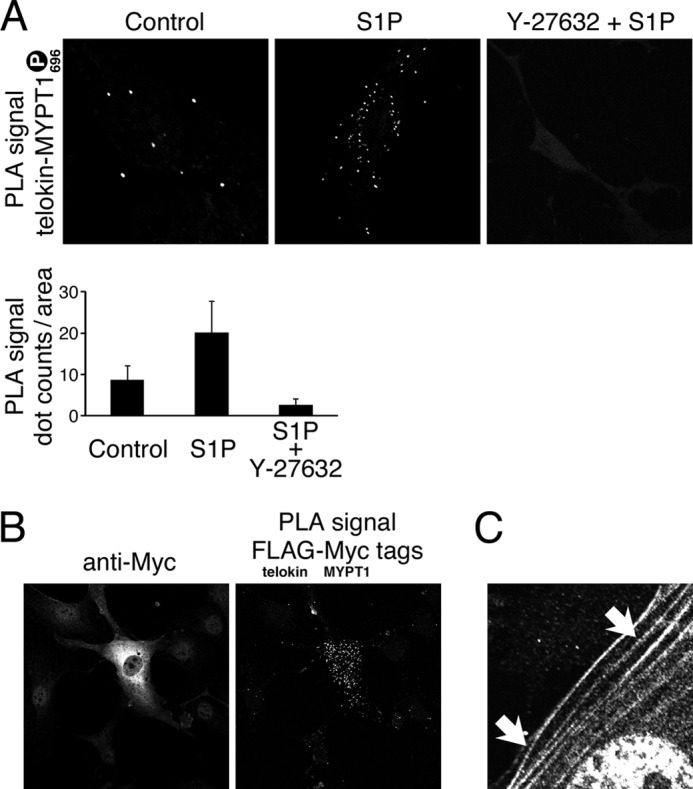
Telokin and phosphorylated MYPT1 are in close proximity in SM cells. PLA and indirect immunofluorescence staining of overexpressed telokin and exogenous MYPT1 and endogenous MYPT1 phosphorylated at Thr-696 are shown. A, PLA using anti-FLAG and anti-pMYP Thr-696 antibodies in myosin light chain kinase/telokin-null mouse aortic SMCs overexpressing FLAG-telokin following treatment with or without Rho kinase inhibitor Y-27632 prior to stimulation by sphingosine 1-phosphate (S1P). The PLA signal indicates close proximity between endogenous phospho-MYPT1 at Thr-696 and overexpressed FLAG-telokin. The graph shows means ± S.E. for 15 cells under each condition. B, indirect immunofluorescence staining and PLA using anti-FLAG and anti-Myc antibodies verifying expression of Myc-MYPT1 and close proximity of FLAG-telokin and Myc-MYPT1 co-expressed in mouse embryonic fibroblast cells. C, indirect immunofluorescence staining with phospho-Thr-696 antibody showing periodic distribution along stress fibers in SMCs following stimulation by sphingosine 1-phosphate as in A.
The interaction of full-length MYPT1/PP1 with telokin was also demonstrated using SPR (Fig. 8). In this experiment, direct binding of S13D telokin with thiophospho-MYPT1/PP1 was examined. Phosphorylated full-length MYPT1 was immobilized on three of the cells on a CM3 chip by direct binding to the matrix. An additional blank cell was used to measure nonspecific binding between telokin and the CM3 matrix. 70, 350, and 700 μm of full-length thiophosphorylated MYPT1 was passed over three different cells resulting in 552, 1732, and 2365 resonance unit (RU) bound. Following washing away of unbound proteins, 6.5 μl (5 μm) of S13D telokin was injected onto each cell resulting in different amounts of bound S13D telokin. S13D telokin bound to the blank cells (without MYPT1) resulted in background signals of 14.4 and 52.4 RU (in the second chip). Upon averaging of SPR signals from S13D injections in each of the cells and subtraction of the respective background, the signals from S13D bound to full-length thiophospho-MYPT1 were as follows: 12 ± 5 and 65 ± 10 RU and 230 correlating with the increasing concentrations of immobilized full-length thiophospho-MYPT1. A positive correlation (Fig. 8) between concentrations of immobilized full-length phosphorylated MYPT1 and bound S13D telokin (after subtraction of background) allows us to conclude that S13D telokin associates with phosphorylated full-length MYPT1 and correlates with the amount of immobilized phosphorylated full-length MYPT1.
