

Background: C5-epimerase converts a glucuronic acid to an iduronic acid residue in the heparan sulfate biosynthetic pathway.
Results: C5-epimerase displays both “reversible” and “irreversible” catalytic modes.
Conclusion: C5-epimerase recognizes the saccharide sequence of the substrate to position the iduronic acid.
Significance: The biphasic catalytic mode of C5-epimerase reveals a unique control mechanism in the biosynthesis of heparan sulfate.
Keywords: Biosynthesis, Carbohydrate, Heparan Sulfate, Heparin, Oligosaccharide
Abstract
Heparan sulfate (HS), a highly sulfated polysaccharide, is biosynthesized through a pathway involving several enzymes. C5-epimerase (C5-epi) is a key enzyme in this pathway. C5-epi is known for being a two-way catalytic enzyme, displaying a “reversible” catalytic mode by converting a glucuronic acid to an iduronic acid residue, and vice versa. Here, we discovered that C5-epi can also serve as a one-way catalyst to convert a glucuronic acid to an iduronic acid residue, displaying an “irreversible” catalytic mode. Our data indicated that the reversible or irreversible catalytic mode strictly depends on the saccharide substrate structures. The biphasic mode of C5-epi offers a novel mechanism to regulate the biosynthesis of HS with the desired biological functions.
Introduction
Heparan sulfate (HS)4 is an essential glycan that is found on the mammalian cell surface and in the extracellular matrix. HS participates in a wide range of physiological and pathophysiological functions, including embryonic development, inflammatory responses, blood coagulation, and viral/bacterial infections (1). HS consists of a disaccharide repeating unit of glucuronic acid (GlcA) or iduronic acid (IdoA) and glucosamine, each of which is capable of carrying sulfo groups. The biosynthetic pathway of HS includes HS polymerases, sulfotransferases, and C5-epi. Collective actions of these enzymes result in matured HS with biological functions (supplemental Fig. S1). Uniquely distributed sulfo groups and IdoA residues play critical roles in determining the functions of HS (2); however, it remains unclear how to regulate the biosynthesis of these specific sulfated saccharide sequences in vivo.
Understanding the biosynthetic mechanism also aids in developing a chemoenzymatic method to synthesize HS-based drugs. Heparin, a commonly used anticoagulant drug, is a special form of HS with higher levels of sulfation and IdoA. Heparin is currently isolated from animal tissues through a long and poorly regulated supply chain. Worldwide distribution of contaminated heparin in 2007 has raised the concerns over the safety of animal-sourced heparins (3). A cost-effective method to prepare synthetic heparin is desirable. Using HS biosynthetic enzymes, we developed a chemoenzymatic approach to synthesize structurally defined HS and heparin oligosaccharides in high efficiency, which significantly expands the synthetic capability by a purely chemical approach (4, 5).
C5-epi converts a GlcA unit to an IdoA by forming a putative carbanion intermediate at the C5-position of a GlcA unit (see Fig. 1A) (6). Only a single isoform of C5-epi is in human genome, and C5-epi knock-out mice are neonatal lethal, suggesting its essential physiological roles in vivo (7). C5-epi reportedly is a two-way catalytic enzyme, performing both the forward and reverse epimerization. Namely, the IdoA unit reverts back to a GlcA unit in the presence of C5-epi (8). The nature of the reaction renders unique challenges to study the mode of action of C5-epi. To date, three approaches have been reported to measure the activity of C5-epi (9–11). Because all these methods utilized structurally heterogeneous polysaccharide substrates and the products were identified by a disaccharide analysis, none of these approaches is capable of locating the GlcA residues participated in the epimerization beyond a disaccharide domain. Therefore, the understanding for the mechanism of action of C5-epi was limited, especially the effects of neighboring saccharide structures on the action of C5-epi.
FIGURE 1.

The reaction catalyzed by C5-epi and the schematic presentation for determining the activity of C5-epi. A shows the reaction catalyzed by C5-epi. A trisaccharide segment of polysaccharide is shown. C5-epi removes the proton from C5 of the GlcA residue to form a putative carbanion intermediate. An H2O molecule then reacts with the carbanion to form an IdoA residue. Conversely, C5-epi can catalyze the reverse reaction, namely to convert an IdoA residue to a GlcA residue. B shows the schematic presentation for determining the activity and reversibility of C5-epi. The assay was developed based on the reaction mechanism of C5-epi. Here, an oligosaccharide was used as a substrate. After incubation with C5-epi in D2O, the obtained epi-oligosaccharide had one unit increase in Mr (MW), indicating that a single GlcA residue was converted to an IdoA residue. Tandem MS was used to locate the site of epimerization because the epimerized residue should carry a deuterium. The reversibility of C5-epi was determined by incubating the epi-oligosaccharide with H2O. If the Mr of the product reduces by one unit, the reaction was proved to be “reversible.” The Mr values of the oligosaccharides were determined by ESI-MS. The keys for shorthand representations of carbohydrate residues are shown in the box.