FIGURE 8.
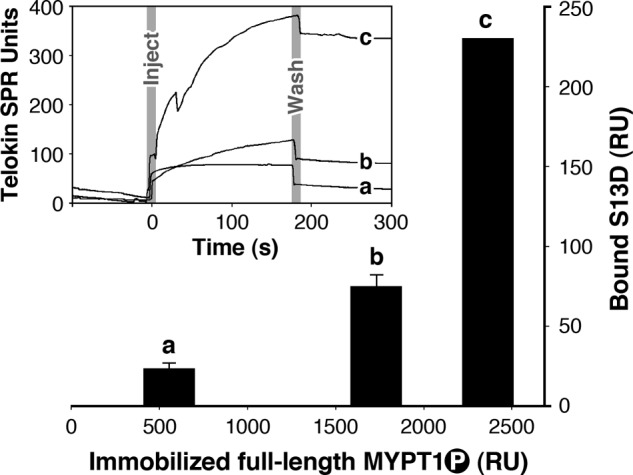
Amplitudes and sensorgrams (inset) showing S13D telokin binding to increasing concentrations of phosphorylated full-length MYPT1. 5 μm S13D telokin was flowed over 70, 350, and 700 μm of full-length thiophosphorylated MYPT1 immobilized on three different cells of a CM3 chip. Background signal was estimated from S13D telokin bound to other blank cells (without MYPT1). Upon averaging of SPR signals from S13D injections and subtraction of the respective backgrounds, the signals from S13D bound to full-length thiophospho-MYPT1 were as follows: 12 ± 5 and 65 ± 10 RU and 230 correlating with the increasing concentrations of immobilized full-length thiophospho-MYPT1.
We also tested the ability of telokin to increase the binding of phosphorylated and inhibited MLCP to phosphorylated myosin using SPR. In the absence of telokin, less myosin binds to phospho-FLAG-MYPT1 (10 versus 16 RU), although in the presence of telokin similar amounts of myosin (23 versus 24 RU) were bound to unphosphorylated and thiophosphorylated MYPT1 consistent with findings in Fig. 6.
However, due to our inability to dissociate our protein of interest from the CM5 matrix despite trying multiple approaches, we were unable to determine the dissociation/association rate constants. Although these results are not optimal, they do lend further support to our other data and hypotheses and are described in the supplemental material.
DISCUSSION
The cyclic nucleotide-dependent protein kinase substrate telokin, consisting of an immunoglobulin-like fold and acidic tail, is present in gastrointestinal and some vascular SMs at concentrations equivalent to the content of myosin. Telokin promotes relaxation through activation of MLCP activity through an unknown mechanism. The simplest explanation would have been if telokin reversed the inhibitory phosphorylation of the Thr-696/853 sites possibly by inhibition of ROCK activity. This was not the case, as in this study the magnitude of telokin-induced relaxation did not decrease with inhibition of ROCK nor did a decrease in phosphorylation of MYPT1 Thr-696/853 accompany telokin-induced relaxation of force. Telokin was without effect on PP1C activity. Furthermore, the 50% greater 8-Br-cGMP-induced relaxation in WT versus telokin-null SM was also not associated with a difference in the status of MYPT1 phosphorylation. Instead, our findings demonstrate that the dominant active form of telokin binds to phospho-MYPT1 and allows the inactive MLCP to target to phosphomyosin. A critical experiment was the demonstration that S13D telokin increased the rate of Pi release from phosphorylated myosin by inhibitory thiophosphorylated MYPT1. Altogether, we find that telokin does not accelerate MLCP activity per se but rather de-inhibits suppressed phosphorylated Thr-696/Thr-853 MLCP activity through enhanced binding of phospho-MYPT1 to phospho- but not unphosphomyosin.
A surprising finding was the dissociation between Thr-853 phosphorylation and force (Figs. 1 and 4). In the first case, ROCK inhibition did not induce any relaxation despite the fact that MYPT1 Thr-853 phosphorylation was reduced. In the second case, S13D telokin-induced relaxation of GST-MYPT1(654–880) fragment-induced force was accompanied by an increase in MYPT1 Thr-853 phosphorylation. Note that unlike Thr-853 phosphorylation, Thr-696 phosphorylation did not change in either case. This raises the possibility that the Thr-853 site serves to target the MYPT1 to myosin rather than regulating its activity, which may be predominantly through the Thr-696 site. It is also possible that the two phosphorylation sites act in concert and may be regulated by different kinases and phosphatases. Resolution of these findings will require further study. The main point for these experiments is the demonstration that telokin-induced relaxation is not due to dephosphorylation and disinhibition of MYPT1 and furthermore that ROCK is upstream of telokin and not regulating telokin-induced relaxation.
In vitro studies have shown binding between MLCP and myosin using nonphosphorylated chicken gizzard MYPT1-PP1C complex and myosin (43), but this binding was dramatically reduced after phosphorylation by ROCK-II at Thr-696 and Thr-853 or solely at Thr-853 (12). This finding plus evidence that phosphorylated MYPT1 translocates to the SM cell membrane upon Ca2+ sensitization (19–21) provided an additional mechanism whereby MLCP activity is decreased. Our data now show that telokin can reverse this dissociation and increase the affinity of inhibited phosphorylated MYPT1 for phosphorylated myosin in mammalian SM.
As with our observation that telokin enhances both the activity (Fig. 5) and the binding of phospho-MYPT1 to phosphomyosin (Figs. 6 and 8), binding of MYPT1 to phosphorylated myosin has also been shown to induce a greater than 2-fold increase in phosphatase activity during mitosis (44). In this case MYPT1 has undergone an activatory phosphorylation at Ser-430, but whether this reflects a conformational change in MYPT1/PP1C resulting in better access or affinity for phospho-MLC20 is not known. Likewise, it was not clear whether the S13D telokin-induced increase in binding between the GST-MYPT1(654–880) fragment and thiophosphomyosin (Fig. 6, B and C) reflects telokin directly binding to MYPT1 and modifying its conformational state or, alternatively, whether telokin associated with phosphorylated myosin modulates the interface between MYPT1 and its substrate thus increasing their mutual affinity. Our SPR and PLA data demonstrate binding of S13D telokin to full-length phospho-MYPT1 in support of the first mechanism. However, in view of the high concentration of telokin, equivalent to the myosin concentration in some SM, and the low 1–2 μm content of MYPT1 in SM, it is likely that both mechanisms may contribute. For example, telokin may serve as a scaffold for myosin and MYPT1 in the same way that scaffold proteins have been shown to increase mitogen-activated protein kinase activation in response to physiological stimuli (45, 46).
A rebinding model based on our findings is presented in Fig. 9. As shown, upon phosphorylation at Thr-696/Thr-853, MYPT1 undergoes a conformational change limiting access of PP1C to phosphorylated MLC20. These changes can be reversed in the presence of telokin. In other words, the active site of the endogenous phosphorylated MLCP becomes accessible to phosphomyosin in the presence of telokin. This mechanism results in an acceleration of MLC20 dephosphorylation and thus contributes to cyclic nucleotide-induced relaxation of SM stimulated by adrenergic, vasoactive intestinal peptide (VIP) or NO in the gut or β-adrenergic or NO stimulation of vascular beds that express telokin.
FIGURE 9.

Rebinding model summarizing the mechanism of telokin-mediated disinhibition of MLCP activity. Telokin does not accelerate MLCP activity per se. Based on our findings, two possible mechanisms are shown. Telokin binds to phosphorylated MYPT1 and/or to phosphorylated myosin and de-inhibits suppressed phosphorylated Thr-696/Thr-853 MLCP activity. This mechanism results in an acceleration of MLC20 dephosphorylation and contributes to cyclic nucleotide-induced relaxation of SMs that express telokin.
Other binding studies have shown that telokin binds to the S1-S2 junction of myosin and suppresses myosin folding into the 10 S conformation (39, 40, 47). If the 10 S conformation of isolated myosin molecules represents an unphosphorylated myosin head conformation of in vivo myosin filaments, then telokin may shift the heads to a 6 S state, but this would not account for the ability of telokin to increase Pi release from phosphomyosin. MLC20 phosphorylation has been reported to move the myosin heads away from the filament backbone (48–50). Thus, telokin could indirectly modify the myosin head orientation through decreasing MLC20 phosphorylation.