In this article, we report an advanced method to characterize C5-epi using a series of structurally defined oligosaccharide substrates coupled with tandem mass spectrometry technique. This method permits us to locate the number and position of epimerization at the oligosaccharide levels with molecular precision. On contrary to the convention view, we uncovered that C5-epi also serves as a one-way catalyst to convert a GlcA to an IdoA residue irreversibly, displaying an irreversible catalytic mode. C5-epi recognizes the N-sulfation pattern of the substrates to display reversible or irreversible catalytic mode of C5-epi. The biphasic mode of C5-epi offers a novel mechanism to regulate the biosynthesis of HS with the desired biological functions.
EXPERIMENTAL PROCEDURES
Expression and Purification of C5-epi
Recombinant C5-epi was expressed and purified as described previously (10). Briefly, a fusion protein of maltose-binding protein and the human C5-epi catalytic domain (Glu53–Asn617) was constructed in pMal c2x vector (New England BioLabs). The expression was carried out in Origami-B DE3 cells (Novagen), which contained pGro7 (Takara, Japan) plasmid expressing chaperonin proteins GroEL and GroES of Escherichia coli. The purification of C5-epi was completed using an amylose-agarose (New England Biolabs) column following the protocol provided by the manufacturer. To carry out the epimerization reaction in D2O buffer, D2O-exchanged C5-epi was prepared. To this end, the harvested bacteria pellet were suspended in the buffer that was prepared in D2O (>99% D, Spectra Stable Isotopes). In addition, the elution buffers for amylose-agarose column purification were also prepared in D2O.
The activity of purified C5-epi (including D2O-exchanged C5-epi) was measured by coupling the reaction of C5-epi and 2-O-sulfotransferase (2-OST) followed by a disaccharide analysis (12). In this assay, C5-epi (1.5 μg) was incubated with 2 μg of N-sulfo heparosan in the buffer containing 50 mm MES (pH 7.0), Triton X-100, and 1 mm CaCl2 at 37 °C for 30 min. To the reaction mixture, 0.6 μg of 2-OST and 3′-phosphoadenosine 5′-phospho[35S]sulfate ([35S]PAPS) (5–7 × 105 cpm) were added. The reaction was incubated at 37 °C for two additional hours. The 35S-labeled polysaccharide was purified by a DEAE column and was then subjected to nitrous acid degradation (at pH 1.5) to yield the disaccharides. The identities of the resultant disaccharides were determined by reverse-phase ion pairing HPLC using a C18-column as described previously (13). As a fully active C5-epi, >90% of resultant disaccharides from the degraded polysaccharide have a structure of IdoA2S-AnMan, where AnMan represents 2,5-anhydromannitol.
Preparation of Oligosaccharide Substrates
A total of eight oligosaccharides, differing in the size of the oligosaccharides and the distribution of N-sulfo groups, were prepared in this study. The preparation of the substrates followed essentially the same procedures described in our previous work (14–16). The synthesis initiated from a disaccharide of GlcA-AnMan, which was prepared from nitrous acid, degraded heparosan. The elongation from disaccharide to the desired size of oligosaccharide substrates was completed by KfiA (N-acetyl glucosaminyl transferase of E. coli K5 strain) and PmHS2 (heparosan synthase 2 from Pasteurella multocida). In a typical elongation reaction from the disaccharide to tetrasaccharide, 6 mg of GlcA-AnMan was incubated with 18 μmol of UDP-GlcNTFA (UDP-N-trifluoroacetyl glucosamine) and 2 mg of KfiA in 40 ml buffer containing 25 mm Tris-HCl, pH 7.2, 10 mm MgCl2 at room temperature overnight. Upon the complete consumption of UDP-GlcNTFA, 2 mg of PmHS2, and 27 μmol of UDP-GlcUA were added into the reaction mixture and allowed to incubate overnight at room temperature. The product was purified by a BioGel P-2 column (0.75 × 200 cm), which was equilibrated with 0.1 m ammonium bicarbonate at a flow rate of 4 ml/h. The product fraction was located by electrospray ionization mass spectrometry (ESI-MS) analysis. For the additional elongation reaction to the hexa-, octa-, and decasaccharides, the conditions were identical essentially to the above, whereas the reaction volumes were adjusted to the smaller sizes depending on the amount of substrates. The conversion of GlcNTFA to GlcNS was performed under an alkaline condition to remove the trifluoroacetyl group followed by N-sulfation using N-sulfotransferase and PAPS. We completed the synthesis of milligrams of each oligosaccharide substrates. The structures of the oligosaccharides were confirmed by ESI-MS analysis.