cGMP/PKG signaling is known to attenuate Ca2+ signaling through inositol trisphosphate receptors and BK channels (51) and ROCK signaling through the inhibitory phosphorylation of RhoA (52). It is also known that Ca2+ sensitivity of permeabilized SM strips is reduced in response to the activation of the cGMP signal (24, 25). This cGMP-mediated Ca2+ desensitization is independent of Ca2+ channels, but RhoA/ROCK signaling may play a role under conditions where the RhoA pathway has been activated. Our finding of telokin-induced re-activation of MLCP demonstrates an additional mode of cGMP-induced Ca2+ desensitization. Furthermore, telokin unlike RhoA is also phosphorylated by cAMP making it a potential target for β-adrenergic or VIP-mediated relaxation. For example, relaxation induced by electrical field stimulation activating nonadrenergic noncholinergic neurons releasing neurotransmitters, such as VIP, in gastric fundus SM is accompanied by phosphorylation of telokin (53). Also, during hypoxia telokin dephosphorylation correlates with an increase in MLC20 phosphorylation and vasoconstriction in small pulmonary vessels that express high concentrations of telokin (54). Interestingly, accumulating evidence suggests that MYPT1 is spontaneously phosphorylated at Thr-696 in various SM tissues, and the level is sometimes barely changed under physiological conditions. Based on enzymology using the purified enzyme, MLCP is still partially active when MYPT1 is phosphorylated at Thr-696 by ROCK (18, 55). Thus, MYPT1 is phosphorylated but active enough to produce relaxation, even in the absence of G-protein activation. Our data show that telokin targets this inactive MLCP fraction in SM cells and can eliminate the negative regulation. This re-activation pathway is independent of Ca2+ and ROCK and other kinases that phosphorylate MYPT1. The direct binding of PKG-1 is also suggested to cause activation of MLCP and is necessary for the regulation of vascular tone (29, 30, 56). Further study is necessary to elucidate the biochemical link between PKG and telokin, both of which bind to MYPT1.
Interestingly, the stoichiometry of binding between MYPT1 and S13D telokin in the SPR assay is likely physiologically relevant. We found that the ratio of SPR signals corresponding to MYPT1 and S13D telokin at the highest immobilized MYPT1 concentration was close to 10 (2365/230 RU). Considering that the amplitude of the SPR signal is strictly proportional to the mass of the protein(s) immobilized on the matrix and that the mass of S13D telokin is ∼10-fold less than that of MYPT1, the apparent stoichiometry of the complex MYPT1·S13D telokin in the SPR experiments is close to 1:1. Telokin content of rabbit ileum is 27 ± 4.6 μm (31). Therefore, in situ binding between phosphorylated MYPT1, released from myosin (12, 19, 20), and free telokin released from phosphorylated myosin (47) is quite probable during the normal contractile cycle in telokin-expressing SMs, despite the modest affinity of telokin for MLCP.
Regarding the physiological role of telokin, we propose that the differential expression of telokin and MYPT1 isoforms in different SMs may contribute to their specialized responses to NO, adrenergic agents, and cyclic nucleotides. The four MYPT1 isoforms arise from alternative mRNA splicing resulting in the presence or absence of a C-terminal LZ domain or a central insert region (57). Direct stimulation of MLCP in arterial SM is reported to occur through interaction between LZ motifs PKG1α and MYPT1 (29) and/or amino acids 888–982 of MYPT1 and fragment PKG1α (30). Activation of LZ-positive MYPT1 is selective for PKG1α. Expression of LZ-positive isoforms differs in different SMs, and high expression correlates with NO/cGMP-induced relaxation (58). Telokin is also differentially expressed, being highest in gastrointestinal SMs and portal vein. Ileum, used in is study, expresses only low levels of the LZ isoform, in agreement with previous studies (20). Thus, there is a reciprocal relationship between telokin and LZ isoform expression resulting in specialized cyclic nucleotide signaling pathways in different SMs. Furthermore, LZ-positive MYPT1 isoform expression does not decrease with down-regulation of telokin protein (Fig. 3). Therefore, the 50% decrease in cGMP-induced relaxation of force in the telokin-null ileum is not due to a decrease in LZ-positive MYPT1 content. Importantly, based on the present data, telokin only functions through inactive MLCP. It is possible that the sensitivity toward cyclic AMP- and cyclic GMP-telokin signaling is possibly determined by the extent of spontaneously phosphorylated MYPT1 at Thr-696, which has been reported in various SM tissues. Quantitative analysis of MYPT1 phosphorylation- and telokin-induced relaxation will uncover the heterogeneity in the response among SM tissues. Unlike MYPT1, telokin is a substrate of both PKA and PKG and therefore subject to regulation by NOS/NO, VIP, and adrenergic stimulation in gastrointestinal SM. Cyclic AMP can also activate PKG. The contributions of their phosphorylated targets may differ. For example, cGMP phosphorylation of Ser-695 accompanying Ca2+ desensitization in ileal SM (27) does not occur in cerebral vessels (20). Also, MYPT1 isoform expression differs in resistance arteries and large vessels and with changes in blood flow (59). Ultimately, the magnitude and kinetics of cAMP/cGMP-induced SM relaxation in different regions of the gut and in different vascular beds will be influenced by the differential expression of telokin and MYPT1 isoforms. In addition, cAMP and cGMP have effects on other targets such as on RhoA, phosphorylation, and activation of MYPT1 at Ser-695 (27), on Ca2+ uptake sites, and on membrane channels (3, 60).
Acknowledgments
We thank Howard Phipps for excellent help in preparation of the manuscript. SPR analysis was performed in the Biomolecular Research Facility, which is supported by the School of Medicine, University of Virginia. We are grateful to Bartosz Zieba for production of recombinant telokins.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK088905 (to A. V. S. and M. E.) and Grant R01 GM086457 from NIGMS (to A. V. S.).

This article contains supplemental Figs. S1–S4, Experimental Procedures, and additional references.
M. Khasnis and M. Eto, manuscript in preparation.
- SM
- smooth muscle
- MLC20
- 20-kDa myosin regulatory light chain
- ROCK
- Rho-associated coiled coil-containing protein kinase
- MLCP
- myosin light chain phosphatase
- LZ
- leucine zipper
- PLA
- proximity ligation assay
- SPR
- surface plasmon resonance
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- ATPγS
- adenosine 5′-O-(thiotriphosphate)
- SMC
- SM cell
- VIP
- vasoactive intestinal peptide
- RU
- resistant unit.
REFERENCES
- 1. Puetz S., Lubomirov L. T., Pfitzer G. (2009) Regulation of smooth muscle contraction by small GTPases. Physiology 24, 342–356 [DOI] [PubMed] [Google Scholar]
- 2. Somlyo A. P., Somlyo A. V. (1994) Signal transduction and regulation in smooth muscle. Nature 372, 231–236 [DOI] [PubMed] [Google Scholar]
- 3. Somlyo A. P., Somlyo A. V. (2003) Ca2+ sensitivity of smooth muscle and nonmuscle myosin II. Modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 [DOI] [PubMed] [Google Scholar]
- 4. Eto M., Ohmori T., Suzuki M., Furuya K., Morita F. (1995) A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J. Biochem. 118, 1104–1107 [DOI] [PubMed] [Google Scholar]
- 5. Ito M., Nakano T., Erdodi F., Hartshorne D. J. (2004) Myosin phosphatase. Structure, regulation, and function. Mol. Cell. Biochem. 259, 197–209 [DOI] [PubMed] [Google Scholar]
- 6. Kawano S., Kuruma A., Hirayama Y., Hiraoka M. (1999) Anion permeability and conduction of adenine nucleotides through a chloride channel in cardiac sarcoplasmic reticulum. J. Biol. Chem. 274, 2085–2092 [DOI] [PubMed] [Google Scholar]
- 7. Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 8. Koyama M., Ito M., Feng J., Seko T., Shiraki K., Takase K., Hartshorne D. J., Nakano T. (2000) Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 475, 197–200 [DOI] [PubMed] [Google Scholar]
- 9. MacDonald J. A., Borman M. A., Murányi A., Somlyo A. V., Hartshorne D. J., Haystead T. A. (2001) Identification of the endogenous smooth muscle myosin phosphatase-associated kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 2419–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murányi A., MacDonald J. A., Deng J. T., Wilson D. P., Haystead T. A., Walsh M. P., Erdodi F., Kiss E., Wu Y., Hartshorne D. J. (2002) Phosphorylation of the myosin phosphatase target subunit by integrin-linked kinase. Biochem. J. 366, 211–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takizawa N., Koga Y., Ikebe M. (2002) Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem. Biophys. Res. Commun. 297, 773–778 [DOI] [PubMed] [Google Scholar]
- 12. Velasco G., Armstrong C., Morrice N., Frame S., Cohen P. (2002) Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr-850 induces its dissociation from myosin. FEBS Lett. 527, 101–104 [DOI] [PubMed] [Google Scholar]
- 13. Mori D., Hori M., Murata T., Ohama T., Kishi H., Kobayashi S., Ozaki H. (2011) Synchronous phosphorylation of CPI-17 and MYPT1 is essential for inducing Ca2+ sensitization in intestinal smooth muscle. Neurogastroenterol. Motil. 23, 1111–1122 [DOI] [PubMed] [Google Scholar]