The preparation of UDP-GlcNTFA was started from glucosamine (Sigma), which was first converted to GlcNTFA by reacting with S-ethyl trifluorothioacetate (Sigma-Aldrich) followed the protocol described previously (16). The resultant GlcNTFA was converted to GlcNTFA-1-phosphate using N-acetylhexosamine 1-kinase (17). The plasmid expressing N-acetylhexosamine 1-kinase was a generous gift from Professor Peng George Wang (Georgia State University, Atlanta, GA), and the expression of the enzyme was carried out in E. coli as reported (17). The UDP-GlcNTFA synthesis was completed by transforming GlcNTFA-1-phosphate using glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridyltransferase as described (16). The protocols for the expression of other HS biosynthetic enzymes and the preparation of PAPS were described elsewhere (16, 18). Additional methods are presented under supplemental “Methods.”
RESULTS AND DISCUSSION
For the current work, eight structurally defined substrates were synthesized using a chemoenzymatic approach (Table 1) (4). An MS-based method to monitor the reaction was devised, which allowed us to pinpoint the epimerization site (EPS) and to examine the reversibility of C5-epi. A substrate is incubated with C5-epi and D2O, leading to an epimerized product with deuterium incorporated. One unit of increase in molecular weight (Mr) for epi-oligosaccharide suggests that one GlcA residue is converted. Using a tandem MS technique, we located the residue that carries the deuterium in the epi-oligosaccharide, thus identifying the EPS site. The reversibility of the epimerization is studied by incubating the deuterated epi-oligosaccharide with C5-epi in H2O followed by MS analysis (Fig. 1B).
TABLE 1.
Reactivity and reaction modes of C5-epi toward the substrates with different structures
| Name | Structure of substrate and site(s) of epimerizationa | Calculated Mr | Measured Mr | Mr after C5-epi treatment in D2O | Reaction mode |
|---|---|---|---|---|---|
| Octa-1b | GlcA-GlcNS-GlcA-GlcNS-GlcA-GlcNAc-GlcA-AnMan | 1554.3 | 1554.5 ± 0.4 | 1556.4 ± 0.3 | Reversible |
| Octa-2c | GlcA-GlcNS-GlcA-GlcNS-GlcA-GlcNS-GlcA-AnMan | 1592.3 | 1592.4 ± 0.4 | 1594.3 ± 0.5 | Reversible |
| Octa-3d | GlcA-GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-AnMan | 1554.3 | 1554.5 ± 0.4 | 1555.4 ± 0.4 | Irreversible |
| Octa-4e | GlcA-GlcNAc-GlcA-GlcNS-GlcA-GlcNAc-GlcA-AnMan | 1516.3 | 1516.6 ± 0.4 | 1517.6 ± 0.5 | Irreversible |
| Octa-5 | GlcA-GlcNAc-GlcA-GlcNAc-GlcA-GlcNAc-GlcA-AnMan | 1478.2 | 1478.5 ± 0.2 | 1478.6 ± 0.4 | No reaction |
| Octa-6f | GlcA-GlcNH2-GlcA-GlcNS-GlcA-GlcNS-GlcA-AnMan | 1512.3 | 1512.2 ± 0.4 | 1513.1 ± 0.3 | Reversible |
| Hexa-7g | GlcA-GlcNS-GlcA-GlcNS-GlcA-AnMan | 1175.0 | 1175.1 ± 0.1 | 1175.9 ± 0.1 | Reversible |
| Deca-8h | GlcA-GlcNAc-GlcA-GlcNS-GlcA(irreversible site)-GlcNS-GlcA (reversible site)-GlcNS-GlcA-AnMan | 1971.6 | 1971.2 ± 0.4 | 1973.2 ± 0.3 | Reversible and irreversible |
a The EPS sites are in boldface type.
b The data from the tandem MS analysis demonstrate that Octa-2 contains two EPS sites are shown in supplemental Fig S2.
c The data from the tandem MS analysis, which demonstrate that Octa-3 contains one irreversible EPS, are shown in Fig 3.
d The data from the tandem MS analysis, which demonstrate that Octa-4 contains one irreversible EPS, are shown in supplemental Fig S3.
e The data from the tandem MS analysis, which demonstrate that Octa-6 contains one reversible EPS, are shown in supplemental Fig S4.
f Only partial epimerization products were observed. The reaction was proved to be reversible.
g The data from the tandem MS analysis, which demonstrate that Deca-8 contains one irreversible EPS and one reversible EPS, are shown in supplemental Fig S4.
h The data from the tandem MS analysis, which demonstrate that Octa-1 contains two EPS sites, are shown in Fig 2.
The reversibility of C5-epi was confirmed using Octa-1 as a substrate, which has a structure of GlcA-GlcNS-GlcA-GlcNS-GlcA-GlcNAc-GlcA-AnMan (Fig. 2). The Mr of epi-Octa-1 was increased to 1556.4 ± 0.4 from 1554.5 ± 0.3 (Table 1 and Fig. 2) when it was treated with C5-epi in the presence of D2O. Furthermore, the incubation of epi-Octa-1 with C5-epi in H2O resulted in the decrease of its Mr by 1.9 units (from 1556.4 ± 0.4 to 1554.5 ± 0.5). Our data suggest that two GlcA units in Octa-1 were susceptible to C5-epi modification, and both EPS sites were reversible. These results were consistent with the previous report by Lindahl and co-workers (8).
FIGURE 2.

MS and MS/MS analysis of Octa-1 treated with D2O buffer with and without C5-epi. Octa-1 was incubated with D2O buffer with and without C5-epi. The products were then subjected to MS and MS/MS analysis. A showed the ESI-MS spectrum of d-labeled epi-Octa-1, which was prepared by incubating Octa-1 with D2O-exhanged C5-epi in D2O buffer. B showed the ESI-MS spectrum of Octa-1 that was treated D2O buffer without C5-epi. C showed the MS/MS spectrum of d-labeled epi-Octa-1. Precursor ion selection was at [M − 3H]3−, m/z 517.90. D showed the MS/MS spectrum of Octa-1 that was treated with D2O buffer only. Precursor ion selection was at [M − 3H]3−, m/z 517.30. The fragmentation pattern of MS/MS analysis is depicted on the top of C and D. Both the fragment B5 (m/z value of 505.39) and C6 (m/z value of 615.88) of d-labeled epi-Octa-1 are ∼2 units higher than those of Octa-1 (the m/z value of 504.37 and 614.86), suggesting that the d-labeled residues are located at residue C and E in d-labeled epi-Octa-1. The product ions in the MS/MS data were labeled according to Domon-Costello nomenclature (25).
Interestingly, C5-epi displayed an irreversible reaction mode when a different substrate (Octa-3) was used, which has the structure of GlcA-GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-AnMan. Incubation of Octa-3 with C5-epi in D2O resulted in d-labeled epi-Octa-3. d-labeled epi-Octa-3 exhibited the Mr of 1555.4 ± 0.4 (Fig. 3A) with an increase of 0.9 units (Fig. 3B), suggesting that a single GlcA residue was converted to IdoA. The Mr of epi-Octa-3 remained unchanged after incubation with C5-epi, suggesting that C5-epi was unable to reverse the reaction. To further confirm the structure of d-labeled epi-Octa-3, a series of experiments were conducted. Tandem MS analysis was employed to prove that the d-labeled residue is located at residue E. For example, the fragment Y5 of epi-Octa-3 (m/z value of 499.23 for the doubly charged ion) is ∼1.3 units higher than that of Octa-3 (m/z value of 498.57) (Fig. 3, C and D). NMR analyses of epi-Octa-3 confirmed the presence of IdoA at residue E (supplemental Table S1). The presence of an IdoA in epi-Octa-3 was also demonstrated after modifying epi-Octa-3 with 2-O-sulfotransferase followed by a disaccharide analysis (supplemental data). High resolution ESI-MS analysis of d-labeled epi-Octa-3 showed a signal at a mass/charge ratio of 517.1121, consistent with [M − 3H]3− of the anticipated structure (calculated mass/charge ratio, 517.1040).