- 14. Hilgers R. H., Todd J., Jr., Webb R. C. (2007) Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J. Hypertens. 25, 1687–1697 [DOI] [PubMed] [Google Scholar]
- 15. Seko T., Ito M., Kureishi Y., Okamoto R., Moriki N., Onishi K., Isaka N., Hartshorne D. J., Nakano T. (2003) Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 92, 411–418 [DOI] [PubMed] [Google Scholar]
- 16. Guilluy C., Sauzeau V., Rolli-Derkinderen M., Guérin P., Sagan C., Pacaud P., Loirand G. (2005) Inhibition of RhoA/Rho kinase pathway is involved in the beneficial effect of sildenafil on pulmonary hypertension. Br. J. Pharmacol. 146, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kandabashi T., Shimokawa H., Miyata K., Kunihiro I., Kawano Y., Fukata Y., Higo T., Egashira K., Takahashi S., Kaibuchi K., Takeshita A. (2000) Inhibition of myosin phosphatase by up-regulated Rho-kinase plays a key role for coronary artery spasm in a porcine model with interleukin-1β. Circulation 101, 1319–1323 [DOI] [PubMed] [Google Scholar]
- 18. Ichikawa K., Ito M., Hartshorne D. J. (1996) Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. J. Biol. Chem. 271, 4733–4740 [DOI] [PubMed] [Google Scholar]
- 19. Bolz S. S., Vogel L., Sollinger D., Derwand R., de Wit C., Loirand G., Pohl U. (2003) Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation 107, 3081–3087 [DOI] [PubMed] [Google Scholar]
- 20. Neppl R. L., Lubomirov L. T., Momotani K., Pfitzer G., Eto M., Somlyo A. V. (2009) Thromboxane A2-induced bi-directional regulation of cerebral arterial tone. J. Biol. Chem. 284, 6348–6360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shin H. M., Je H. D., Gallant C., Tao T. C., Hartshorne D. J., Ito M., Morgan K. G. (2002) Differential association and localization of myosin phosphatase subunits during agonist-induced signal transduction in smooth muscle. Circ. Res. 90, 546–553 [DOI] [PubMed] [Google Scholar]
- 22. Khromov A., Choudhury N., Stevenson A. S., Somlyo A. V., Eto M. (2009) Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J. Biol. Chem. 284, 21569–21579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ikebe M., Inagaki M., Naka M., Hidaka H. (1988) Correlation of conformation and phosphorylation and dephosphorylation of smooth muscle myosin. J. Biol. Chem. 263, 10698–10704 [PubMed] [Google Scholar]
- 24. Lee M. R., Li L., Kitazawa T. (1997) Cyclic GMP causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J. Biol. Chem. 272, 5063–5068 [DOI] [PubMed] [Google Scholar]
- 25. Wu X., Somlyo A. V., Somlyo A. P. (1996) Cyclic GMP-dependent stimulation reverses G-protein-coupled inhibition of smooth muscle myosin light chain phosphate. Biochem. Biophys. Res. Commun. 220, 658–663 [DOI] [PubMed] [Google Scholar]
- 26. Nakamura K., Koga Y., Sakai H., Homma K., Ikebe M. (2007) cGMP-dependent relaxation of smooth muscle is coupled with the change in the phosphorylation of myosin phosphatase. Circ. Res. 101, 712–722 [DOI] [PubMed] [Google Scholar]
- 27. Wooldridge A. A., MacDonald J. A., Erdodi F., Ma C., Borman M. A., Hartshorne D. J., Haystead T. A. (2004) Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of serine 695 in response to cyclic nucleotides. J. Biol. Chem. 279, 34496–34504 [DOI] [PubMed] [Google Scholar]
- 28. Vetterkind S., Lee E., Sundberg E., Poythress R. H., Tao T. C., Preuss U., Morgan K. G. (2010) Par-4. A new activator of myosin phosphatase. Mol. Biol. Cell 21, 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., Mendelsohn M. E. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Iα. Science 286, 1583–1587 [DOI] [PubMed] [Google Scholar]