FIGURE 3.

MS and MS/MS analysis of Octa-3 with and without the treatment of C5-epi. Octa-3 was incubated in the D2O buffer with and without C5-epi. After incubation, the products were subjected to MS and MS/MS analysis. A shows the ESI-MS spectrum of Octa-3 that was treated with D2O buffer and D2O-exchanged C5-epi. B shows the ESI-MS spectrum of Octa-3 that was treated in D2O buffer without C5-epi. C shows the MS/MS spectrum of d-labeled epi-Octa-3. Precursor ion selection was at [M − 3H]3−, m/z 517.70. D shows the MS/MS spectrum of Octa-3. Precursor ion selection was at [M − 3H]3−, m/z 517.40. The fragmentation pattern of MS/MS analysis is depicted on the top of C and D. Both the fragment Y5 (m/z value of 499.23) and C6 (m/z value of 575.33) of d-labeled epi-Octa-3 are ∼1.3 units higher than those of Octa-3 (m/z value of 498.57 and 574.67), suggesting that the d-labeled residue is located at residue E in d-labeled epi-Octa-3. The product ions in the MS/MS data were labeled according to Domon-Costello nomenclature (25).
Subjecting additional oligosaccharides to the analysis revealed a relationship between the reaction mode of C5-epi and the structures of substrates (Table 1). C5-epi appears to recognize the flanking saccharide sequence at the nonreducing end of the EPS residue. Oligosaccharides containing a nonreducing end GlcNS residue immediately adjacent to the EPS residue are reactive to C5-epi. In contrast, when the GlcNS is replaced with GlcNAc (e.g. Octa-5), the oligosaccharide is no longer a substrate of C5-epi, proving the critical role of GlcNS at the nonreducing end of the EPS (8). Whether C5-epi exhibits an irreversible or a reversible mode depends on the residue at the mode of reaction recognition site (MRRS) that is three residues away from the EPS site toward the nonreducing end (Fig. 4A). If a GlcNAc is present at the MRRS site, C5-epi displays an irreversible reaction mode (as for Octa-3 and Octa-4). If a GlcNS or a GlcNH2 residue is present at the MRRS site (e.g. Ocat-1, Octa-2, and Octa-6) or the MRRS site is unoccupied (i.e. Hexa-7 or the second EPS site (EPS2) for Octa-1 and -2), C5-epi displays a reversible reaction mode (Fig. 4A).
FIGURE 4.

Reaction mode of C5-epi in response to different substrates. A summarizes possible reactions of C5-epi under the single reaction mode scenario. The designated EPS is at the residue 0. The residue −1 must be a GlcNS residue to serve as an EPS site. The structure of the saccharide residue at the MRRS determines the mode of reaction of C5-epi. If the MRRS site (residue −3) is a GlcNS or GlcNH2 residue or unoccupied, C5-epi displays a reversible reaction mode (like reaction a, c, and b). If a GlcNAc residue is at the MRRS site, C5-epi displays an irreversible reaction mod (like reaction d). If residue −1 is a GlcNAc residue, residue 0 is not an EPS site. B shows mixed reaction modes displayed by C5-epi when Deca-8 was used as a substrate. Two EPS sites were present in Deca-8. EPS1 is a reversible site, whereas EPS2 is an irreversible site. The gray shaded box indicates the saccharide residues recognized by C5-epi. The yellow shaded box indicates the saccharide residues recognized by C5-epi during the mixed reaction modes. The keys for shorthand representations of carbohydrate residues are shown in the box.
To further strengthen our conclusion, we synthesized a decasaccharide substrate (Deca-8), permitting C5-epi to display mixed reaction modes (Fig. 4B). Here, two EPS sites were constructed at the residue 0 (EPS1) and residue −2 (EPS2), respectively. Two MRRS sites were also introduced: a GlcNS and a GlcNAc residue were placed at residue −3 and residue −5, which should make a reversible EPS1 site and an irreversible EPS2 site, respectively. Indeed, our data confirmed that EPS1 is a reversible site, whereas EPS2 is irreversible (supplemental Fig. S5).
Lastly, we tested the role of irreversible reaction mode of C5-epi in contributing to the biosynthesis of HS that binds to antithrombin (AT). HS or heparin forms a 1:1 complex with AT, which deactivates the activities of factor Xa and IIa in the blood coagulation cascade, to exhibit the anticoagulant activity. Anticoagulant HS and heparin isolated from natural sources have an AT-binding pentasaccharide with a structure of -GlcNAc6S-GlcA-GlcNS3S±6S-IdoA2S-GlcNS6S- (where GlcNS3S+6S represents N-sulfo glucosamine 3-O-sulfate with or without 6-O-sulfate) (19, 20). The IdoA2S residue in the pentasaccharide domain is known to be essential for high AT-binding affinity (21).
The uncovered irreversible reaction mode of C5-epi provides insight for the natural selection for a GlcNAc6S (not a GlcNS6S) residue as part of the AT-binding site for its role in positioning the IdoA residue. The GlcNAc residue is expected to serve as a MRRS site to direct C5-epi to synthesize an irreversible IdoA residue in the AT-binding pentasaccharide site, thus increasing the efficiency for the biosynthesis of AT-binding HS. To prove this assertion, the synthesis of AT-binding decasaccharides was completed using three decasaccharide substrates, including Deca-9, Deca-10, and Deca-11(supplemental Fig. S6). Deca-9 had no IdoA residue; Deca-10 contained two reversible IdoA residues; and Deca-11 had one irreversible IdoA residue at EPS2 site and one reversible IdoA residue at EPS1 site. The irreversible IdoA residue at EPS2 site in Deca-11 was achieved by exposing Deca-10 to C5-epi modification. These decasaccharides were modified by O-sulfotransferases to produce O-sulfated decasaccharides and were then fractionated by AT affinity columns to determine the amount of AT-binding site in the products. The results revealed that 40% O-sulfated Deca-11 bound to the AT affinity column, whereas the amount of the AT-binding portion for Deca-10 was determined to be 16%, a significant decrease in comparing to that of Deca-11 (Fig. 5). As expected, only 4% of O-sulfated Deca-8 bound to AT affinity column because it lacked an IdoA residue. Taken together, our data suggest that an irreversible IdoA residue at EPS2 site enhances the biosynthesis of AT-binding site.
FIGURE 5.
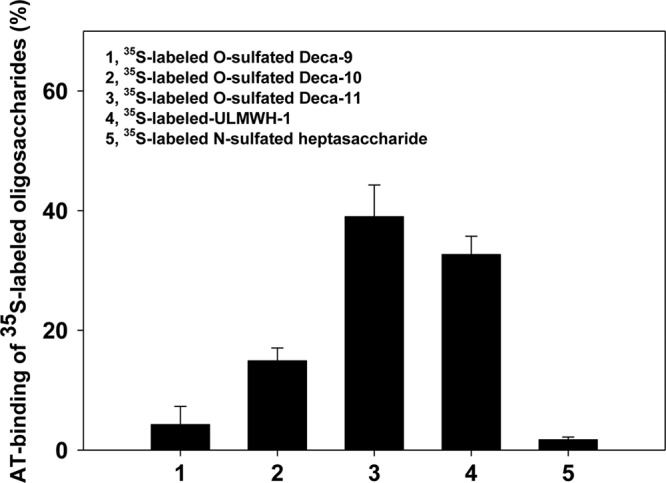
The binding of 35S-labeled decasaccharides to AT. 35S-labeled O-sulfated Deca-9, O-sulfated Deca-10, and O-sulfated Deca-11 (∼1 × 104 cpm of each sample) were incubated with 5 μg of human AT in 50 μl of binding buffer containing 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Mn2+, 1 mm Mg2+, 1 mm Ca2+, 10 μm dextran sulfate, 0.0004% Triton X-100, and 0.02% sodium azide for 30 min at 22 °C, respectively. Concanavalin A-Sepharose gel (Sigma, 200 μl of 1:1 slurry) was then added, and the reaction was shaken at 22 °C for 2 h. The beads were then washed by 3 × 1 ml binding buffer, and the amount of 35S-labeled radioactivity was determined by scintillation counting. The 35S-labeled ultra-low molecular weight heparin1 (ULMWH-1) has the structure of GlcNAc6S-GlcA-[3-O-35S]GlcNS6S3S-IdoA2S-GlcNS6S-GlcA-AnMan. The 35S-labeled N-[35S]-sulfated heptasaccharide has the structure of GlcNAc-GlcA-GlcNS-GlcA-GlcNS-GlcA-AnMan. ULMWH-1 and the N-sulfated heptasaccharide were used as the positive and negative control in the AT-binding experiment, respectively. The synthesis and characterizations of both saccharides were described elsewhere (14). The data presented represent the average of these six independent determinations ± S.D.