- 30. Given A. M., Ogut O., Brozovich F. V. (2007) MYPT1 mutants demonstrate the importance of aa 888–928 for the interaction with PKGIα. Am. J. Physiol. Cell Physiol. 292, C432–C439 [DOI] [PubMed] [Google Scholar]
- 31. Choudhury N., Khromov A. S., Somlyo A. P., Somlyo A. V. (2004) Telokin mediates Ca2+ desensitization through activation of myosin phosphatase in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 25, 657–665 [DOI] [PubMed] [Google Scholar]
- 32. Khromov A. S., Wang H., Choudhury N., McDuffie M., Herring B. P., Nakamoto R., Owens G. K., Somlyo A. P., Somlyo A. V. (2006) Smooth muscle of telokin-deficient mice exhibits increased sensitivity to Ca2+ and decreased cGMP-induced relaxation. Proc. Natl. Acad. Sci. U.S.A. 103, 2440–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu X., Haystead T. A., Nakamoto R. K., Somlyo A. V., Somlyo A. P. (1998) Acceleration of myosin light chain dephosphorylation and relaxation of smooth muscle by telokin. Synergism with cyclic nucleotide-activated kinase. J. Biol. Chem. 273, 11362–11369 [DOI] [PubMed] [Google Scholar]
- 34. Gallagher P. J., Herring B. P. (1991) The carboxyl terminus of the smooth muscle myosin light chain kinase is expressed as an independent protein, telokin. J. Biol. Chem. 266, 23945–23952 [PMC free article] [PubMed] [Google Scholar]
- 35. Herring B. P., Smith A. F. (1996) Telokin expression is mediated by a smooth muscle cell-specific promoter. Am. J. Physiol. 270, C1656–C1665 [DOI] [PubMed] [Google Scholar]
- 36. Ito M., Dabrowska R., Guerriero V., Jr., Hartshorne D. J. (1989) Identification in turkey gizzard of an acidic protein related to the C-terminal portion of smooth muscle myosin light chain kinase. J. Biol. Chem. 264, 13971–13974 [PubMed] [Google Scholar]
- 37. MacDonald J. A., Walker L. A., Nakamoto R. K., Gorenne I., Somlyo A. V., Somlyo A. P., Haystead T. A. (2000) Phosphorylation of telokin by cyclic nucleotide kinases and the identification of in vivo phosphorylation sites in smooth muscle. FEBS Lett. 479, 83–88 [DOI] [PubMed] [Google Scholar]
- 38. Walker L. A., MacDonald J. A., Liu X., Nakamoto R. K., Haystead T. A., Somlyo A. V., Somlyo A. P. (2001) Site-specific phosphorylation and point mutations of telokin modulate its Ca2+-desensitizing effect in smooth muscle. J. Biol. Chem. 276, 24519–24524 [DOI] [PubMed] [Google Scholar]
- 39. Silver D. L., Vorotnikov A. V., Watterson D. M., Shirinsky V. P., Sellers J. R. (1997) Sites of interaction between kinase-related protein and smooth muscle myosin. J. Biol. Chem. 272, 25353–25359 [DOI] [PubMed] [Google Scholar]
- 40. Masato T., Numata T., Katoh T., Morita F., Yazawa M. (1997) Cross-linking of telokin to chicken gizzard smooth muscle myosin. J. Biochem. 121, 225–230 [PubMed] [Google Scholar]
- 41. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 42. Somlyo A. V., Wang H., Choudhury N., Khromov A. S., Majesky M., Owens G. K., Somlyo A. P. (2004) Myosin light chain kinase knockout. J. Muscle Res. Cell Motil. 25, 241–242 [DOI] [PubMed] [Google Scholar]
- 43. Johnson D., Cohen P., Chen M. X., Chen Y. H., Cohen P. T. (1997) Identification of the regions on the M110 subunit of protein phosphatase 1M that interact with the M21 subunit and with myosin. Eur. J. Biochem. 244, 931–939 [DOI] [PubMed] [Google Scholar]