Conclusions
Although the biosynthesis of HS is not a template-driven process, our results support the notion that HS biosynthetic pathway adopts an exquisite way to fine tune the extents of modifications through substrate control (22). Those enzymes involved in modifying the highly sulfated substrates (supplemental Fig. S1), i.e. 3-O-sulfotransferase, are able to distinguish the saccharide sequences with complicated sulfation patterns (23). In contrast, the mechanism used by those enzymes that modify the nonsulfated or low sulfated substrates, including N-deacetylase/N-sulfotransferase and C5-epi, is perceived to be subtle because the polysaccharide substrates have relatively simple repetitive structures. In the initial N-sulfation step, N-deacetylase/N-sulfotransferase introduces the GlcNS residues consecutively, rather than randomly, along the polysaccharide substrate (24). Esko and Selleck (20) hypothesized that the N-sulfation step introduces the codes to direct the extent in the subsequent modification steps. Indeed, our findings demonstrate that C5-epi recognizes the distribution of GlcNS residues in the substrate and displays distinctive catalytic modes, namely translating the N-sulfation code into the positions of GlcA/IdoA residues. Our data clearly show the impact of the irreversible mode of C5-epi on the efficiency for the biosynthesis of anticoagulant HS. Further investigation will unfold the full implication of the biphasic mode possessed by C5-epi in controlling the biosynthesis of HS.
Acknowledgments
We thank Drs. Ulf Lindahl and Ji-ping Li (Uppsala University, Uppsala, Sweden) for insightful discussion during the preparation of this article. We also thank Elizabeth Pempe (University of North Carolina) for critically reading the manuscript and Kermit Johnson and Dr. Daniel Holmes (both from Michigan State University) for expert help with NMR data acquisition.
This work was supported in part by National Institutes of Health Grants AI50050, HL094463, and GM072667 and National Science Foundation Grant CHE-1111550.

This article contains supplemental “Methods,” Table S1, Figs. S1–S6, and additional references.
- HS
- heparan sulfate
- C5-epi
- C5-epimerase
- AnMan
- 2,5-anhydromannitol
- GlcA
- glucuronic acid
- IdoA
- iduronic acid
- ESI
- electrospray ionization
- EPS
- epimerization site
- MRRS
- mode of reaction recognition site
- AT
- antithrombin.
REFERENCES
- 1. Bishop J. R., Schuksz M., Esko J. D. (2007) Heparan sulfate proteoglycans fine-tune mammalian physiology. Nature 446, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 2. Gama C. I., Tully S. E., Sotogaku N., Clark P. M., Rawat M., Vaidehi N., Goddard W. A., 3rd, Nishi A., Hsieh-Wilson L. C. (2006) Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat. Chem. Biol. 2, 467–473 [DOI] [PubMed] [Google Scholar]
- 3. Liu H., Zhang Z., Linhardt R. J. (2009) Lessons learned from the contamination of heparin. Nat. Prod. Rep. 26, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu Y., Masuko S., Takieddin M., Xu H., Liu R., Jing J., Mousa S. A., Linhardt R. J., Liu J. (2011) Chemoenzymatic synthesis of homogeneous ultralow molecular weight heparins. Science 334, 498–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Z., Xu Y., Yang B., Tiruchinapally G., Sun B., Liu R., Dulaney S., Liu J., Huang X. (2010) Preactivation-based, one-pot combinatorial synthesis of heparin-like hexasaccharides for the analysis of heparin-protein interactions. Chem. Eur. J. 16, 8365–8375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hagner-McWhirter A., Hannesson H. H., Campbell P., Westley J., Rodén L., Lindahl U., Li J. P. (2000) Biosynthesis of heparin/heparan sulfate: kinetic studies of the glucuronyl C5-epimerase with N-sulfated derivatives of the Escherichia coli K5 capsular polysaccharide as substrates. Glycobiology 10, 159–171 [DOI] [PubMed] [Google Scholar]
- 7. Feyerabend T. B., Li J. P., Lindahl U., Rodewald H. R. (2006) Heparan sulfate C5-epimerase is essential for heparin biosynthesis in mast cells. Nat. Chem. Biol. 2, 195–196 [DOI] [PubMed] [Google Scholar]
- 8. Li J., Hagner-McWhirter A., Kjellén L., Palgi J., Jalkanen M., Lindahl U. (1997) Biosynthesis of heparin/heparan sulfate. cDNA cloning and expression of d-glucuronyl C5-epimerase from bovine lung. J. Biol. Chem. 272, 28158–28163 [DOI] [PubMed] [Google Scholar]
- 9. Campbell P., Hannesson H. H., Sandbäck D., Rodén L., Lindahl U., Li J. P. (1994) Biosynthesis of heparin/heparan sulfate. Purification of the d-glucuronyl C5-epimerase from bovine liver. J. Biol. Chem. 269, 26953–26958 [PubMed] [Google Scholar]
- 10. Li K., Bethea H. N., Liu J. (2010) Using engineered 2-O-sulfotransferase to determine the activity of heparan sulfate C5-epimerase and its mutants. J. Biol. Chem. 285, 11106–11113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Babu P., Victor X. V., Nelsen E., Nguyen T. K., Raman K., Kuberan B. (2011) Hydrogen/deuterium exchange-LC-MS approach to characterize the action of heparan sulfate C5-epimerase. Anal. Bioanal. Chem. 401, 237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen J., Jones C. L., Liu J. (2007) Using an enzymatic combinatorial approach to identify anticoagulant heparan sulfate structures. Chem. Biol. 14, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kobayashi M., Sugumaran G., Liu J., Shworak N. W., Silbert J. E., Rosenberg R. D. (1999) Molecular cloning and characterization of a human uronyl 2-sulfotransferase that sulfates iduronyl and glucuronyl residues in dermatan/chondroitin sulfate. J. Biol. Chem. 274, 10474–10480 [DOI] [PubMed] [Google Scholar]
- 14. Bethea H. N., Xu D., Liu J., Pedersen L. C. (2008) Redirecting the substrate specificity of heparan sulfate 2-O-sulfotransferase by structurally guided mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 18724–18729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu Y., Wang Z., Liu R., Bridges A. S., Huang X., Liu J. (2012) Directing the biological activities of heparan sulfate oligosaccharides using a chemoenzymatic approach. Glycobiology 22, 96–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu R., Xu Y., Chen M., Weïwer M., Zhou X., Bridges A. S., DeAngelis P. L., Zhang Q., Linhardt R. J., Liu J. (2010) Chemoenzymatic design of heparan sulfate oligosaccharides. J. Biol. Chem. 285, 34240–34249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao G., Guan W., Cai L., Wang P. G. (2010) Enzymatic route to preparative-scale synthesis of UDP-GlcNAc/GalNAc, their analogues and GDP-fucose. Nat. Protoc. 5, 636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou X., Chandarajoti K., Pham T. Q., Liu R., Liu J. (2011) Expression of heparan sulfate sulfotransferases in Kluyveromyces lactis and preparation of 3'-phosphoadenosine-5'-phosphosulfate. Glycobiology 21, 771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guerrini M., Guglieri S., Casu B., Torri G., Mourier P., Boudier C., Viskov C. (2008) Antithrombin-binding octasaccharides and role of extensions of the active pentasaccharide sequence in the specificity and strength of interaction. Evidence for very high affinity induced by an unusual glucuronic acid residue. J. Biol. Chem. 283, 26662–26675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Esko J. D., Selleck S. B. (2002) Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Ann. Rev. Biochem. 71, 435–471 [DOI] [PubMed] [Google Scholar]
- 21. Das S. K., Mallet J. M., Esnault J., Driguez P. A., Duchaussoy P., Sizun P., Herault J. P., Herbert J. M., Petitou M., Sinaÿ P. (2001) Synthesis of conformationally locked l-iduronic acid derivatives: Direct evidence for a critical role of the skew-boat 2S0 conformer in the activation of antithrombin by heparin. Chemistry 7, 4821–4834 [DOI] [PubMed] [Google Scholar]
- 22. Kreuger J., Spillmann D., Li J. P., Lindahl U. (2006) Interactions between heparan sulfate and proteins: The concept of specificity. J. Cell Biol. 174, 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu D., Moon A. F., Song D., Pedersen L. C., Liu J. (2008) Engineering sulfotransferases to modify heparan sulfate. Nat. Chem. Biol. 4, 200–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sheng J., Liu R., Xu Y., Liu J. (2011) The dominating role of N-deacetylase/N-sulfotransferase 1 in forming domain structures in heparan sulfate. J. Biol. Chem. 286, 19768–19776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Domon B., Costello C. E. (1988) A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 5, 397–409 [Google Scholar]