- 44. Totsukawa G., Yamakita Y., Yamashiro S., Hosoya H., Hartshorne D. J., Matsumura F. (1999) Activation of myosin phosphatase targeting subunit by mitosis-specific phosphorylation. J. Cell Biol. 144, 735–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karandikar M., Cobb M. H. (1999) Scaffolding and protein interactions in MAP kinase modules. Cell Calcium 26, 219–226 [DOI] [PubMed] [Google Scholar]
- 46. Whitmarsh A. J., Davis R. J. (1998) Structural organization of MAP kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem. Sci. 23, 481–485 [DOI] [PubMed] [Google Scholar]
- 47. Shirinsky V. P., Vorotnikov A. V., Birukov K. G., Nanaev A. K., Collinge M., Lukas T. J., Sellers J. R., Watterson D. M. (1993) A kinase-related protein stabilizes unphosphorylated smooth muscle myosin minifilaments in the presence of ATP. J. Biol. Chem. 268, 16578–16583 [PubMed] [Google Scholar]
- 48. Levine R. J., Kensler R. W., Yang Z., Stull J. T., Sweeney H. L. (1996) Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys. J. 71, 898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wendt T., Taylor D., Trybus K. M., Taylor K. (2001) Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. U.S.A. 98, 4361–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu X., Clack B. A., Zhi G., Stull J. T., Cremo C. R. (1999) Phosphorylation-dependent structural changes in the regulatory light chain domain of smooth muscle heavy meromyosin. J. Biol. Chem. 274, 20328–20335 [DOI] [PubMed] [Google Scholar]
- 51. Hofmann F. (2005) The biology of cyclic GMP-dependent protein kinases. J. Biol. Chem. 280, 1–4 [DOI] [PubMed] [Google Scholar]
- 52. Sauzeau V., Le Jeune H., Cario-Toumaniantz C., Smolenski A., Lohmann S. M., Bertoglio J., Chardin P., Pacaud P., Loirand G. (2000) Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 275, 21722–21729 [DOI] [PubMed] [Google Scholar]
- 53. Puetz S., Lubomirov L. T., Neulen A., Chang Z., Solzin J., Aumailley M., Somlyo A., Pfitzer G. (2010) Signaling events involved in cyclic nucleotide-mediated relaxation of murine gastric fundus (GF). Acta Physiol. 198, O-SUN-6-5 [Google Scholar]
- 54. Madden J. A., Dantuma M. W., Sorokina E. A., Weihrauch D., Kleinman J. G. (2008) Telokin expression and the effect of hypoxia on its phosphorylation status in smooth muscle cells from small and large pulmonary arteries. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L1166–L1173 [DOI] [PubMed] [Google Scholar]
- 55. Feng J., Ito M., Kureishi Y., Ichikawa K., Amano M., Isaka N., Okawa K., Iwamatsu A., Kaibuchi K., Hartshorne D. J., Nakano T. (1999) Rho-associated kinase of chicken gizzard smooth muscle. J. Biol. Chem. 274, 3744–3752 [DOI] [PubMed] [Google Scholar]
- 56. Michael S. K., Surks H. K., Wang Y., Zhu Y., Blanton R., Jamnongjit M., Aronovitz M., Baur W., Ohtani K., Wilkerson M. K., Bonev A. D., Nelson M. T., Karas R. H., Mendelsohn M. E. (2008) High blood pressure arising from a defect in vascular function. Proc. Natl. Acad. Sci. U.S.A. 105, 6702–6707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hartshorne D. J., Ito M., Erdödi F. (1998) Myosin light chain phosphatase. Subunit composition, interactions, and regulation. J. Muscle Res. Cell Motil. 19, 325–341 [DOI] [PubMed] [Google Scholar]
- 58. Khatri J. J., Joyce K. M., Brozovich F. V., Fisher S. A. (2001) Role of myosin phosphatase isoforms in cGMP-mediated smooth muscle relaxation. J. Biol. Chem. 276, 37250–37257 [DOI] [PubMed] [Google Scholar]
- 59. Zhang H., Fisher S. A. (2007) Conditioning effect of blood flow on resistance artery smooth muscle myosin phosphatase. Circ. Res. 100, 730–737 [DOI] [PubMed] [Google Scholar]
- 60. Murthy K. S. (2006) Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 68, 345–374 [DOI] [PubMed] [Google Scholar]